EGFR/ErbB Inhibition Promotes OPC Maturation up to Axon Engagement by Co-Regulating PIP2 and MBP

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Compound Treatments
2.3. Gene Silencing and Transfection
2.4. Crude Extract Preparation and Immunoblot Analysis
2.5. Quantitative Immunofluorescence Analysis and Confocal Microscopy
2.6. Evaluation of Cell Engagement on PS Microfibers
2.7. Total RNA Extraction and qPCR
2.8. Bioinformatics and Statistical Methods
3. Results
3.1. Gefitinib Promotes MBP Expression and Oli-neuM Differentiation in Myelinating OLs
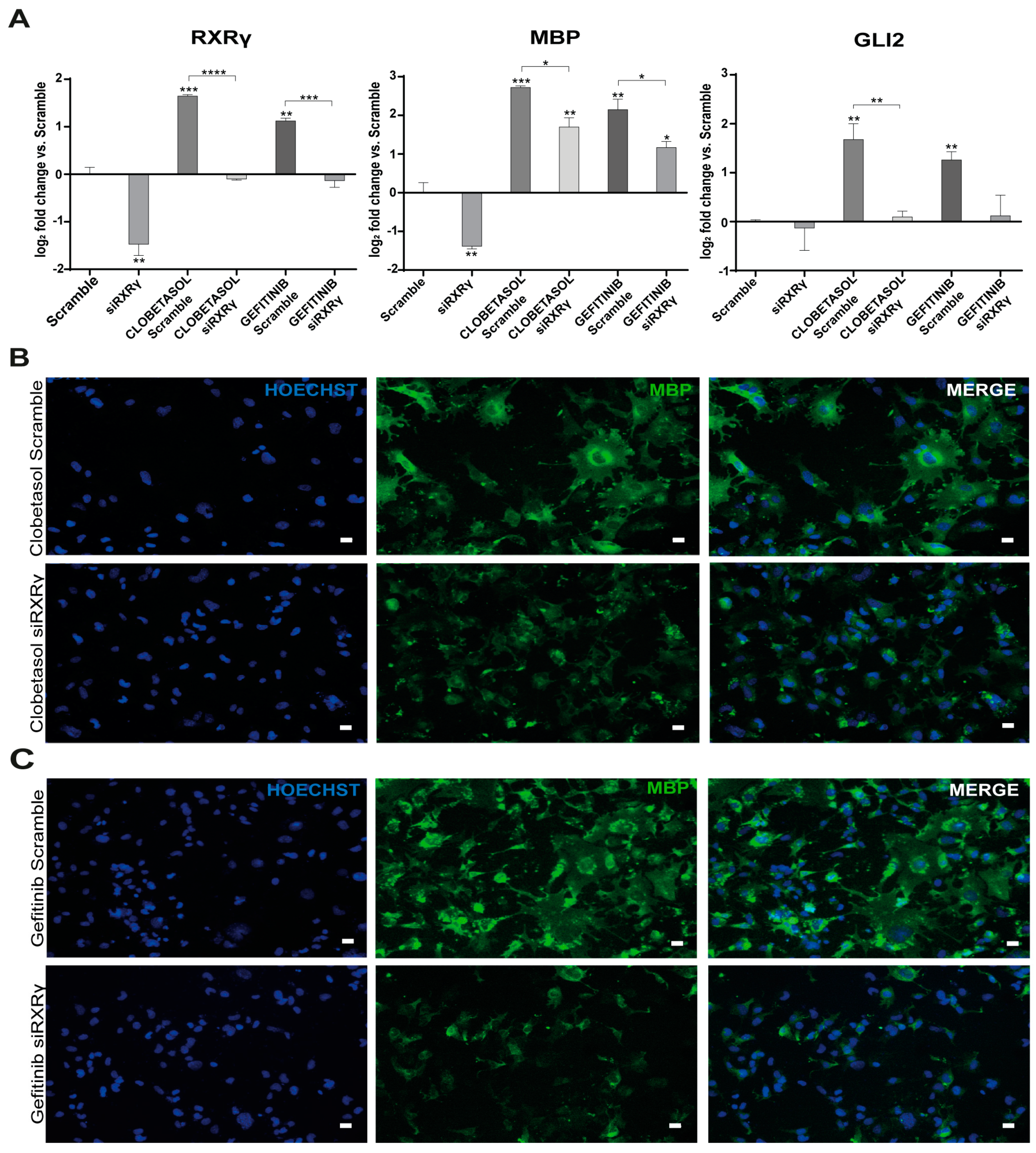
3.2. Gefitinib and Clobetasol Treatment Upregulate RXRγ and Gli2 Expression
3.3. EGFR/ErbB Inhibition Promotes MBP Expression and Gefitinib or siEGFR Silencing Combined with Clobetasol Treatment Potently Stimulates MBP Expression and Oli-neuM Differentiation
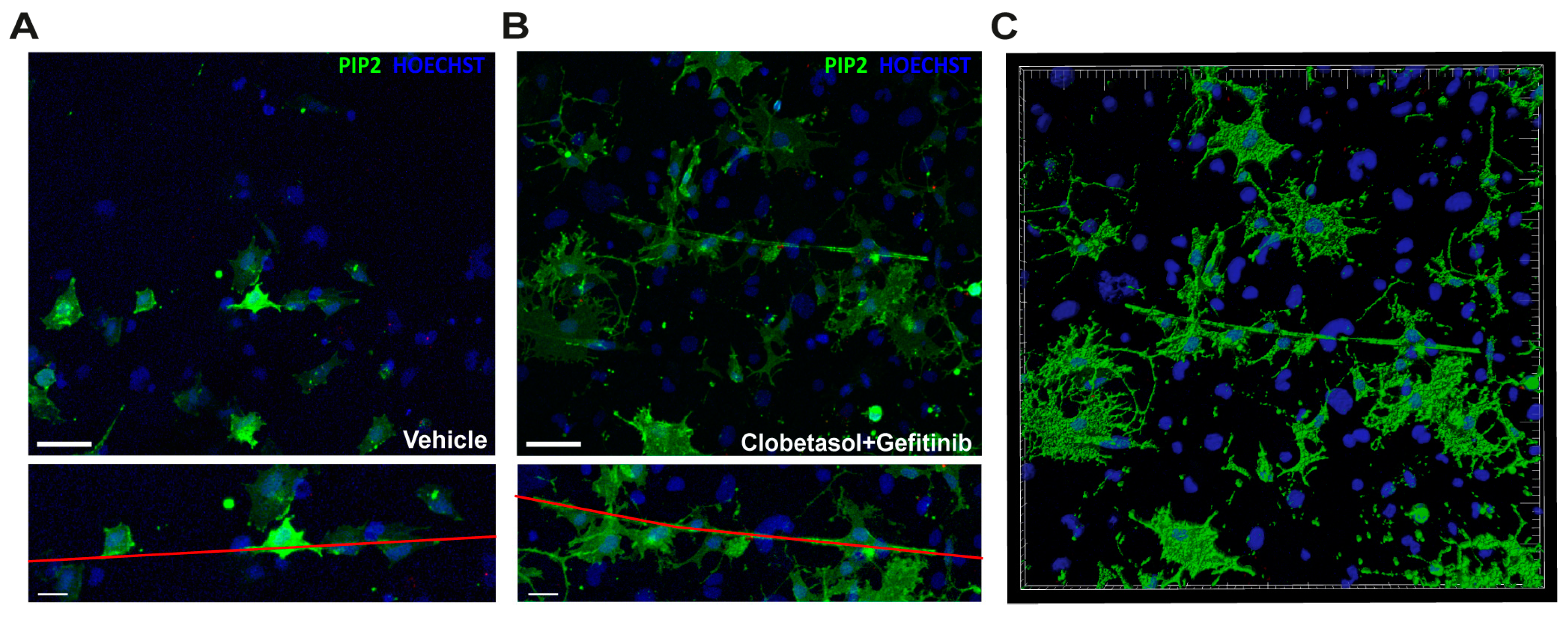
3.4. Clobetasol and Gefitinib Co-Treatment Enhances Oli-neuM Differentiation by Co-Regulating PIP2 Availability with MBP Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plemel, J.R.; Liu, W.Q.; Yong, V.W. Remyelination Therapies: A New Direction and Challenge in Multiple Sclerosis. Nat. Rev. Drug Discov. 2017, 16, 617–634. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Nanescu, S.E.; Psachoulia, K.; Huang, J.K. Oligodendrocyte Regeneration: Its Significance in Myelin Replacement and Neuroprotection in Multiple Sclerosis. Neuropharmacology 2016, 110, 633–643. [Google Scholar] [CrossRef]
- Lopez Juarez, A.; He, D.; Richard Lu, Q. Oligodendrocyte Progenitor Programming and Reprogramming: Toward Myelin Regeneration. Brain Res. 2016, 1638, 209–220. [Google Scholar] [CrossRef]
- Villoslada, P. Neuroprotective Therapies for Multiple Sclerosis and Other Demyelinating Diseases. Mult. Scler. Demyelinating Disord. 2016, 1, 1. [Google Scholar] [CrossRef]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A Regenerative Approach to the Treatment of Multiple Sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef]
- Lariosa-Willingham, K.D.; Rosler, E.S.; Tung, J.S.; Dugas, J.C.; Collins, T.L.; Leonoudakis, D. A High Throughput Drug Screening Assay to Identify Compounds That Promote Oligodendrocyte Differentiation Using Acutely Dissociated and Purified Oligodendrocyte Precursor Cells. BMC Res. Notes 2016, 9. [Google Scholar] [CrossRef]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar Arrays as a High-Throughput Screening Platform for Therapeutics in Multiple Sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef]
