RhoGTPase in Vascular Disease


Abstract
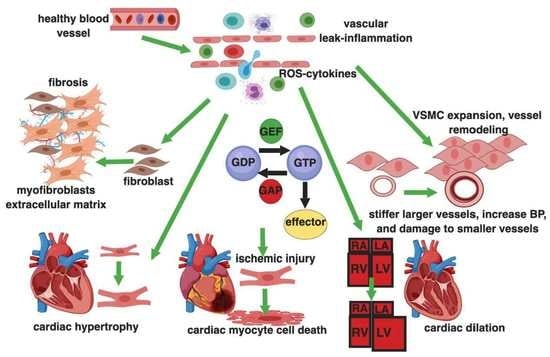
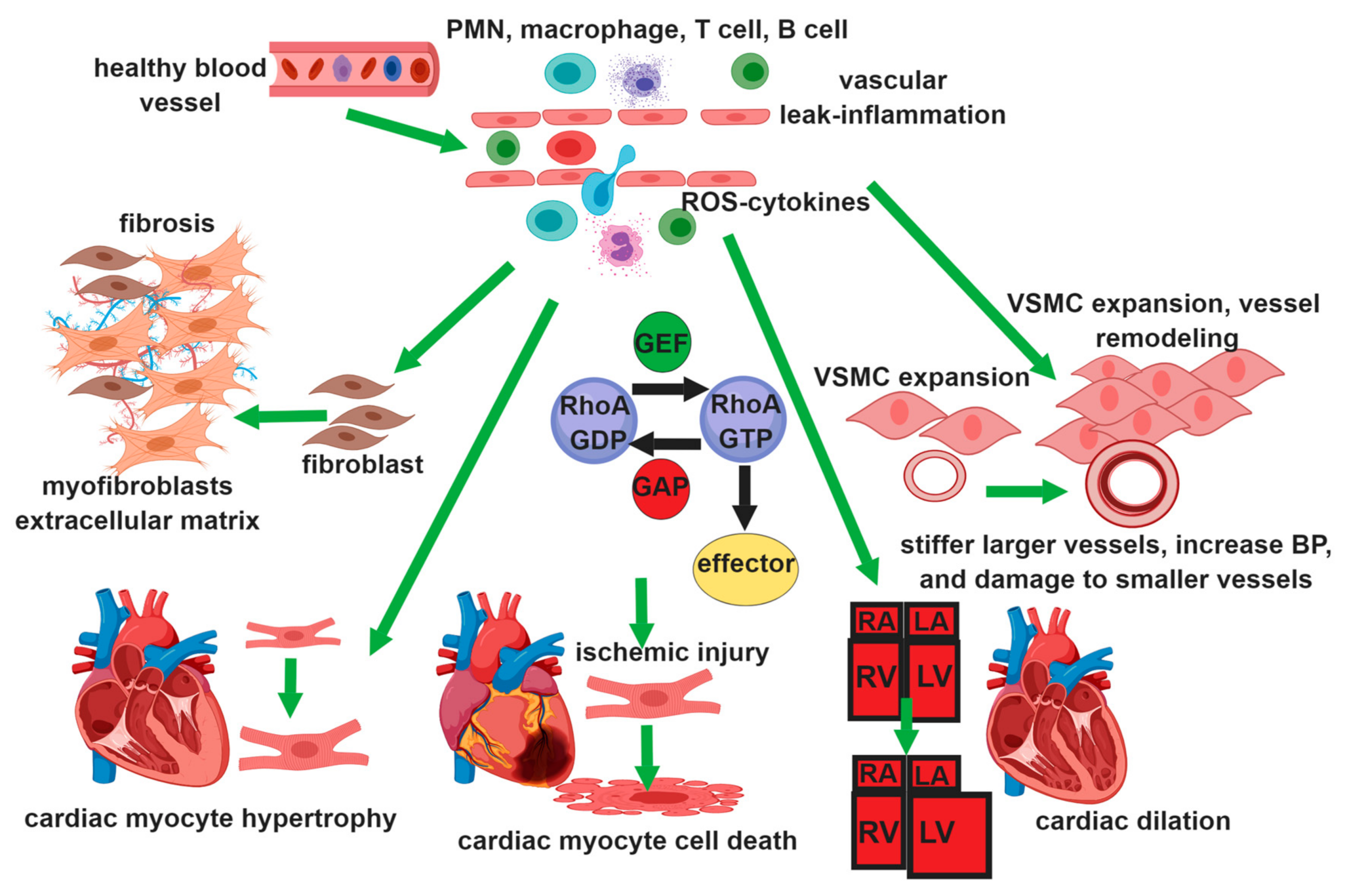
1. Introduction
2. Endothelial Cells
2.1. Role of RhoGTPase in Endothelial Barrier Function
2.2. RhoGTPase in Venous Endothelial Dysfunction
2.3. Role of RhoGTPase in Vascular Inflammation
3. Smooth Muscle Cells
4. Fibroblasts
4.1. Role of RhoGTPase in Vascular Oxidative Stress
4.2. RhoGTPase in Heart and Lung
4.3. Clinical Studies on the Role of Rho GTPase in Vascular Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Duluc, L.; Wojciak-Stothard, B. Rho GTPases in the regulation of pulmonary vascular barrier function. Cell Tissue Res. 2014, 355, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, J.N.; Lucas, R.; Verin, A.D. The Acute Respiratory Distress Syndrome: Mechanisms and Perspective Therapeutic Approaches. Austin J. Vasc. Med. 2015, 2. [Google Scholar]
- Marinkovic, G.; Heemskerk, N.; van Buul, J.D.; de Waard, V. The Ins and Outs of Small GTPase Rac1 in the Vasculature. J. Pharmacol. Exp. Ther. 2015, 354, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Romero, M.J.; Toque, H.A.; Yang, G.; Caldwell, R.B.; Caldwell, R.W. The role of RhoA/Rho kinase pathway in endothelial dysfunction. J. Cardiovasc. Dis. Res. 2010, 1, 165–170. [Google Scholar] [PubMed]
- Ba, W.; Nadif Kasri, N. RhoGTPases at the synapse: An embarrassment of choice. Small GTPases 2017, 8, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Wennerberg, K.; Burridge, K. Serine phosphorylation negatively regulates RhoA in vivo. J. Biol. Chem. 2003, 278, 19023–19031. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell. Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Storck, E.M.; Wojciak-Stothard, B. Rho GTPases in pulmonary vascular dysfunction. Vascul. Pharmacol. 2013, 58, 202–210. [Google Scholar] [CrossRef]
- Shimokawa, H. Rho-kinase as a novel therapeutic target in treatment of cardiovascular diseases. J. Cardiovasc. Pharmacol. 2002, 39, 319–327. [Google Scholar] [CrossRef]
- Shimokawa, H.; Takeshita, A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Loirand, G.; Pacaud, P. Involvement of Rho GTPases and their regulators in the pathogenesis of hypertension. Small GTPase. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bei, Y.; Duong-Quy, S.; Hua-Huy, T.; Dao, P.; Le-Dong, N.N.; Dinh-Xuan, A.T. Activation of RhoA/Rho-kinase pathway accounts for pulmonary endothelial dysfunction in patients with chronic obstructive pulmonary disease. Physiol. Rep. 2013, 1, e00105. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Wittchen, E.S.; Doerschuk, C.M.; Burridge, K. A Rnd3/p190RhoGAP pathway regulates RhoA activity in idiopathic pulmonary fibrosis fibroblasts. Mol. Biol. Cell 2018, 29, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Matsusue, K.; Misawa, M. RhoA, a possible target for treatment of airway hyperresponsiveness in bronchial asthma. J. Pharmacol. Sci. 2010, 114, 239–247. [Google Scholar] [CrossRef]
- Cui, Q.Q.; Feng, X.B.; Wang, Y.S.; Zhao, L. Expression of RhoA in the lung tissue of acute lung injury rats and the influence of RhoA on the expression of IL-8 and IL-10. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2011, 27, 1308–1311. [Google Scholar]
- Miyamoto, S.; Del Re, D.P.; Xiang, S.Y.; Zhao, X.; Florholmen, G.; Brown, J.H. Revisited and revised: Is RhoA always a villain in cardiac pathophysiology? J. Cardiovasc. Transl. Res. 2010, 3, 330–343. [Google Scholar] [CrossRef]
- Zhou, Q.; Liao, J.K. Rho kinase: An important mediator of atherosclerosis and vascular disease. Curr. Pharm. Des. 2009, 15, 3108–3115. [Google Scholar] [CrossRef]
- Loirand, G.; Rolli-Derkinderen, M.; Pacaud, P. RhoA and resistance artery remodeling. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1051–H1056. [Google Scholar] [CrossRef]
- Aghajanian, A.; Wittchen, E.S.; Campbell, S.L.; Burridge, K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS ONE 2009, 4, e8045. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.M.; Brown, J.H. G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol. Pharmacol. 2015, 88, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Sward, K.; Mita, M.; Wilson, D.P.; Deng, J.T.; Susnjar, M.; Walsh, M.P. The role of RhoA and Rho-associated kinase in vascular smooth muscle contraction. Curr. Hypertens. Rep. 2003, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Li, D.B.; Yang, G.J.; Xu, H.W.; Fu, Z.X.; Wang, S.W.; Hu, S.J. Regulation on RhoA in vascular smooth muscle cells under inflammatory stimulation proposes a novel mechanism mediating the multiple-beneficial action of acetylsalicylic acid. Inflammation 2013, 36, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Dong, Z.; Han, W.; Kondrikov, D.; Su, Y. The role of RhoA and cytoskeleton in myofibroblast transformation in hyperoxic lung fibrosis. Free Radic. Biol. Med. 2013, 61, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef] [PubMed]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef]
