Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells


Abstract
1. Introduction
2. The Metabolic State of TICs
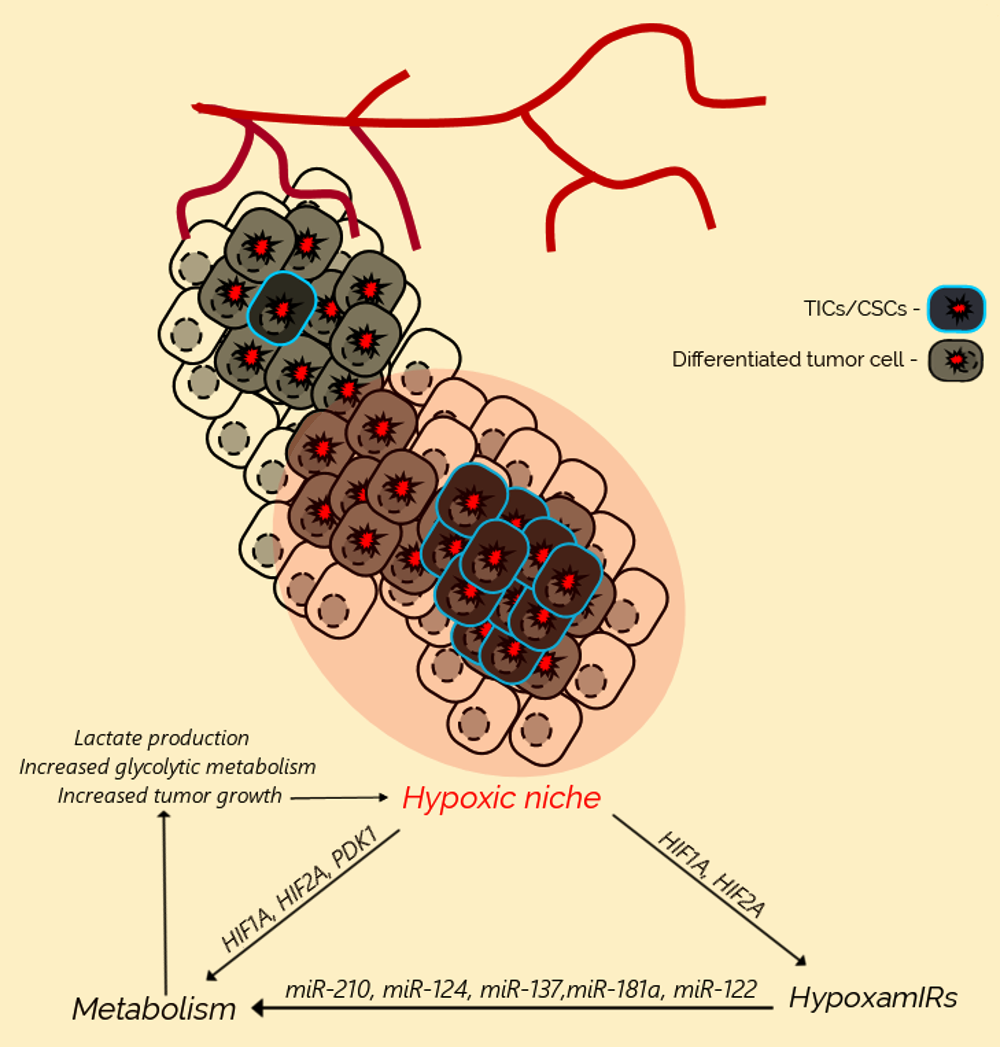
3. Hypoxia Promotes Cancer Initiation and Progression
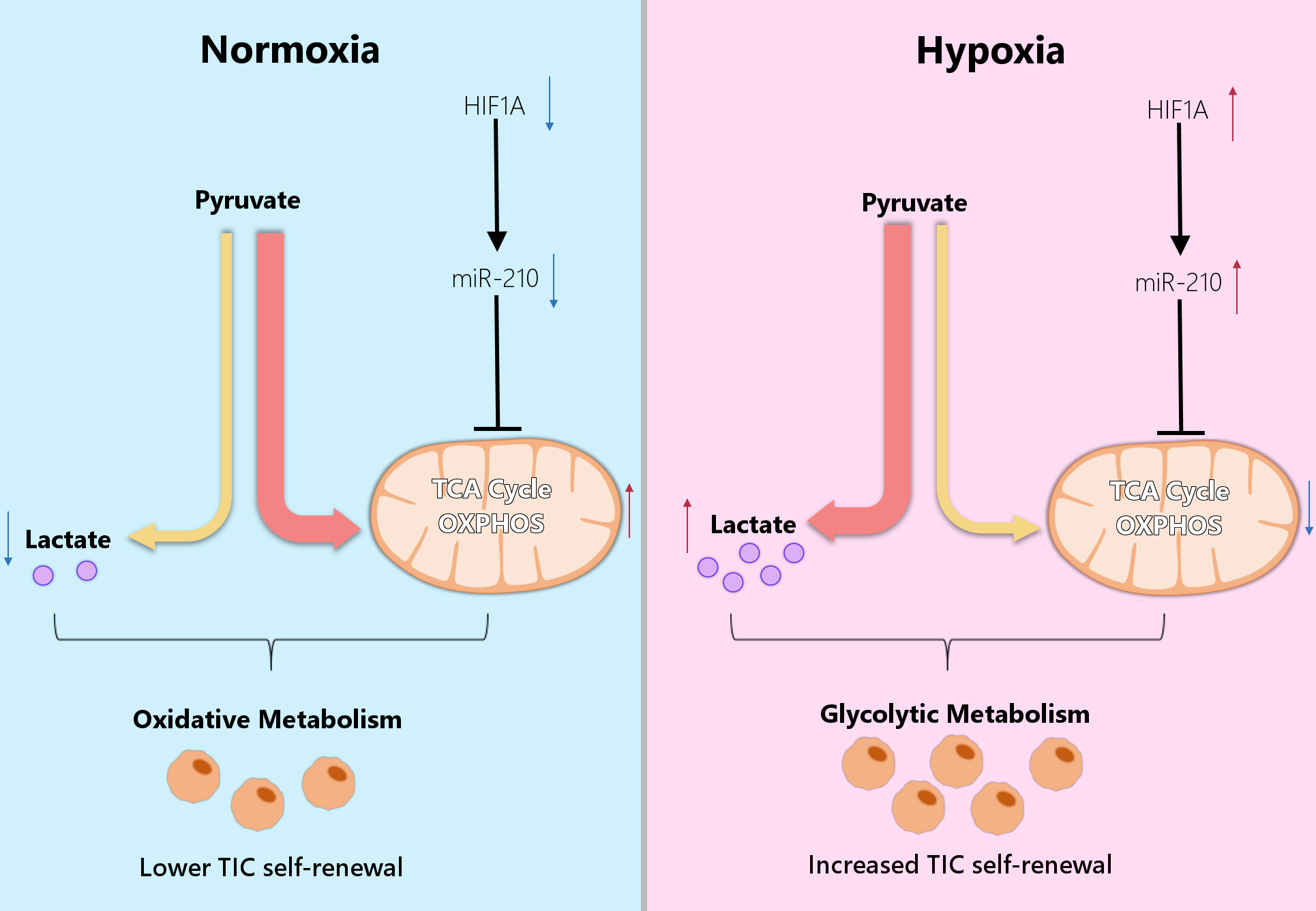
4. Metabolic Reprogramming of Cancer Cells Under Hypoxia
5. MicroRNAs Regulate Metabolic Reprogramming
6. Lactate Acts as a TIC-Promoting Oncometabolite
7. Clinical Targeting of Lactate Metabolism in TICs
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Cufí, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; López-Bonet, E.; Alarcón, T.; Vazquez-Martin, A. The Warburg effect version 2.0: Metabolic reprogramming of cancer stem cells. Cell Cycle 2013, 12, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorilation and selectively eradicates quiscent human leukemia stem cells. Cell Stem Cell 2012, 127, 358–366. [Google Scholar]
- Zhang, G.; Yang, P.; Guo, P.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Unraveling the mystery of cancer metabolism in the genesis of tumor-initiating cells and development of cancer. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 49–59. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Mooney, S.M.; Natsumeda, M.; Steiger, H.-J.; Maciaczyk, J. Targeting cancer stem-like cells in glioblastoma and colorectal cancer through metabolic pathways. Int. J. Cancer 2017, 140, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-J.J.; Zhang, S.-S.S.; Guo, X.-L.L.; Sun, K.; Han, Z.-P.P.; Li, R.; Zhao, Q.-D.D.; Deng, W.-J.J.; Xie, X.-Q.Q.; Zhang, J.-W.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; Van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Vincent, Z.; Urakami, K.; Maruyama, K.; Yamaguchi, K.; Kusuhara, M. CD133-positive cancer stem cells from colo205 human colon adenocarcinoma cell line show resistance to chemotherapy and display a specific metabolomic profile. Genes Cancer 2014, 5, 250. [Google Scholar] [PubMed]
- Song, K.; Kwon, H.; Han, C.; Zhang, J.; Dash, S.; Lim, K.; Wu, T. Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: Regulation by MIR-122. Oncotarget 2015, 6, 40822–40835. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Liu, X.; Bu, P.; Lin, C.S.; Rakhilin, N.; Locasale, J.W.; Shen, X. A metabolic signature of colon cancer initiating cells. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 August 2014; IEEE: Piscataway, NJ, USA, 2014; Volume 2014, pp. 4759–4762. [Google Scholar]
- Qureshi-Baig, K.; Ullmann, P.; Rodriguez, F.; Frasquilho, S.; Nazarov, P.V.; Haan, S.; Letellier, E. What do we learn from spheroid culture systems? Insights from tumorspheres derived from primary colon cancer tissue. PLoS ONE 2016, 11, e0146052. [Google Scholar] [CrossRef]