- Porcu, G.; Serone, E.; De Nardis, V.; Di Giandomenico, D.; Lucisano, G.; Scardapane, M.; Poma, A.; Ragnini-Wilson, A. Clobetasol and Halcinonide Act as Smoothened Agonists to Promote Myelin Gene Expression and RxRγ Receptor Activation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-Based Modulation of Endogenous Stem Cells Promotes Functional Remyelination in Vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef]
- Patel, S.S.; Tomar, S.; Sharma, D.; Mahindroo, N.; Udayabanu, M. Targeting Sonic Hedgehog Signaling in Neurological Disorders. Neurosci. Biobehav. Rev. 2017, 74, 76–97. [Google Scholar] [CrossRef]
- Zuccaro, E.; Arlotta, P. The Quest for Myelin in the Adult Brain. Nat. Cell Biol. 2013, 15, 572–575. [Google Scholar] [CrossRef]
- Ferent, J.; Zimmer, C.; Durbec, P.; Ruat, M.; Traiffort, E. Sonic Hedgehog Signaling Is a Positive Oligodendrocyte Regulator during Demyelination. J. Neurosci. 2013, 33, 1759–1772. [Google Scholar] [CrossRef]
- Ferent, J.; Cochard, L.; Faure, H.; Taddei, M.; Hahn, H.; Ruat, M.; Traiffort, E. Genetic Activation of Hedgehog Signaling Unbalances the Rate of Neural Stem Cell Renewal by Increasing Symmetric Divisions. Stem Cell Rep. 2014, 3, 312–323. [Google Scholar] [CrossRef]
- Samanta, J.; Salzer, J.L. Myelination: Actin Disassembly Leads the Way. Dev. Cell 2015, 34, 129–130. [Google Scholar] [CrossRef]
- Del Giovane, A.; Ragnini-Wilson, A. Targeting Smoothened as a New Frontier in the Functional Recovery of Central Nervous System Demyelinating Pathologies. Int. J. Mol. Sci. 2018, 19, 3677. [Google Scholar] [CrossRef]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA Interaction Is a Druggable Target for Hedgehog-dependent Tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef]
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 Mobilizes Endogenous Neural Stem Cells for Remyelination. Nature 2015, 526, 448–452. [Google Scholar] [CrossRef]
- Wang, L.C.; Almazan, G. Cdon, a Cell Surface Protein, Mediates Oligodendrocyte Differentiation and Myelination. Glia 2016, 64, 1021–1033. [Google Scholar] [CrossRef]
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin Gene Regulatory Factor Is a Critical Transcriptional Regulator Required for CNS Myelination. Cell 2009, 138, 172–185. [Google Scholar] [CrossRef]
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor Skill Learning Requires Active Central Myelination. Science 2014, 346, 318–322. [Google Scholar] [CrossRef]