- Birukov, K.G. Small GTPases in mechanosensitive regulation of endothelial barrier. Microvasc. Res. 2009, 77, 46–52. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. (Oxf.) 2018, 222. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M.; Garcia Ponce, A.; Vadillo, E.; Pelayo, R.; Rossaint, J.; Zarbock, A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. 2017, 74, 1985–1997. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, G.M.; Zhu, Y.; Peng, X.Y.; Li, T.; Liu, L.M. Role of connexin 43 in vascular hyperpermeability and relationship to Rock1-MLC20 pathway in septic rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1323–L1332. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Mehta, D.; Malik, A.B. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium. 2016, 60, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Brinks, H.L.; Eckhart, A.D. Regulation of GPCR signaling in hypertension. Biochim. Biophys. Acta 2010, 1802, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.D.; Kaiser, M.A.; Ghaderi Najafabadi, M.; Koplev, S.; Zhao, Y.; Douglas, G.; Kyriakou, T.; Andrews, S.; Rajmohan, R.; Watkins, H.; et al. JCAD, a Gene at the 10p11 Coronary Artery Disease Locus, Regulates Hippo Signaling in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Nunes, K.P.; Rigsby, C.S.; Webb, R.C. RhoA/Rho-kinase and vascular diseases: What is the link? Cell Mol. Life Sci. 2010, 67, 3823–3836. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Leiper, J. Rho GTPases and hypoxia in pulmonary vascular endothelial cells. Methods Enzymol. 2008, 439, 267–283. [Google Scholar] [PubMed]
- Bryant, A.J.; Scott, E.W. “A small leak will sink a great ship”: Hypoxia-inducible factor and group III pulmonary hypertension. Receptors Clin. Investig. 2016, 3, e1213. [Google Scholar]
- Cercone, M.A.; Schroeder, W.; Schomberg, S.; Carpenter, T.C. EphA2 receptor mediates increased vascular permeability in lung injury due to viral infection and hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L856–L863. [Google Scholar] [CrossRef]
- Irwin, D.C.; McCord, J.M.; Nozik-Grayck, E.; Beckly, G.; Foreman, B.; Sullivan, T.; White, M.; Crossno, T.J.J.; Bailey, D.; Flores, S.C.; et al. A potential role for reactive oxygen species and the HIF-1alpha-VEGF pathway in hypoxia-induced pulmonary vascular leak. Free Radic. Biol. Med. 2009, 47, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Morote-Garcia, J.C.; Rosenberger, P.; Kuhlicke, J.; Eltzschig, H.K. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 2008, 111, 5571–5580. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Faigle, M.; Grenz, A.; Laucher, S.; Thompson, L.F.; Eltzschig, H.K. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 2008, 111, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Woodward, H.N.; Anwar, A.; Riddle, S.; Taraseviciene-Stewart, L.; Fragoso, M.; Stenmark, K.R.; Gerasimovskaya, E.V. PI3K, Rho, and ROCK play a key role in hypoxia-induced ATP release and ATP-stimulated angiogenic responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L954–L964. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.; Collado, A.; Escudero, P.; Rius, C.; Gonzalez, C.; Servera, E.; Piqueras, L.; Sanz, M.J. Cigarette Smoke Increases Endothelial CXCL16-Leukocyte CXCR6 Adhesion In Vitro and In Vivo. Potential Consequences in Chronic Obstructive Pulmonary Disease. Front Immunol. 2017, 8, 1766. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Zebda, N.; Oskolkova, O.; Afonyushkin, T.; Berdyshev, E.; Tian, Y.; Meng, F.; Sarich, N.; Bochkov, V.N.; Wang, J.M.; et al. Anti-Inflammatory Effects of OxPAPC Involve Endothelial Cell-Mediated Generation of LXA4. Circ. Res. 2017, 121, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Meliton, A.; Meng, F.; Tian, Y.; Shah, A.A.; Birukova, A.A.; Birukov, K.G. Role of Krev Interaction Trapped-1 in Prostacyclin-Induced Protection against Lung Vascular Permeability Induced by Excessive Mechanical Forces and Thrombin Receptor Activating Peptide 6. Am. J. Respir. Cell Mol. Biol. 2015, 53, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Zagranichnaya, T.; Alekseeva, E.; Bokoch, G.M.; Birukov, K.G. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J. Cell Physiol. 2008, 215, 715–724. [Google Scholar] [CrossRef]
- Rikitake, Y.; Liao, J.K. Rho GTPases, statins, and nitric oxide. Circ Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef]
- Liu, P.Y.; Liu, Y.W.; Lin, L.J.; Chen, J.H.; Liao, J.K. Evidence for statin pleiotropy in humans: Differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation 2009, 119, 131–138. [Google Scholar] [CrossRef]
- Bruder-Nascimento, T.; Callera, G.; Montezano, A.C.; Antunes, T.T.; He, Y.; Cat, A.N.; Ferreira, N.S.; Barreto, P.A.; Olivon, V.C.; Tostes, R.C.; et al. Renoprotective Effects of Atorvastatin in Diabetic Mice: Downregulation of RhoA and Upregulation of Akt/GSK3. PLoS ONE 2016, 11, e0162731. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Barabutis, N.; Birmpas, C.; Dimitropoulou, C.; Thangjam, G.; Cherian-Shaw, M.; Dennison, J.; Catravas, J.D. Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1410–L1419. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Ravindran, K.; Kuebler, W.M. Novel regulators of endothelial barrier function. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L924–L935. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Yan, B.P.; Liao, J.K.; Lam, Y.Y.; Yip, G.W.; Yu, C.M. Rho-kinase inhibition: A novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov. Today 2010, 15, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.W.; Cushman, M.; Rosamond, W.D.; Heckbert, S.R.; Polak, J.F.; Folsom, A.R. Cardiovascular risk factors and venous thromboembolism incidence: The longitudinal investigation of thromboembolism etiology. Arch. Intern. Med. 2002, 162, 1182–1189. [Google Scholar] [CrossRef]