- Ullmann, P.; Qureshi-Baig, K.; Rodriguez, F.; Ginolhac, A.; Nonnenmacher, Y.; Ternes, D.; Weiler, J.; Gäbler, K.; Bahlawane, C.; Hiller, K.; et al. Hypoxia-responsive miR-210 promotes self-renewal capacity of colon tumor-initiating cells by repressing ISCU and by inducing lactate production. Oncotarget 2016, 7, 65454. [Google Scholar] [CrossRef]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M.; et al. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Chandel, N. Navigating Metabolism; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2014. [Google Scholar]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-T.; Lin, Y.-W.; Chiu, H.-M.; Chiang, B.-H. Curcumin Induces Apoptosis of Colorectal Cancer Stem Cells by Coupling with CD44 Marker. J. Agric. Food Chem. 2016, 64, 2247–2253. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S.; et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J. Transl. Med. 2015, 13, 210. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Chaneton, B.; Smith, M.; Van Den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2013, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Harris, B.H.L.; Barberis, A.; West, C.M.L.; Buffa, F.M. Gene Expression Signatures as Biomarkers of Tumour Hypoxia. Clin. Oncol. (R. Coll. Radiol). 2015, 27, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Rajaganeshan, R.; Prasad, R.; Guillou, P.J.; Poston, G.; Scott, N.; Jayne, D.G. The role of hypoxia in recurrence following resection of Dukes’ B colorectal cancer. Int. J. Colorectal Dis. 2008, 23, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Müller, C.I.; Reis, H.; Alakus, H.; Winde, G.; Baba, H.A.; Wohlschlaeger, J.; Jasani, B.; Fandrey, J.; Schmid, K.W. Combined analysis of hypoxia-inducible factor 1 alpha and metallothionein indicates an aggressive subtype of colorectal carcinoma. Int. J. Colorectal Dis. 2009, 24, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.; Harris, A.L.; Tekkis, P.P.; Turley, H.; Silver, A.; McDonald, P.J.; Talbot, I.C.; Glynne-Jones, R.; Northover, J.M.A.; Guenther, T. Hypoxia-inducible factor-1alpha and -2alpha are expressed in most rectal cancers but only hypoxia-inducible factor-1alpha is associated with prognosis. Br. J. Cancer 2009, 100, 1666–1673. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. BioEssays 2016, 38, S65–S74. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.A.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef]
- Jogi, A.; Ora, I.; Nilsson, H.; Lindeheim, A.; Makino, Y.; Poellinger, L.; Axelson, H.; Pahlman, S.; Jögi, A.; Øra, I.; et al. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef]
- Axelson, H.; Fredlund, E.; Ovenberger, M.; Landberg, G.; Påhlman, S. Hypoxia-induced dedifferentiation of tumor cells—A mechanism behind heterogeneity and aggressiveness of solid tumors. Semin. Cell Dev. Biol. 2005, 16, 554–563. [Google Scholar] [CrossRef]
- Ma, Y.; Liang, D.; Liu, J.; Axcrona, K.; Kvalheim, G.; Stokke, T.; Nesland, J.M.; Suo, Z. Prostate cancer cell lines under hypoxia exhibit greater stem-like properties. PLoS ONE 2011, 6, e29170. [Google Scholar] [CrossRef]
