Mitophagy in Cancer: A Tale of Adaptation

Abstract
1. Introduction
2. Molecular Mechanisms Leading to Mitophagy

2.1. Canonical Mitophagy Pathways
2.1.1. PINK1/Parkin-Mediated Mitophagy
2.1.2. BNIP3/NIX-Mediated Mitophagy
2.1.3. FUNDC1-Mediated Mitophagy
2.2. Non-Canonical Mitophagy Pathways
2.2.1. Lipid-Mediated Mitophagy
2.2.2. AMBRA1-Mediated Mitophagy
2.2.3. BCL2L13-Mediated Mitophagy
2.2.4. FKBP8-Mediated Mitophagy
2.2.5. Rab-Mediated Mitophagy
3. Mitophagy and Cancer
3.1. Mitophagy Modulators and Cancer Metabolism
3.1.1. Mitophagy and Its Contribution to the Warburg Effect
3.1.2. Mitophagy and OXPHOS
3.1.3. Mitophagy and Iron Metabolism
3.2. Mitophagy and Cancer Stem Cells
3.3. Non-Autonomous Effects of Mitophagy: Mitochondrial Transfer
3.4. Mitophagy, Innate Immunity and Inflammation
4. Mitophagy and Anti-Cancer Therapies
Mitophagy and Cancer-Related Side Effects
5. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGER | Advanced Glycosylation End-Product Specific Receptor |
| AIM2 | Absent In Melanoma 2 |
| AMBRA1 | Autophagy and Beclin 1 Regulator 1 |
| AMBRA1ActA | AMBRA1 fusion protein that localizes in the mitochondria |
| AMPK | AMP-activated protein kinase |
| ARIH1 | Ariadne RBR E3 Ubiquitin Protein Ligase 1 |
| Atg32 | autophagy-related protein 32 |
| ATG5 | autophagy-related protein 5 |
| ATG8 | autophagy-related protein 8 |
| ATG9 | autophagy-related protein 9 |
| ATP | adenosin triphosphate |
| B16 | murine melanoma cell line |
| BCL-2 | B-Cell CLL/Lymphoma 2 |
| BCL2L13/BCL-RAMBO | BCL2 Like 13 |
| Beclin1 | Coiled-Coil Myosin-Like BCL2-Interacting Protein |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| BNIP3L/NIX | BNIP3-like |
| B-RAF | V-Raf Murine Sarcoma Viral Oncogene Homolog B |
| C57Bl6 | mouse strain |
| CAFs | cancer associated fibroblasts |
| CD44 | Cluster of differentiation 44 |
| CD8 | Cluster of Differentiation 8 |
| CDH6 | cadherin6 |
| Cer18 | Ceramide 18 |
| cGAS | Cyclic GMP-AMP Synthase |
| CL | Cardiolipin |
| c-Myc | Proto-Oncogene C-Myc |
| CpG | regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide |
| CSCs | Cancer stem cells |
| DAMPs | damage-associated molecular pattern |
| DNA | Deoxyribonucleic acid |
| DRP1 | Dynamin-related protein 1 |
| EMT | epithelial–mesenchymal transition |
| ER | endoplasmic reticulum |
| ESCRT | endosomal sorting complex |
| F-actin | Filamentous Actin |
| FAO | Fatty acid β-oxidation |
| FIS1 | Fission, Mitochondrial 1 |
| FKBP8/FKBP38 | FK506-binding protein 8 |
| FOXOa3 | Forkhead Box O3 isoform A |
| FUNDC1 | FUN14 Domain Containing 1 |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| GABARAPL1 | GABARAP-like 1 |
| GABARAPL2 | GABARAP-like 2 |
| HBx | Hepatitis B virus X protein |
| HeLa | human cervical cancer cell line |
| HIF1α | Hypoxia-Inducible Factor 1 alpha |
| HMGB1 | High Mobility Group Box 1 |
| HUWE1 | HECT, UBA And WWE Domain Containing E3 Ubiquitin Protein Ligase 1 |
| IECs | Intestinal epithelial cells |
| IGF-1 | insulin-like growth factor 1 |
| IKKα | Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Alpha |
| IL-18 | Interleukin 18 |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| IMM | Inner mitochondrial membranes |
| IMS | Inter-membrane space |
| INF2 | Inverted formin 2 |
| K-RAS | Kirsten Rat Sarcoma Viral Oncogene |
| LC3A | Microtubule-associated proteins 1A/1B light chain 3A |
| LC3B | Microtubule-associated proteins 1A/1B light chain 3B |
| LC3B-II | lipidated from of LC3B |
| LC3C | Microtubule-associated proteins 1A/1B light chain 3C |
| LIR | LC3-interacting domain |
| LON | Lon Peptidase 1 |
| MAMs | ER-mitochondria contact sites |
| MAPK | Mitogen-Activated Protein Kinase |
| MARCH5 | Membrane Associated Ring-CH-Type Finger 5 |
| MAVS | mitochondria-associated adaptor protein |
| MCF-7 | human breast cancer cell line |
| MDA5 | Melanoma Differentiation-Associated Protein 5 |
| Mff | Mitochondrial Fission Factor |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| MiD49 | mitochondrial dynamics protein of 49 kDa |
| MiD51 | mitochondrial dynamics protein of 51 kDa |
| Miro1 | Mitochondrial Rho GTPase 1 |
| Miz1 | Zinc Finger and BTB Domain Containing 17 |
| MSCs | mesenchymal stem cells |
| mtDNA | mitochondrial DNA |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| NANOG | Homeobox Transcription Factor Nanog |
| NDPK-D | Nucleoside Diphosphate Kinase isoform D |
| NF-kB | Nuclear Factor Kappa Beta |
| NK | Natural Killer |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| OCT4 | Octamer-Binding Protein 4 |
| OIP5 | Opa-interacting protein 5 |
| OMA1 | OMA1 Zinc Metallopeptidase |