- Toba, M.; Nagaoka, T.; Morio, Y.; Sato, K.; Uchida, K.; Homma, N.; Takahashi, K. Involvement of Rho kinase in the pathogenesis of acute pulmonary embolism-induced polystyrene microspheres in rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L297–L303. [Google Scholar] [CrossRef]
- Takeda, K.; Ichiki, T.; Tokunou, T.; Iino, N.; Fujii, S.; Kitabatake, A.; Shimokawa, H.; Takeshita, A. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 868–873. [Google Scholar] [CrossRef]
- Masumoto, A.; Mohri, M.; Shimokawa, H.; Urakami, L.; Usui, M.; Takeshita, A. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation 2002, 105, 1545–1547. [Google Scholar] [CrossRef]
- Hattori, T.; Shimokawa, H.; Higashi, M.; Hiroki, J.; Mukai, Y.; Kaibuchi, K.; Takeshita, A. Long-term treatment with a specific Rho-kinase inhibitor suppresses cardiac allograft vasculopathy in mice. Circ. Res. 2004, 94, 46–52. [Google Scholar] [CrossRef]
- Carrizzo, A.; Vecchione, C.; Damato, A.; di Nonno, F.; Ambrosio, M.; Pompeo, F.; Cappello, E.; Capocci, L.; Peruzzi, M.; Valenti, V.; et al. Rac1 Pharmacological Inhibition Rescues Human Endothelial Dysfunction. J. Am. Heart Assoc. 2017, 6, e004746. [Google Scholar] [CrossRef]
- Sugimoto, M.; Yamanouchi, D.; Komori, K. Therapeutic approach against intimal hyperplasia of vein grafts through endothelial nitric oxide synthase/nitric oxide (eNOS/NO) and the Rho/Rho-kinase pathway. Surg. Today 2009, 39, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Cario-Toumaniantz, C.; Evellin, S.; Maury, S.; Baron, O.; Pacaud, P.; Loirand, G. Role of Rho kinase signalling in healthy and varicose human saphenous veins. Br. J. Pharmacol. 2002, 137, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bu, X.; Lu, B.; Avraham, H.; Flavell, R.A.; Lim, B. The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPases by an inhibitory function. Mol. Cell Biol. 2002, 22, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Tergaonkar, V. Rho protein GTPases and their interactions with NFkappaB: Crossroads of inflammation and matrix biology. Biosci. Rep. 2014, 34, e00115. [Google Scholar] [CrossRef] [PubMed]
- Honing, H.; van den Berg, T.K.; van der Pol, S.M.; Dijkstra, C.D.; van der Kammen, R.A.; Collard, J.G.; de Vries, H.E. RhoA activation promotes transendothelial migration of monocytes via ROCK. J. Leukoc. Biol. 2004, 75, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.; Ridley, A.J. Rho GTPases and leucocyte-induced endothelial remodelling. Biochem. J. 2005, 385, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Van Helden, S.F.; Anthony, E.C.; Dee, R.; Hordijk, P.L. Rho GTPase expression in human myeloid cells. PLoS ONE 2012, 7, e42563. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.W.; Maass, D.L.; Ballard-Croft, C. Rho-associated kinase modulates myocardial inflammatory cytokine responses. Shock 2005, 24, 53–58. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef]
- Gerasimcik, N.; He, M.; Dahlberg, C.I.M.; Kuznetsov, N.V.; Severinson, E.; Westerberg, L.S. The Small Rho GTPases Rac1 and Rac2 Are Important for T-Cell Independent Antigen Responses and for Suppressing Switching to IgG2b in Mice. Front Immunol. 2017, 8, 1264. [Google Scholar] [CrossRef]
- Konigs, V.; Jennings, R.; Vogl, T.; Horsthemke, M.; Bachg, A.C.; Xu, Y.; Grobe, K.; Brakebusch, C.; Schwab, A.; Bahler, M.; et al. Mouse macrophages completely lacking Rho subfamily GTPases (RhoA, RhoB, and RhoC) have severe lamellipodial retraction defects, but robust chemotactic navigation and altered motility. J. Biol. Chem. 2014, 289, 30772–30784. [Google Scholar] [CrossRef] [PubMed]
- Ricker, E.; Chowdhury, L.; Yi, W.; Pernis, A.B. The RhoA-ROCK pathway in the regulation of T and B cell responses. F1000 Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.D.; Szczur, K.; Harris, C.E.; Berclaz, P.Y. Rho GTPase Rac1 is critical for neutrophil migration into the lung. Blood 2007, 109, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, J.C.; Cancelas, J.A.; Filippi, M.D.; Kalfa, T.A.; Guo, F.; Zheng, Y. Rho GTPases in hematopoiesis and hemopathies. Blood 2010, 115, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, D.; Steward, M.; Macdonald, D.L.; Francis, G.A.; Dinauer, M.C.; Lacy, P. Rac2 is critical for neutrophil primary granule exocytosis. Blood 2004, 104, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.T.; Strengert, M.; Hayes, P.; El-Benna, J.; Brakebusch, C.; Kubica, M.; Knaus, U.G. RhoA determines disease progression by controlling neutrophil motility and restricting hyperresponsiveness. Blood 2014, 123, 3635–3645. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef]
- Pleines, I.; Hagedorn, I.; Gupta, S.; May, F.; Chakarova, L.; van Hengel, J.; Offermanns, S.; Krohne, G.; Kleinschnitz, C.; Brakebusch, C.; et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 2012, 119, 1054–1063. [Google Scholar] [CrossRef]
- Noma, K.; Rikitake, Y.; Oyama, N.; Yan, G.; Alcaide, P.; Liu, P.Y.; Wang, H.; Ahl, D.; Sawada, N.; Okamoto, R.; et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J. Clin. Invest. 2008, 118, 1632–1644. [Google Scholar] [CrossRef]