- Yeung, T.M.; Gandhi, S.C.; Bodmer, W.F. Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4382–4387. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; Mclendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Wu, Q.; Rivera, M.; Minhas, S.; Lathia, J.D.; Sloan, A.E.; Iliopoulos, O.; Hjelmeland, A.B.; Rich, J.N. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. 2012, 19, 428–439. 2012, 19, 428–439. [Google Scholar]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef]
- Wu, M.-Z.; Chen, S.-F.; Nieh, S.; Benner, C.; Ger, L.-P.; Jan, C.-I.; Ma, L.; Chen, C.-H.; Hishida, T.; Chang, H.-T.; et al. Hypoxia Drives Breast Tumor Malignancy through a TET-TNFα-p38-MAPK Signaling Axis. Cancer Res. 2015, 75, 3912–3924. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Korotchkina, L.G. Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 2006, 34, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C. V HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by Hypoxia-inducible Factor-1: INTERACTION BETWEEN H-ras AND HYPOXIA. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.; et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Iizuka, N.; Tsunedomi, R.; Hamamoto, Y.; Miyamoto, T.; Iida, M.; Tokuhisa, Y.; Sakamoto, K.; Takashima, M.; Tamesa, T.; et al. Glycolysis module activated by hypoxia-inducible factor 1alpha is related to the aggressive phenotype of hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 725–731. [Google Scholar] [PubMed]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kappel, A.; Keller, A. MiRNA assays in the clinical laboratory: Workflow, detection technologies and automation aspects. Clin. Chem. Lab. Med. 2017, 55, 636–647. [Google Scholar] [CrossRef]
- Gao, P.; Sun, L.; He, X.; Cao, Y.; Zhang, H. MicroRNAs and the Warburg Effect: New Players in an Old Arena. Curr. Gene Ther. 2012, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Manley, J.; Lee, J.; Singh, S.R. The emerging roles of microRNAs in cancer metabolism. Cancer Lett. 2015, 356, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Jacobsen, A.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-143 down-regulates Hexokinase 2 in colon cancer cells. BMC Cancer 2012, 12, 232. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, H.; Liu, Y.; Wu, J.; Wang, C.; Hou, X.; Chen, X.; Yang, G.; Zhao, L.; Che, H.; et al. MiR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013, 333, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, X.; Zhou, Y.; Hu, Y. MiR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352. [Google Scholar] [CrossRef]