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.W.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin Regulatory Factor Drives Remyelination in Multiple Sclerosis. Acta Neuropathol. 2017, 134, 403–422. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Kippert, A.; Saab, A.S.; Werner, H.B.; Lang, T.; Nave, K.-A.; Simons, M. Phosphatidylinositol 4,5-Bisphosphate-Dependent Interaction of Myelin Basic Protein with the Plasma Membrane in Oligodendroglial Cells and Its Rapid Perturbation by Elevated Calcium. J. Neurosci. 2009, 29, 4794–4807. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Sánchez, P.; Schmitt, S.; Snaidero, N.; Mitkovski, M.; Velte, C.; Bruckner, B.R.; Alexopoulos, I.; Czopka, T.; Rhee, J.S.; et al. Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Dev. Cell 2015, 34, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Fu, M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS Myelin Wrapping Is Driven by Actin Disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Bechler, M.E.; Byrne, L.; ffrench-Constant, C. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. 2015, 25, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Hoyos, D.; Jagielska, A.; Homan, K.A.; Du, H.; Busbee, T.; Anderson, D.G.; Fang, N.X.; Lewis, J.A.; Vliet, K.J.V. Engineered 3D-Printed Artificial Axons. Sci. Rep. 2018, 8, 478. [Google Scholar] [CrossRef] [PubMed]
- Káradóttir, R.T.; Stockley, J.H. Deconstructing Myelination: It All Comes down to Size. Nat. Methods 2012, 9, 883–884. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Urbanski, M.M.; Melendez-Vasquez, C.V. Preparation of Matrices of Variable Stiffness for the Study of Mechanotransduction in Schwann Cell Development. Methods Mol. Biol. 2018, 1739, 281–297. [Google Scholar] [CrossRef]
- Chang, K.-J.; Redmond, S.A.; Chan, J.R. Remodeling Myelination: Implications for Mechanisms of Neural Plasticity. Nat. Neurosci. 2016, 19, 190–197. [Google Scholar] [CrossRef]
- Yao, X.; Su, T.; Verkman, A.S. Clobetasol Promotes Remyelination in a Mouse Model of Neuromyelitis Optica. Acta Neuropathol. Commun. 2016, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, J.; Bond, M.C.; Chen, M.; Ren, X.-R.; Lyerly, H.K.; Barak, L.S.; Chen, W. Identification of Select Glucocorticoids as Smoothened Agonists: Potential Utility for Regenerative Medicine. Proc. Natl. Acad. Sci. USA 2010, 107, 9323–9328. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Rai, A.K.; Kaushik, V.; Brünnert, D.; Chahar, K.R.; Pandey, J.; Goyal, P. Identification of Gefitinib Off-Targets Using a Structure-Based Systems Biology Approach; Their Validation with Reverse Docking and Retrospective Data Mining. Sci. Rep. 2016, 6, 33949. [Google Scholar] [CrossRef] [PubMed]