- Grimm, M.; Tischner, D.; Troidl, K.; Albarran Juarez, J.; Sivaraj, K.K.; Ferreiros Bouzas, N.; Geisslinger, G.; Binder, C.J.; Wettschureck, N. S1P2/G12/13 Signaling Negatively Regulates Macrophage Activation and Indirectly Shapes the Atheroprotective B1-Cell Population. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 37–48. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Wu, C.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Kloc, M.; Ghobrial, R.M. Macrophage/monocyte-specific deletion of Ras homolog gene family member A (RhoA) downregulates fractalkine receptor and inhibits chronic rejection of mouse cardiac allografts. J. Heart Lung Transplant. 2017, 36, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Nigro, P.; Matoba, T.; O’Dell, M.R.; Cui, Z.; Shi, X.; Mohan, A.; Yan, C.; Abe, J.; Illig, K.A.; et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat. Med. 2009, 15, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Cassis, L. Angiotensin II and abdominal aortic aneurysms. Curr. Hypertens. Rep. 2004, 6, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 2000, 105, 1605–1612. [Google Scholar] [CrossRef]
- Wang, Y.X.; da Cunha, V.; Martin-McNulty, B.; Vincelette, J.; Li, W.; Choy, D.F.; Halks-Miller, M.; Mahmoudi, M.; Schroeder, M.; Johns, A.; et al. Inhibition of Rho-kinase by fasudil attenuated angiotensin II-induced cardiac hypertrophy in apolipoprotein E deficient mice. Eur. J. Pharmacol. 2005, 512, 215–222. [Google Scholar] [CrossRef]
- Hizume, T.; Morikawa, K.; Takaki, A.; Abe, K.; Sunagawa, K.; Amano, M.; Kaibuchi, K.; Kubo, C.; Shimokawa, H. Sustained elevation of serum cortisol level causes sensitization of coronary vasoconstricting responses in pigs in vivo: A possible link between stress and coronary vasospasm. Circ. Res. 2006, 99, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Sulciner, D.J.; Irani, K.; Yu, Z.X.; Ferrans, V.J.; Goldschmidt-Clermont, P.; Finkel, T. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol. Cell Biol. 1996, 16, 7115–7121. [Google Scholar] [CrossRef]
- Aslan, J.E.; McCarty, O.J. Rho GTPases in platelet function. J. Thromb. Haemost. 2013, 11, 35–46. [Google Scholar] [CrossRef]
- Yaoita, N.; Shirakawa, R.; Fukumoto, Y.; Sugimura, K.; Miyata, S.; Miura, Y.; Nochioka, K.; Miura, M.; Tatebe, S.; Aoki, T.; et al. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.A. mTOR and vascular remodeling in lung diseases: Current challenges and therapeutic prospects. FASEB J. 2013, 27, 1796–1807. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Hu, D.; Yang, Y.; Chen, Z.; Fu, M.; Wang, D.W.; Xu, X.; Tu, L. EETs reduces LPS-induced hyperpermeability by targeting GRP78 mediated Src activation and subsequent Rho/ROCK signaling pathway. Oncotarget 2017, 8, 50958–50971. [Google Scholar] [CrossRef] [PubMed]
- Soulet, C.; Sauzeau, V.; Plantavid, M.; Herbert, J.M.; Pacaud, P.; Payrastre, B.; Savi, P. Gi-dependent and -independent mechanisms downstream of the P2Y12 ADP-receptor. J. Thromb. Haemost. 2004, 2, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Itoh, H.; Louie, O.; Kubota, K.; Kent, K.C. The signaling protein Rho is necessary for vascular smooth muscle migration and survival but not for proliferation. Surgery 2002, 132, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Loirand, G.; Pacaud, P. The role of Rho protein signaling in hypertension. Nat. Rev. Cardiol. 2010, 7, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Braun-Dullaeus, R.C.; Sedding, D.G. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat. Med. 2002, 8, 1249. [Google Scholar] [CrossRef]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef]
- Sakurada, S.; Takuwa, N.; Sugimoto, N.; Wang, Y.; Seto, M.; Sasaki, Y.; Takuwa, Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ. Res. 2003, 93, 548–556. [Google Scholar] [CrossRef]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.P.; Somlyo, A.V.; Hautmann, M.; Somlyo, A.P.; Owens, G.K. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 2001, 276, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Lee, J.J.; Stoll, S.; Ma, B.; Costa, K.D.; Qiu, H. Rho Kinase Regulates Aortic Vascular Smooth Muscle Cell Stiffness Via Actin/SRF/Myocardin in Hypertension. Cell Physiol. Biochem. 2017, 44, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Long, X.; Hendershot, A.; Miano, J.M.; Sottile, J. Fibronectin matrix polymerization regulates smooth muscle cell phenotype through a Rac1 dependent mechanism. PLoS ONE 2014, 9, e94988. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.J.; Cherng, W.J. Coronary Vasospastic Angina: Current Understanding and the Role of Inflammation. Acta Cardiol. Sin. 2013, 29, 1–10. [Google Scholar] [PubMed]
- Yasue, H.; Mizuno, Y.; Harada, E. Coronary artery spasm—Clinical features, pathogenesis and treatment. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M. Role of Rac1 GTPase in salt-sensitive hypertension. Curr. Opin. Nephrol. Hypertens. 2013, 22, 148–155. [Google Scholar] [CrossRef]
- Wirth, A.; Benyo, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Orsy, P.; Horvath, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Hajicek, N.; Kozasa, T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 2009, 17, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Bregeon, J.; Toumaniantz, G.; Rolli-Derkinderen, M.; Retailleau, K.; Loufrani, L.; Henrion, D.; Scalbert, E.; Bril, A.; Torres, R.M.; et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010, 16, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.L.; Chadeuf, G.; Heurtebise-Chretien, S.; Prieur, X.; Quillard, T.; Goueffic, Y.; Vaillant, N.; Rio, M.; Castan, L.; Durand, M.; et al. Leukocyte RhoA exchange factor Arhgef1 mediates vascular inflammation and atherosclerosis. J. Clin. Invest. 2017, 127, 4516–4526. [Google Scholar] [CrossRef]