- Nanbakhsh, A.; Visentin, G.; Olive, D.; Janji, B.; Mussard, E.; Dessen, P.; Meurice, G.; Zhang, Y.; Louache, F.; Bourhis, J.-H.; et al. miR-181a modulates acute myeloid leukemia susceptibility to natural killer cells. Oncoimmunology 2015, 4, e996475. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Cui, L.; Mei, Z.; Liu, M.; Zhang, D. miR-181a mediates metabolic shift in colon cancer cells via the PTEN/AKT pathway. FEBS Lett. 2014, 588, 1773–1779. [Google Scholar] [CrossRef]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-regulated mevalonate metabolism maintains brain tumor–initiating cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef]
- Lu, Y.X.; Yuan, L.; Xue, X.L.; Zhou, M.; Liu, Y.; Zhang, C.; Li, J.P.; Zheng, L.; Hong, M.; Li, X.N. Regulation of colorectal carcinoma Stemness, Growth, and metastasis by an miR-200c-Sox2-negative feedback loop mechanism. Clin. Cancer Res. 2014, 20, 2631–2642. [Google Scholar] [CrossRef]
- Cha, Y.; Han, M.J.; Cha, H.J.; Zoldan, J.; Burkart, A.; Jung, J.H.; Jang, Y.; Kim, C.H.; Jeong, H.C.; Kim, B.G.; et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 2017, 19, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, J.; Xu, C.; Tang, S.C.; Ren, H. The insights of Let-7 miRNAs in oncogenesis and stem cell potency. J. Cell. Mol. Med. 2016, 20, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Serguienko, A.; Grad, I.; Wennerstrøm, A.B.; Meza-Zepeda, L.A.; Thiede, B.; Stratford, E.W.; Myklebost, O.; Munthe, E. Metabolic reprogramming of metastatic breast cancer and melanoma by let-7a microRNA. Oncotarget 2015, 6, 2451. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Bai, R.; Yang, K.; Tian, Z. Erratum: MiR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1α regulation. Oncogenesis 2017, 6, e318. [Google Scholar] [CrossRef]
- Sun, P.; Hu, J.-W.; Xiong, W.-J.; Mi, J. miR-186 regulates glycolysis through Glut1 during the formation of cancer-associated fibroblasts. Asian Pac. J. Cancer Prev. 2014, 15, 4245–4250. [Google Scholar] [CrossRef]
- Okuda, H.; Xing, F.; Pandey, P.R.; Sharma, S.; Watabe, M.; Pai, S.K.; Mo, Y.-Y.; Iiizumi-Gairani, M.; Hirota, S.; Liu, Y.; et al. miR-7 Suppresses Brain Metastasis of Breast Cancer Stem-Like Cells By Modulating KLF4. Cancer Res. 2013, 73, 1434–1444. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cell 2014, 32, 2858–2868. [Google Scholar] [CrossRef]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Oh, P.S.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P.N. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFβr2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar] [CrossRef]
- Zhai, S.; Zhao, L.; Lin, T.; Wang, W. Downregulation of miR-33b promotes non-small cell lung cancer cell growth through reprogramming glucose metabolism miR-33b regulates non-small cell lung cancer cell growth. J. Cell. Biochem. 2019, 120, 6651–6660. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Chen, J.H.; Wang, L.; Walters, J.; Shin, Y.J.; Goerger, J.P.; Sun, J.; Witherspoon, M.; Rakhilin, N.; et al. A microRNA miR-34a Regulated Bimodal Switch targets Notch in Colon Cancer Stem Cells. Cell Stem Cell 2013, 12, 602. [Google Scholar] [CrossRef]