- Joubert, L.; Foucault, I.; Sagot, Y.; Bernasconi, L.; Duval, F.; Alliod, C.; Frossard, M.-J.; Gobert, R.P.; Curchod, M.-L.; Salvat, C.; et al. Chemical Inducers and Transcriptional Markers of Oligodendrocyte Differentiation. J. Neurosci. Res. 2010, 88, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Koprivica, V.; Cho, K.-S.; Park, J.B.; Yiu, G.; Atwal, J.; Gore, B.; Kim, J.A.; Lin, E.; Tessier-Lavigne, M.; Chen, D.F.; et al. EGFR Activation Mediates Inhibition of Axon Regeneration by Myelin and Chondroitin Sulfate Proteoglycans. Science 2005, 310, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Starossom, S.C.; Campo Garcia, J.; Woelfle, T.; Romero-Suarez, S.; Olah, M.; Watanabe, F.; Cao, L.; Yeste, A.; Tukker, J.J.; Quintana, F.J.; et al. Chi3l3 Induces Oligodendrogenesis in an Experimental Model of Autoimmune Neuroinflammation. Nat. Commun. 2019, 10, 217. [Google Scholar] [CrossRef]
- Amir-Levy, Y.; Mausner-Fainberg, K.; Karni, A. Treatment with Anti-EGF Ab Ameliorates Experimental Autoimmune Encephalomyelitis via Induction of Neurogenesis and Oligodendrogenesis. Mult. Scler. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Gonzalez-Perez, O.; Quiñones-Hinojosa, A. Dose-Dependent Effect of EGF on Migration and Differentiation of Adult Subventricular Zone Astrocytes. Glia 2010, 58, 975–983. [Google Scholar] [CrossRef]
- Gonzalez-Perez, O.; Alvarez-Buylla, A. Oligodendrogenesis in the Subventricular Zone and the Role of Epidermal Growth Factor. Brain Res. Rev. 2011, 67, 147–156. [Google Scholar] [CrossRef]
- Abliz, A.; Deng, W.; Sun, R.; Guo, W.; Zhao, L.; Wang, W. Wortmannin, PI3K/AKT signaling pathway inhibitor, attenuates thyroid injury associated with severe acute pancreatitis in rats. Int. J. Clin. Exp. Pathol. 2015, 8, 13821–13833. [Google Scholar]
- Cantrell, W.; Huang, Y.; Menchaca, A.A.; Kulik, G.; Welker, M.E. Synthesis of PI3 Kinase Inhibition activity of a Wortmannin-Leucine Derivative. Molecules 2018, 23, 1792. [Google Scholar] [CrossRef]
- Shin, M.; Yang, E.G.; Song, H.K.; Jeon, H. Insulin activates EGFR by stimulating its interaction with IGF-1R in low-EGFR-expressing TNBC cells. Sci. Technol. 2006, 48, 136–791. [Google Scholar] [CrossRef] [PubMed]