- Carbone, M.L.; Bregeon, J.; Devos, N.; Chadeuf, G.; Blanchard, A.; Azizi, M.; Pacaud, P.; Jeunemaitre, X.; Loirand, G. Angiotensin II activates the RhoA exchange factor Arhgef1 in humans. Hypertension 2015, 65, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Lenhart, K.C.; Bird, K.E.; Suen, A.A.; Rojas, M.; Kakoki, M.; Li, F.; Smithies, O.; Mack, C.P.; Taylor, J.M. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat. Commun. 2013, 4, 2910. [Google Scholar] [CrossRef] [PubMed]
- Seko, T.; Ito, M.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Onishi, K.; Isaka, N.; Hartshorne, D.J.; Nakano, T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 2003, 92, 411–418. [Google Scholar] [CrossRef]
- Eto, Y.; Shimokawa, H.; Hiroki, J.; Morishige, K.; Kandabashi, T.; Matsumoto, Y.; Amano, M.; Hoshijima, M.; Kaibuchi, K.; Takeshita, A. Gene transfer of dominant negative Rho kinase suppresses neointimal formation after balloon injury in pigs. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1744–H1750. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, N.; Ogata, T.; Naito, D.; Miyagawa, K.; Taniguchi, T.; Hamaoka, T.; Maruyama, N.; Kasahara, T.; Nishi, M.; Matoba, S.; et al. MURC deficiency in smooth muscle attenuates pulmonary hypertension. Nat. Commun. 2016, 7, 12417. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Matoba, T.; Ito, A.; Tanaka, H.; Kishi, T.; Hayashidani, S.; Abe, K.; Takeshita, A.; Shimokawa, H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 2005, 91, 391–392. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Mohri, M.; Inokuchi, K.; Ito, A.; Hirakawa, Y.; Masumoto, A.; Hirooka, Y.; Takeshita, A.; Shimokawa, H. Anti-ischemic effects of fasudil, a specific Rho-kinase inhibitor, in patients with stable effort angina. J. Cardiovasc. Pharmacol. 2007, 49, 117–121. [Google Scholar] [CrossRef]
- Jin, H.G.; Yamashita, H.; Nagano, Y.; Fukuba, H.; Hiji, M.; Ohtsuki, T.; Takahashi, T.; Kohriyama, T.; Kaibuchi, K.; Matsumoto, M. Hypoxia-induced upregulation of endothelial small G protein RhoA and Rho-kinase/ROCK2 inhibits eNOS expression. Neurosci. Lett. 2006, 408, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.A.; Novoa, E.; Lecuona, E.; Sun, H.; Sznajder, J.I. Role of the small GTPase RhoA in the hypoxia-induced decrease of plasma membrane Na,K-ATPase in A549 cells. J. Cell Sci. 2007, 120, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Fagan, K.A.; Jones, P.L.; McMurtry, I.F. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br. J. Pharmacol. 2008, 155, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Karoor, V.; Fini, M.A.; Loomis, Z.; Sullivan, T.; Hersh, L.B.; Gerasimovskaya, E.; Irwin, D.; Dempsey, E.C. Sustained Activation of Rho GTPases Promotes a Synthetic Pulmonary Artery Smooth Muscle Cell Phenotype in Neprilysin Null Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, D.; Liu, L.; Xia, W.; Li, F. Rho signaling pathway enhances proliferation of PASMCs by suppressing nuclear translocation of Smad1 in PAH. Exp. Ther. Med. 2019, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sforza, E.; Roche, F. Chronic intermittent hypoxia and obstructive sleep apnea: An experimental and clinical approach. Hypoxia (Auckl.) 2016, 4, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Yasue, H.; Nakagawa, H.; Itoh, T.; Harada, E.; Mizuno, Y. Coronary artery spasm--clinical features, diagnosis, pathogenesis, and treatment. J. Cardiol. 2008, 51, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Ponticos, M.; Smith, B.D. Extracellular matrix synthesis in vascular disease: Hypertension, and atherosclerosis. J. Biomed. Res. 2014, 28, 25–39. [Google Scholar] [PubMed]
- Stenmark, K.R.; Nozik-Grayck, E.; Gerasimovskaya, E.; Anwar, A.; Li, M.; Riddle, S.; Frid, M. The adventitia: Essential role in pulmonary vascular remodeling. Compr. Physiol. 2011, 1, 141–161. [Google Scholar] [PubMed]
- Forte, A.; Della Corte, A.; De Feo, M.; Cerasuolo, F.; Cipollaro, M. Role of myofibroblasts in vascular remodelling: Focus on restenosis and aneurysm. Cardiovasc. Res. 2010, 88, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Coen, M.; Gabbiani, G.; Bochaton-Piallat, M.L. Myofibroblast-mediated adventitial remodeling: An underestimated player in arterial pathology. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Kuivaniemi, H.; Ryer, E.J.; Elmore, J.R.; Tromp, G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert. Rev. Cardiovasc. Ther. 2015, 13, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.C. Physiology and homeostasis of extracellular matrix: Cardiovascular adaptation and remodeling. Pathophysiology 2000, 7, 177–182. [Google Scholar] [CrossRef]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [PubMed]
- Usuki, J.; Matsuda, K.; Azuma, A.; Kudoh, S.; Gemma, A. Sequential analysis of myofibroblast differentiation and transforming growth factor-beta1/Smad pathway activation in murine pulmonary fibrosis. J. Nippon Med. Sch. 2012, 79, 46–59. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Doyle, J.J.; Gerber, E.E.; Dietz, H.C. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012, 586, 2003–2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chu, Y.; Zhu, D.; Yan, C.; Liu, J.; Ji, K.; Gao, P. Perivascular gene transfer of dominant-negative N19RhoA attenuates neointimal formation via inhibition of TGF-beta1-Smad2 signaling in rats after carotid artery balloon injury. Biochem. Biophys. Res. Commun. 2009, 389, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, e93187. [Google Scholar] [CrossRef]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef]