- Yu, X.F.; Zou, J.; Bao, Z.J.; Dong, J. miR-93 suppresses proliferation and colony formation of human colon cancer stem cells. World J. Gastroenterol 2011, 17, 4711–4717. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Y.; Chen, Z. MiR-125a/b regulates the activation of cancer stem cells in paclitaxel-resistant colon cancer. Cancer Investig. 2013, 31, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.-L.; Jiang, J.-K.; Yang, S.-H.; Huang, T.-S.; Lan, H.-Y.; Teng, H.-W.; Yang, C.-Y.; Tsai, Y.-P.; Lin, C.-H.; Wang, H.-W.; et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Kong, X.; Lv, L.; Gao, J. MiR-155 targets TP53INP1 to regulate liver cancer stem cell acquisition and self-renewal. FEBS Lett. 2015, 589, 500–506. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Rotem, A.; Struhl, K. Inhibition of miR-193a expression by Max and RXRα activates K-Ras and PLAU to mediate distinct aspects of cellular transformation. Cancer Res. 2011, 71, 5144–5153. [Google Scholar] [CrossRef] [PubMed]
- Saumet, A.; Vetter, G.; Bouttier, M.; Antoine, E.; Roubert, C.; Orsetti, B.; Theillet, C.; Lecellier, C.H. Estrogen and retinoic acid antagonistically regulate several microRNA genes to control aerobic glycolysis in breast cancer cells. Mol. Biosyst. 2012, 8, 3242–3253. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.F.; Hara, T.; Francis, P.; Li, X.L.; Bilke, S.; Zhu, Y.; Pineda, M.; Subramanian, M.; Bodmer, W.F.; Lal, A. The CDX1–microRNA-215 axis regulates colorectal cancer stem cell differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, E1550–E1558. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, P.; Nurmik, M.; Schmitz, M.; Rodriguez, F.; Weiler, J.; Qureshi-Baig, K.; Felten, P.; Nazarov, P.V.; Nicot, N.; Zuegel, N.; et al. Tumor suppressor miR-215 counteracts hypoxia-induced colon cancer stem cell activity. Cancer Lett. 2019, 450, 32–41. [Google Scholar] [CrossRef]
- Xu, X.T.; Xu, Q.; Tong, J.L.; Zhu, M.M.; Nie, F.; Chen, X.; Xiao, S.D.; Ran, Z.H. MicroRNA expression profiling identifies miR-328 regulates cancer stem cell-like SP cells in colorectal cancer. Br. J. Cancer 2012, 106, 1320–1330. [Google Scholar] [CrossRef]
- Bitarte, N.; Bandres, E.; Boni, V.; Zarate, R.; Rodriguez, J.; Gonzalez-Huarriz, M.; Lopez, I.; Sola, J.J.; Alonso, M.M.; Fortes, P.; et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells 2011, 29, 1661–1671. [Google Scholar] [CrossRef]