- Szentpetery, Z.; Balla, A.; Kim, Y.J.; Lemmon, M.A.; Balla, T. Live cell imaging with protein domains capable of recognizing phosphatidylinositol 4,5-bisphosphate; a comparative study. BMC Cell Biol. 2009, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Porcu, G.; Parsons, A.B.; Di Giandomenico, D.; Lucisano, G.; Mosca, M.G.; Boone, C.; Ragnini-Wilson, A. Combined P21-Activated Kinase and Farnesyltransferase Inhibitor Treatment Exhibits Enhanced Anti-Proliferative Activity on Melanoma, Colon and Lung Cancer Cell Lines. Mol. Cancer 2013, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Gherardini, P.F.; Paoluzi, S.; Saez-Rodriguez, J.; Helmer-Citterich, M.; Ragnini-Wilson, A.; Castagnoli, L.; Cesareni, G. Mapping the Human Phosphatome on Growth Pathways. Mol. Syst. Biol. 2012, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Hubler, Z.; Allimuthu, D.; Bederman, I.; Elitt, M.S.; Madhavan, M.; Allan, K.C.; Shick, H.E.; Garrison, E.; Karl, M.T.; Factor, D.C.; et al. Accumulation of 8,9-Unsaturated Sterols Drives Oligodendrocyte Formation and Remyelination. Nature 2018, 560, 372. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Anti-inflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- White, R.; Gonsior, C.; Krämer-Albers, E.-M.; Stöhr, N.; Hüttelmaier, S.; Trotter, J. Activation of Oligodendroglial Fyn Kinase Enhances Translation of MRNAs Transported in HnRNP A2–Dependent RNA Granules. J. Cell Biol. 2008, 181, 579–586. [Google Scholar] [CrossRef]
- White, R.; Krämer-Albers, E.-M. Axon-Glia Interaction and Membrane Traffic in Myelin Formation. Front. Cell Neurosci. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Emery, B.; Lu, Q.R. Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 2015, 7, a020461. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kelland, E.E.; hong Kim, J.; Lund, B.T.; Chang, X.; Wang, K.; Weiner, L.P. The influence of retinoic acid on the human oligodendrocyte precursor cells by RNA-sequencing. Biochem. Biophys. Rep. 2017, 9, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Jarjour, A.A.; Nait Oumesmar, B.; Kerninon, C.; Williams, A.; Krezel, W.; Kagechika, H.; Bauer, J.; Zhao, C.; Baron-Van Evercooren, A.; et al. Retinoid X Receptor Gamma Signaling Accelerates CNS Remyelination. Nat. Neurosci. 