- Carlin, C.M.; Peacock, A.J.; Welsh, D.J. Fluvastatin inhibits hypoxic proliferation and p38 MAPK activity in pulmonary artery fibroblasts. Am. J. Respir. Cell Mol. Biol. 2007, 37, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Recinos, A., 3rd; LeJeune, W.S.; Sun, H.; Lee, C.Y.; Tieu, B.C.; Lu, M.; Hou, T.; Boldogh, I.; Tilton, R.G.; Brasier, A.R. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis 2007, 194, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Pagano, P.J. The reactive adventitia: Fibroblast oxidase in vascular function. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yan, H.; Guo, M.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species in vascular formation and development. Oxid. Med. Cell Longev. 2013, 2013, 374963. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwala, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Zmudka, K.; et al. Mechanisms of oxidative stress in human aortic aneurysms—Association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Ngo, D.T.; Chapman, M.J.; Ali, O.A.; Chirkov, Y.Y.; Horowitz, J.D. Pathogenesis of aortic stenosis: Not just a matter of wear and tear. Am. J. Cardiovasc. Dis. 2011, 1, 185–199. [Google Scholar] [PubMed]
- Heo, J.; Campbell, S.L. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J. Biol. Chem. 2005, 280, 31003–31010. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P.; Taddei, L.; Giannoni, E.; Camici, G.; Manao, G.; Raugei, G.; Ramponi, G. The low M(r) protein-tyrosine phosphatase is involved in Rho-mediated cytoskeleton rearrangement after integrin and platelet-derived growth factor stimulation. J. Biol. Chem. 2000, 275, 4640–4646. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Zhou, B.; Cox, A.D.; Campbell, S.L. Rho GTPases, oxidation, and cell redox control. Small GTPases 2014, 5, e28579. [Google Scholar] [CrossRef]
- Lum, H.; Roebuck, K.A. Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell Physiol. 2001, 280, C719–C741. [Google Scholar] [CrossRef] [PubMed]
- Alom-Ruiz, S.P.; Anilkumar, N.; Shah, A.M. Reactive oxygen species and endothelial activation. Antioxid. Redox. Signal. 2008, 10, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Boueiz, A.; Hassoun, P.M. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc. Res. 2009, 77, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.L.; Sessa, W.C. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 2013, 126, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Chandra, S.; Toque, H.A.; Bhatta, A.; Rojas, M.; Caldwell, R.B.; Caldwell, R.W. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc. Res. 2013, 97, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanthan, S.; Zieba, B.J.; Ge, Z.D.; Momotani, K.; Zheng, X.; Lund, H.; Artamonov, M.V.; Maas, J.E.; Szabo, A.; Zhang, D.X.; et al. Rap1b in smooth muscle and endothelium is required for maintenance of vascular tone and normal blood pressure. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1486–1494. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef]
- Satoh, K.; Fukumoto, Y.; Shimokawa, H. Rho-kinase: Important new therapeutic target in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H287–H296. [Google Scholar] [CrossRef]
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The role of reactive oxygen species in microvascular remodeling. Int. J. Mol. Sci. 2014, 15, 23792–23835. [Google Scholar] [CrossRef]
- Breton-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef]
- Haurani, M.J.; Pagano, P.J. Adventitial fibroblast reactive oxygen species as autocrine and paracrine mediators of remodeling: Bellwether for vascular disease? Cardiovasc. Res. 2007, 75, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kapoor, M.; Leask, A. Rac1 expression by fibroblasts is required for tissue repair in vivo. Am. J. Pathol. 2009, 174, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, J.; Hu, K.; Tang, S.; Zhou, X.; Xu, L.; Li, Y.; Yu, S. The role of the Nox4-derived ROS-mediated RhoA/Rho kinase pathway in rat hypertension induced by chronic intermittent hypoxia. Sleep Breath. 2017, 21, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Yada, T.; Shimokawa, H.; Hiramatsu, O.; Kajita, T.; Shigeto, F.; Tanaka, E.; Shinozaki, Y.; Mori, H.; Kiyooka, T.; Katsura, M.; et al. Beneficial effect of hydroxyfasudil, a specific Rho-kinase inhibitor, on ischemia/reperfusion injury in canine coronary microcirculation in vivo. J. Am. Coll. Cardiol. 2005, 45, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Ikegaki, I.; Asano, T.; Shimokawa, H. Antiischemic properties of fasudil in experimental models of vasospastic angina. Jpn. J. Pharmacol. 2001, 87, 34–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Utsunomiya, T.; Satoh, S.; Ikegaki, I.; Toshima, Y.; Asano, T.; Shimokawa, H. Antianginal effects of hydroxyfasudil, a Rho-kinase inhibitor, in a canine model of effort angina. Br. J. Pharmacol. 2001, 134, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Ikegaki, I.; Toshima, Y.; Watanabe, A.; Asano, T.; Shimokawa, H. Effects of Rho-kinase inhibitor on vasopressin-induced chronic myocardial damage in rats. Life Sci. 2002, 72, 103–112. [Google Scholar] [CrossRef]