- Xiao, Y.; Sun, Y.; Liu, G.; Zhao, J.; Gao, Y.; Yeh, S.; Gong, L.; Chang, C. Androgen receptor (AR)/miR-520f-3p/SOX9 signaling is involved in altering hepatocellular carcinoma (HCC) cell sensitivity to the Sorafenib therapy under hypoxia via increasing cancer stem cells phenotype. Cancer Lett. 2019, 444, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Lu, Y.X.; Chen, D.L.; Tian, T.; Mo, H.Y.; Wei, X.L.; Liao, J.W.; Wang, F.; Zeng, Z.L.; Pelicano, H.; et al. Redox regulation of stem-like cells though the CD44v-xCT axis in colorectal cancer: Mechanisms and therapeutic implications. Theranostics 2016, 6, 1160–1175. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Yagi, S.; Ito, T.; Lowenstein, C.J. Microrna-22 regulates hypoxia signaling in colon cancer cells. PLoS ONE 2011, 6, e20291. [Google Scholar] [CrossRef] [PubMed]
- Mathew, L.K.; Lee, S.S.; Skuli, N.; Rao, S.; Keith, B.; Nathanson, K.L.; Lal, P.; Simon, M.C. Restricted expression of miR-30c-2-3p and miR-30a-3p in clear cell renal cell carcinomas enhances HIF2α activity. Cancer Discov. 2014, 4, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pu, J.; Qi, T.; Qi, M.; Yang, C.; Li, S.; Huang, K.; Zheng, L.; Tong, Q. MicroRNA-145 inhibits the growth, invasion, metastasis and angiogenesis of neuroblastoma cells through targeting hypoxia-inducible factor 2 alpha. Oncogene 2014, 33, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Taniguchi, K.; Sugito, N.; Kuranaga, Y.; Inamoto, T.; Takahara, K.; Takai, T.; Yoshikawa, Y.; Kiyama, S.; Akao, Y.; et al. MiR-145 negatively regulates Warburg effect by silencing KLF4 and PTBP1 in bladder cancer cells. Oncotarget 2017, 8, 33064–33077. [Google Scholar] [CrossRef]
- Qin, Q.; Furong, W.; Baosheng, L. Multiple functions of hypoxia-regulated miR-210 in cancer. J. Exp. Clin. Cancer Res. 2014, 33, 50. [Google Scholar] [CrossRef]
- Wang, F.; Xiong, L.; Huang, X.; Zhao, T.; Wu, L.; Liu, Z.; Ding, X.; Liu, S.; Wu, Y.; Zhao, Y.; et al. miR-210 suppresses BNIP3 to protect against the apoptosis of neural progenitor cells. Stem Cell Res. 2013, 11, 657–667. [Google Scholar] [CrossRef]
- Chio, C.-C.; Lin, J.-W.; Cheng, H.-A.; Chiu, W.-T.; Wang, Y.-H.; Wang, J.-J.; Hsing, C.-H.; Chen, R.-M. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2013, 87, 459–468. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Lu, Y.; Xu, S.; Mao, L.; Zhang, L.; Duan, W.; Liu, C.; Pi, H.; Zhang, Y.; Zhong, M.; et al. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. 2014, 5, e1090. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Nakayama, Y.; Umeda, M.; Miyazaki, K. Induction of 103-kDa gelatinase/type IV collagenase by acidic culture conditions in mouse metastatic melanoma cell lines. J. Biol. Chem. 1992, 267, 11424–11430. [Google Scholar] [PubMed]
- Li, X.; Yu, X.; Dai, D.; Song, X.; Xu, W. The altered glucose metabolism in tumor and a tumor acidic microenvironment associated with extracellular matrix metalloproteinase inducer and monocarboxylate transporters. Oncotarget 2016, 7, 23141–23155. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Bettum, I.J.; Gorad, S.S.; Barkovskaya, A.; Pettersen, S.; Moestue, S.A.; Vasiliauskaite, K.; Tenstad, E.; Øyjord, T.; Risa, Ø.; Nygaard, V.; et al. Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett. 2015, 366, 71–83. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef]