2011, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Giner, X.C.; Cotnoir-White, D.; Mader, S.; Lévesque, D. Selective Ligand Activity at Nur/Retinoid X Receptor Complexes Revealed by Dimer-Specific Bioluminescence Resonance Energy Transfer-Based Sensors. FASEB J. 2015, 29, 4256–4267. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Mangelberger, D.; Kern, D.; Loipetzberger, A.; Eberl, M.; Aberger, F. Cooperative Hedgehog-EGFR Signaling. Front. Biosci. (Landmark Ed) 2012, 17, 90–99. [Google Scholar] [CrossRef]
- Riobo, N.A.; Saucy, B.; DiLizio, C.; Manning, D.R. Activation of Heterotrimeric G Proteins by Smoothened. Proc. Natl. Acad. Sci. USA 2006, 103, 12607–12612. [Google Scholar] [CrossRef]
- Illiano, M.; Sapio, L.; Salzillo, A.; Capasso, L.; Caiafa, I.; Chiosi, E.; Spina, A.; Naviglio, S. Forskolin Improves Sensitivity to Doxorubicin of Triple Negative Breast Cancer Cells via Protein Kinase A-Mediated ERK1/2 Inhibition. Biochem. Pharmacol. 2018, 152, 104–113. [Google Scholar] [CrossRef]
- Michailidis, I.E.; Rusinova, R.; Georgakopoulos, A.; Chen, Y.; Iyengar, R.; Robakis, N.K.; Logothetis, D.E.; Baki, L. Phosphatidylinositol-4,5-Bisphosphate Regulates Epidermal Growth Factor Receptor Activation. Pflug. Arch. Eur. J. Physiol. 2011, 461, 387–397. [Google Scholar] [CrossRef]
- Bissonnette, R.P.; Fan, B.; Roegner, K.; Ng, S.; Corpuz, M.; Prudente, R. Cooperative antitumor activity between the retinoid X receptor (RXR)-selective agonist bexarotene and EGFR-tyrosine kinase inhibitors in preclinical models of NSCLC. Int. J. Clin. Oncol. 2006, 18, 17073. [Google Scholar]
- Gianni, M.; Kopf, E.; Bastien, J.; Oulad-Abdelghani, M.; Garattini, E.; Chambon, P.; Rochette-Egly, C. Down-Regulation of the Phosphatidylinositol 3-Kinase/Akt Pathway Is Involved in Retinoic Acid-Induced Phosphorylation, Degradation, and Transcriptional Activity of Retinoic Acid Receptor Gamma 2. J. Biol. Chem. 2002, 277, 24859–24862. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, A.G.; Errea, O.; van Wijngaarden, P.; Gonzalez, G.A.; Kerninon, C.; Jarjour, A.A.; Lewis, J.L.; Jones, C.A.; Nait-Oumesmar, B.; Zhao, C.; et al. Vitamin D receptor–retinoid X receptor heterodimer signaling regulates oligodendrocyte progenitor cell differentiation. J. Cell Biol. 2015, 211, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C. ErbB receptors and the development of the nervous system. Exp. Cell Res. 2009, 315, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., II. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rico, B.; Marín, O. Neuregulin signaling, cortical circuitry development and schizophrenia. Curr. Opin. Genet. Dev. 2011, 21, 262–270. [Google Scholar] [CrossRef]