- Wei, L.; Imanaka-Yoshida, K.; Wang, L.; Zhan, S.; Schneider, M.D.; DeMayo, F.J.; Schwartz, R.J. Inhibition of Rho family GTPases by Rho GDP dissociation inhibitor disrupts cardiac morphogenesis and inhibits cardiomyocyte proliferation. Development 2002, 129, 1705–1714. [Google Scholar] [PubMed]
- Zhou, X.; Zheng, Y. Cell type-specific signaling function of RhoA GTPase: Lessons from mouse gene targeting. J. Biol. Chem. 2013, 288, 36179–36188. [Google Scholar] [CrossRef]
- Sah, V.P.; Minamisawa, S.; Tam, S.P.; Wu, T.H.; Dorn, G.W., 2nd; Ross, J., Jr.; Chien, K.R.; Brown, J.H. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J. Clin. Invest. 1999, 103, 1627–1634. [Google Scholar] [CrossRef]
- Xiang, S.Y.; Vanhoutte, D.; Del Re, D.P.; Purcell, N.H.; Ling, H.; Banerjee, I.; Bossuyt, J.; Lang, R.A.; Zheng, Y.; Matkovich, S.J.; et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J. Clin. Invest. 2011, 121, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.A.; Bower, H.S.; Baxter, G.F. Rho kinase activation plays a major role as a mediator of irreversible injury in reperfused myocardium. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2598–H2606. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Horinaka, S.; Mita, S.; Nakano, S.; Honda, T.; Yoshida, K.; Kobayashi, T.; Matsuoka, H. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc. Res. 2002, 55, 757–767. [Google Scholar] [CrossRef]
- Guan, P.; Liang, Y.; Wang, N. Fasudil alleviates pressure overload-induced heart failure by activating Nrf2-mediated antioxidant responses. J. Cell Biochem. 2018, 119, 6452–6460. [Google Scholar] [CrossRef]
- Shi, J.; Wei, L. Rho kinases in cardiovascular physiology and pathophysiology: The effect of fasudil. J. Cardiovasc. Pharmacol. 2013, 62, 341–354. [Google Scholar] [CrossRef]
- Arita, R.; Hata, Y.; Nakao, S.; Kita, T.; Miura, M.; Kawahara, S.; Zandi, S.; Almulki, L.; Tayyari, F.; Shimokawa, H.; et al. Rho kinase inhibition by fasudil ameliorates diabetes-induced microvascular damage. Diabetes 2009, 58, 215–226. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y.J. RhoA/Rho kinase: A novel therapeutic target in diabetic complications. Chin. Med. J. (Engl.) 2010, 123, 2461–2466. [Google Scholar]
- Sunamura, S.; Satoh, K.; Kurosawa, R.; Ohtsuki, T.; Kikuchi, N.; Elias-Al-Mamun, M.; Shimizu, T.; Ikeda, S.; Suzuki, K.; Satoh, T.; et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E7129–E7138. [Google Scholar] [CrossRef]
- Loirand, G. Rho Kinases in Health and Disease: From Basic Science to Translational Research. Pharmacol. Rev. 2015, 67, 1074–1095. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.W.; Yang, Y.; Zhang, L.; Wei, L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J. Mol. Cell Cardiol. 2010, 49, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Yang, X.; Lin, X.; Yang, T.; Yi, X.; Dai, Y.; Guo, J.; Li, T.; Shi, J.; Wei, L.; et al. Rnd3 haploinsufficient mice are predisposed to hemodynamic stress and develop apoptotic cardiomyopathy with heart failure. Cell Death Dis. 2014, 5, e1284. [Google Scholar] [CrossRef]
- Lay, A.J.; Coleman, P.R.; Formaz-Preston, A.; Ting, K.K.; Roediger, B.; Weninger, W.; Schwartz, M.A.; Vadas, M.A.; Gamble, J.R. ARHGAP18: A Flow-Responsive Gene That Regulates Endothelial Cell Alignment and Protects Against Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e010057. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.; Wei, L.; Shi, J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011, 7, 657–671. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Aoki, T.; Fukumoto, Y.; Shimokawa, H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 2012, 60, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Dobashi, K.; Iizuka, K.; Horie, T.; Suzuki, K.; Tukagoshi, H.; Nakazawa, T.; Nakazato, Y.; Mori, M. Contribution of small GTPase Rho and its target protein rock in a murine model of lung fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 210–217. [Google Scholar] [CrossRef]
- Knipe, R.S.; Tager, A.M.; Liao, J.K. The Rho kinases: Critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacol. Rev. 2015, 67, 103–117. [Google Scholar] [CrossRef]
- Tsou, P.S.; Haak, A.J.; Khanna, D.; Neubig, R.R. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: Focus on Rho GTPase-regulated gene transcription. Am. J. Physiol. Cell Physiol. 2014, 307, C2–C13. [Google Scholar] [CrossRef]
- Krishna, S.M.; Moxon, J.V.; Golledge, J. A review of the pathophysiology and potential biomarkers for peripheral artery disease. Int. J. Mol. Sci. 2015, 16, 11294–11322. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Hirooka, Y.; Masumoto, A.; Ito, K.; Kimura, Y.; Inokuchi, K.; Tagawa, T.; Shimokawa, H.; Takeshita, A.; Sunagawa, K. Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation 2005, 111, 2741–2747. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.; Ohtomo, E.; Fasudil Ischemic Stroke Study Group. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Vicari, R.M.; Chaitman, B.; Keefe, D.; Smith, W.B.; Chrysant, S.G.; Tonkon, M.J.; Bittar, N.; Weiss, R.J.; Morales-Ballejo, H.; Thadani, U.; et al. Efficacy and safety of fasudil in patients with stable angina: A double-blind, placebo-controlled, phase 2 trial. J. Am. Coll. Cardiol. 2005, 46, 1803–1811. [Google Scholar] [CrossRef]