- Xie, H.; Hanai, J.-I.; Ren, J.-G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase—A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.; Lin, Z.; Witkiewicz, A.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle 2010, 9, 3276–3296. [Google Scholar] [CrossRef]
- Curtis, M.; Kenny, H.A.; Ashcroft, B.; Mukherjee, A.; Johnson, A.; Zhang, Y.; Helou, Y.; Batlle, R.; Liu, X.; Gutierrez, N.; et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019, 29, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Ratner, S. Lymphocytes stimulated with recombinant human interleukin-2: Relationship between motility into protein matrix and in vivo localization in normal and neoplastic tissues of mice. J. Natl. Cancer Inst. 1990, 82, 612–616. [Google Scholar] [CrossRef]
- Lugini, L.; Matarrese, P.; Tinari, A.; Lozupone, F.; Federici, C.; Iessi, E.; Gentile, M.; Luciani, F.; Parmiani, G.; Rivoltini, L.; et al. Cannibalism of Live Lymphocytes by Human Metastatic but Not Primary Melanoma Cells. Cancer Res. 2006, 66, 3629–3638. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Mendler, A.N.; Hu, B.; Prinz, P.U.; Kreutz, M.; Gottfried, E.; Noessner, E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int. J. Cancer 2012, 131, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family-Role and regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Sapio, L.; Spina, A.; Naviglio, D.; Calogero, A.; Naviglio, S. Lactic dehydrogenase and cancer: An overview. Front. Biosci. (Landmark Ed.) 2015, 20, 1234–1249. [Google Scholar] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C. V c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- WARBURG, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Lactate dehydrogenase 5: An old friend and a new hope in the war on cancer. Cancer Lett. 2015, 358, 1–7. [Google Scholar] [CrossRef]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zheng, J.; Chen, Z.; Liu, Y.; Dura, B.; Kwak, M.; Xavier-Ferrucio, J.; Lu, Y.C.; Zhang, M.; Roden, C.; et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Ferrer-Luna, R.; Rooj, A.K.; Mineo, M.; Ricklefs, F.; Takeda, Y.S.; Nowicki, M.O.; Salińska, E.; Nakano, I.; Lee, H.; et al. MicroRNA Signatures and Molecular Subtypes of Glioblastoma: The Role of Extracellular Transfer. Stem Cell Rep. 2017, 8, 1497–1505. [Google Scholar] [CrossRef]
- Di Francesco, A.M.; Toesca, A.; Cenciarelli, C.; Giordano, A.; Gasbarrini, A.; Puglisi, M.A. Metabolic Modification in Gastrointestinal Cancer Stem Cells: Characteristics and Therapeutic Approaches. J. Cell. Physiol. 2016, 231, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, K.; Zheng, N.; Qiu, Y.; Xie, G.; Su, M.; Jia, W.; Li, H. Metformin suppressed the proliferation of LoVo cells and induced a time-dependent metabolic and transcriptional alteration. Sci. Rep. 2015, 5, 17423. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| miRNA | Target Gene | Role of miRNA | Tumor Type | Reference |