- Cruz-Rangel, S.; Hernández-Benítez, R.; Vázquez-Juárez, E.; López-Dominguez, A.; Pasantes-Morales, H. Potentiation by Thrombin of Hyposmotic Glutamate and Taurine Efflux from Cultured Astrocytes: Signalling Chains. Neurochem. Res. 2008, 33, 1518–1524. [Google Scholar] [CrossRef]
- Beyer, B.A.; Fang, M.; Sadrian, B.; Montenegro-Burke, J.R.; Plaisted, W.C.; Kok, B.P.C.; Saez, E.; Kondo, T.; Siuzdak, G.; Lairson, L.L. Metabolomics-based discovery of a metabolite that enhances oligodendrocyte maturation. Nat. Chem. Biol. 2018, 14, 22–28. [Google Scholar] [CrossRef]
- Fidler, M.J.; Basu, S.; Buckingham, L.; Walters, K.; McCormack, S.; Batus, M.; Coon, J.; Bonomi, P. Utility of Insulin-like Growth Factor Receptor-1 Expression in Gefitinib-Treated Patients with Non-Small Cell Lung Cancer. Anticancer Res. 2012, 32, 1705–1710. [Google Scholar]
- Shi, D.; Lv, X.; Zhang, Z.; Yang, X.; Zhou, Z.; Zhang, L.; Zhao, Y. Smoothened oligomerization/higher order clustering in lipid rafts is essential for high Hedgehog activity transduction. J. Biol. Chem. 2013, 288, 12605–12614. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nocita, E.; Del Giovane, A.; Tiberi, M.; Boccuni, L.; Fiorelli, D.; Sposato, C.; Romano, E.; Basoli, F.; Trombetta, M.; Rainer, A.; et al. EGFR/ErbB Inhibition Promotes OPC Maturation up to Axon Engagement by Co-Regulating PIP2 and MBP. Cells 2019, 8, 844. https://doi.org/10.3390/cells8080844
Nocita E, Del Giovane A, Tiberi M, Boccuni L, Fiorelli D, Sposato C, Romano E, Basoli F, Trombetta M, Rainer A, et al. EGFR/ErbB Inhibition Promotes OPC Maturation up to Axon Engagement by Co-Regulating PIP2 and MBP. Cells. 2019; 8(8):844. https://doi.org/10.3390/cells8080844
Chicago/Turabian StyleNocita, Emanuela, Alice Del Giovane, Marta Tiberi, Laura Boccuni, Denise Fiorelli, Carola Sposato, Elena Romano, Francesco Basoli, Marcella Trombetta, Alberto Rainer, and et al. 2019. "EGFR/ErbB Inhibition Promotes OPC Maturation up to Axon Engagement by Co-Regulating PIP2 and MBP" Cells 8, no. 8: 844. https://doi.org/10.3390/cells8080844
APA StyleNocita, E., Del Giovane, A., Tiberi, M., Boccuni, L., Fiorelli, D., Sposato, C., Romano, E., Basoli, F., Trombetta, M., Rainer, A., Traversa, E., & Ragnini-Wilson, A. (2019). EGFR/ErbB Inhibition Promotes OPC Maturation up to Axon Engagement by Co-Regulating PIP2 and MBP. Cells, 8(8), 844. https://doi.org/10.3390/cells8080844