- Ishikura, K.; Yamada, N.; Ito, M.; Ota, S.; Nakamura, M.; Isaka, N.; Nakano, T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ. J. 2006, 70, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Noma, K.; Goto, C.; Nishioka, K.; Hara, K.; Kimura, M.; Umemura, T.; Jitsuiki, D.; Nakagawa, K.; Oshima, T.; Chayama, K.; et al. Smoking, endothelial function, and Rho-kinase in humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2630–2635. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rankinen, T.; Church, T.; Rice, T.; Markward, N.; Blair, S.N.; Bouchard, C. A major haplotype block at the rho-associated kinase 2 locus is associated with a lower risk of hypertension in a recessive manner: The HYPGENE study. Hypertens. Res. 2008, 31, 1651–1657. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shimizu, T.; Liao, J.K. Rho Kinases and Cardiac Remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.C.; Liu, P.Y.; Lin, H.F.; Lin, W.Y.; Liao, J.K.; Juo, S.H. Two functional polymorphisms of ROCK2 enhance arterial stiffening through inhibiting its activity and expression. J. Mol. Cell Cardiol. 2015, 79, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.Y.L.; Wang, Q.M.; Chasman, D.I.; Ridker, P.M.; Liao, J.K. Gene variations of ROCKs and risk of ischaemic stroke: The Women’s Genome Health Study. Clin. Sci. (Lond.) 2014, 126, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Kim, J.; Cheong, S.; Shin, D.H.; Jang, J.; Lee, C.; Tahk, S.J.; Shin, J.H.; Choi, S.Y.; Yoon, M.H. Rho-associated kinase 2 polymorphism in patients with vasospastic angina. Korean Circ. J. 2012, 42, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Lahm, H.; Schon, P.; Doppler, S.; Dressen, M.; Cleuziou, J.; Deutsch, M.A.; Ewert, P.; Lange, R.; Krane, M. Tetralogy of Fallot and Hypoplastic Left Heart Syndrome—Complex Clinical Phenotypes Meet Complex Genetic Networks. Curr. Genom. 2015, 16, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiu, J.; Zhang, P.; Zhang, J.; Jiang, M.; Ma, Z. Genetic variants in FAM13A and IREB2 are associated with the susceptibility to COPD in a Chinese rural population: A case-control study. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 99, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Mohri, M.; Shimokawa, H.; Hirakawa, Y.; Masumoto, A.; Takeshita, A. Rho-kinase inhibition with intracoronary fasudil prevents myocardial ischemia in patients with coronary microvascular spasm. J. Am. Coll. Cardiol. 2003, 41, 15–29. [Google Scholar] [CrossRef]
- Olson, M.F. Applications for ROCK kinase inhibition. Curr. Opin. Cell. Biol. 2008, 20, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Satoh, K. 2015 ATVB Plenary Lecture: Translational research on rho-kinase in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1756–1769. [Google Scholar] [CrossRef]
- Dee, R.A.; Mangum, K.D.; Bai, X.; Mack, C.P.; Taylor, J.M. Druggable targets in the Rho pathway and their promise for therapeutic control of blood pressure. Pharmacol. Ther. 2019, 193, 121–134. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Trial | Drug | |
|---|---|---|
| NCT03753269 | Early Intracoronary Administration of Fasudil in the Primary PCI of ST-segment-Elevation Myocardial Infarction | Fasudil |
| NCT03404843 | Red Blood Cell ATP Release and Vascular Function in Humans | Fasudil/Saline |
| NCT00120718 | The Effect of Fasudil on Vascular Function in Humans | Fasudil |
| NCT00670202 | Rho Kinase (ROCK) Inhibition in Carotid Atherosclerosis | Fasudil/placebo |
| NCT03391219 | Combined Intravitreal Injection of Bevacizumab and Fasudil Versus Bevacizumab Alone for Macular Edema Secondary to Retinal Vein Occlusion in Previously Treated Patients | Avastin Avastin/fasudil |
| NCT00498615 | A Rho-kinase Inhibitor (Fasudil) in the Treatment of Raynaud’s Phenomenon | Fasudil |
| NCT00560170 | Vascular Effects of Ezetimibe/Simvastatin and Simvastatin on Atherosclerosis | Statin/ROCK biomarker |
| NCT01823081 | Trimetazidine in Pulmonary Artery Hypertension | Trimetazidine/ROCK biomarker |
| NCT01732718 | NCT01732718 Effect of Atorvastatin on Endothelial Dysfunction and Albuminuria in Sickle Cell Disease (ENDO) | Statin ROCK biomarker |
| NCT02754518 | Demonstration of Reverse Remodeling Effects of Entresto (Valsartan/Sacubitril) Using Echocardiography Endocardial Surface Analysis | Entresto ROCK biomarker |
| NCT00839449 | Eicosapentaenoic Acid Cerebral Vasospasm Therapy Study (EVAS) | ROCK biomarker |
| NCT01065753 | Multi-faceted Evaluations Following Weight Reduction in Subjects With Metabolic Syndrome | Rho biomarker |
| NCT00115830 | Rho Kinase in Patients With Atherosclerosis | ROCK biomarker |
| NCT01069042 | Anti-Hypertensive Agent (ACEi) and Heart Function Improvement in Association With Rho Kinase Activity Changes in Human | ROCK biomarker |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strassheim, D.; Gerasimovskaya, E.; Irwin, D.; Dempsey, E.C.; Stenmark, K.; Karoor, V. RhoGTPase in Vascular Disease. Cells 2019, 8, 551. https://doi.org/10.3390/cells8060551
Strassheim D, Gerasimovskaya E, Irwin D, Dempsey EC, Stenmark K, Karoor V. RhoGTPase in Vascular Disease. Cells. 2019; 8(6):551. https://doi.org/10.3390/cells8060551
Chicago/Turabian StyleStrassheim, Derek, Evgenia Gerasimovskaya, David Irwin, Edward C. Dempsey, Kurt Stenmark, and Vijaya Karoor. 2019. "RhoGTPase in Vascular Disease" Cells 8, no. 6: 551. https://doi.org/10.3390/cells8060551
APA StyleStrassheim, D., Gerasimovskaya, E., Irwin, D., Dempsey, E. C., Stenmark, K., & Karoor, V. (2019). RhoGTPase in Vascular Disease. Cells, 8(6), 551. https://doi.org/10.3390/cells8060551