|---|---|---|---|---|
| miR-7 | KLF4, SETDB1 | Inhibits stemness and tumorigenesis by directly targeting KLF4, inhibits metastatic ability of breast TICs, reverses epithelial–mesenchymal transition (EMT) via SETDB1 targeting | Breast (Brain metastasis) | [78,79] |
| miR-21 | TGFBR2 | Induces stemness by activating the Wnt/β-catenin pathway through TGFBR2 downregulation | Colon | [80] |
| miR-33b | MYC | Regulates MYC via the RAS/ERK/miR33b pathway | Glioblastoma | [81] |
| miR-34a | NOTCH1 | Controls symmetric/asymmetric cell division | Colon | [82] |
| miR-93 | HDAC8, TLE4 | Inhibits proliferation and colony formation | Colon | [83] |
| miR-125 | ALDH1A3, MCL1 | Regulates chemoresistance | Colon | [84] |
| miR-146a | NUMB | Controls symmetric/asymmetric cell division | Colon | [85] |
| miR-155 | TP53INP1 | Induces TIC-like phenotype by blocking the tumor suppressor gene TP53INP1 | Liver | [86] |
| miR-193a | PLAU, KRAS | Inhibits tumorigenic potential | Breast, Colon, and Pancreas | [87] |
| miR-200c | BMI1, SOX2 | Regulates chemoresistance and reduces tumorigenic capacity | Colon | [71,72] |
| miR-210 | ISCU, LDHA | Promotes self-renewal of colorectal cancer (CRC) TICs by reducing tricarboxylic acid (TCA) cycle activity and enhancing lactate production. | Colon, Breast | [18,88] |
| miR-215 | BMI1, LGR5 | Promotes differentiation and inhibits stemness | Colon | [89,90] |
| miR-328 | ABCG2, MMP16 | Inhibits drug resistance and cell invasion | Colon | [91] |
| miR-451 | PTGS2, ABCB1 | Represses Wnt activation and chemoresistance | Colon | [92] |
| miR-520f | SOX9 | Induces hypoxia-driven Sorafenib resistance by increasing the number of TIC-like cells | Liver | [93] |
| miR-1297 | SLC7A11 | Impairs cysteine uptake and glutathione production | Colon | [94] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullmann, P.; Nurmik, M.; Begaj, R.; Haan, S.; Letellier, E. Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells. Cells 2019, 8, 528. https://doi.org/10.3390/cells8060528
Ullmann P, Nurmik M, Begaj R, Haan S, Letellier E. Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells. Cells. 2019; 8(6):528. https://doi.org/10.3390/cells8060528
Chicago/Turabian StyleUllmann, Pit, Martin Nurmik, Rubens Begaj, Serge Haan, and Elisabeth Letellier. 2019. "Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells" Cells 8, no. 6: 528. https://doi.org/10.3390/cells8060528
APA StyleUllmann, P., Nurmik, M., Begaj, R., Haan, S., & Letellier, E. (2019). Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells. Cells, 8(6), 528. https://doi.org/10.3390/cells8060528