| OMM | Outer mitochondrial membrane |
| (S/L)-Opa1 | (Short/Long isoform of) Optic Atrophy 1 |
| OPTN | optineurin |
| OXPHOS | Oxidative phosphorylation |
| O2 | Oxygen |
| p53 | Tumor Protein P53 |
| p62/SQSTM1 | Sequestosome 1 |
| Park2 | gene encoding Parkin |
| PARL | Presenilin-associated rhomboid-like |
| PCYT1A | phosphate cytidylyltransferase 1 choline-α |
| PDK2 | Pyruvate Dehydrogenase Kinase 2 |
| PD-L1 | Programmed Cell Death 1 Ligand 1 |
| PGAM5 | phosphoglycerate mutase 5 |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator-1 alpha |
| PHB2 | Prohibitin 2 |
| Pink1 | gene encoding PINK1 |
| PINK1 | Serine/threonine PTEN-induced putative kinase 1 (PINK1) |
| PKM2 | Pyruvate kinase muscle isozyme 2 |
| PP2A | Protein phosphatase 2 A |
| PRRs | pattern recognition receptors |
| PTEN | Phosphatase and Tensin Homolog |
| RABGEF1 | RAB Guanine Nucleotide Exchange Factor 1 |
| RAS | Rat Sarcoma protein, family Small GTP Binding Protein |
| RAS-v12 | RAS mutated in the 12 aminoacid to be constitutively active |
| Rheb | Ras homolog enriched in brain protein |
| RIG-I | Retinoic Acid-Inducible Gene I Protein |
| ROS | Reactive oxygen species |
| SLC25A28/mitoferrin-2 | Solute Carrier Family 25 Member 28 |
| SLC25A37/mitoferrin-1 | Solute Carrier Family 25 Member 37 |
| SOX2 | SRY-Box 2 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| STING | Stimulator of Interferon Genes |
| TBC1D15 | TBC1 Domain Family Member 15 |
| TBC1D17 | TBC1 Domain Family Member 17 |
| TCA | Tricarboxylic acid (cycle) |
| TIM | Translocase of the Inner Membrane |
| TME | Tumor microenvironment |
| TNFα | Tumor Necrosis Factor alpha |
| TOM | Translocase of the Outer Membrane |
| UCP3 | Uncoupling Protein 3 |
| Ulk1 | Unc-51 Like Autophagy Activating Kinase 1 |
| VDAC1 | Voltage Dependent Anion Channel 1 |
| YME1L | YME1-Like Protein 1 |
| ρ0 cells | Cells depleted from mitochondrial content |
References
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Reviews. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Shirihai, O.S.; Song, M.; Dorn, G.W., 2nd. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Macdonald, P.J.; Francy, C.A.; Stepanyants, N.; Lehman, L.; Baglio, A.; Mears, J.A.; Qi, X.; Ramachandran, R. Distinct Splice Variants of Dynamin-related Protein 1 Differentially Utilize Mitochondrial Fission Factor as an Effector of Cooperative GTPase Activity. J. Biol. Chem. 2016, 291, 493–507. [Google Scholar] [CrossRef]
- Francy, C.A.; Clinton, R.W.; Frohlich, C.; Murphy, C.; Mears, J.A. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Sci. Rep. 2017, 7, 10744. [Google Scholar] [CrossRef]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nister, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Et Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Et Biophys. Acta 2014, 1843, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; McQuibban, G.A. The Mitochondrial Rhomboid Protease PARL Is Regulated by PDK2 to Integrate Mitochondrial Quality Control and Metabolism. Cell Rep. 2017, 18, 1458–1472. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Glauser, L.; Sonnay, S.; Stafa, K.; Moore, D.J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 2011, 118, 636–645. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Lopez-Domenech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Schwarten, M.; Mohrluder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Et Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiology. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef]
- Real, P.J.; Benito, A.; Cuevas, J.; Berciano, M.T.; de Juan, A.; Coffer, P.; Gomez-Roman, J.; Lafarga, M.; Lopez-Vega, J.M.; Fernandez-Luna, J.L. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005, 65, 8151–8157. [Google Scholar] [CrossRef]
- Dhingra, R.; Gang, H.; Wang, Y.; Biala, A.K.; Aviv, Y.; Margulets, V.; Tee, A.; Kirshenbaum, L.A. Bidirectional regulation of nuclear factor-kappaB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circ. Heart Fail. 2013, 6, 335–343. [Google Scholar] [CrossRef]
- Frazier, D.P.; Wilson, A.; Graham, R.M.; Thompson, J.W.; Bishopric, N.H.; Webster, K.A. Acidosis regulates the stability, hydrophobicity, and activity of the BH3-only protein Bnip3. Antioxid. Redox Signal. 2006, 8, 1625–1634. [Google Scholar] [CrossRef]
- Hendgen-Cotta, U.B.; Esfeld, S.; Rudi, K.; Miinalainen, I.; Klare, J.P.; Rassaf, T. Cytosolic BNIP3 Dimer Interacts with Mitochondrial BAX Forming Heterodimers in the Mitochondrial Outer Membrane under Basal Conditions. Int. J. Mol. Sci. 2017, 18, 687. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kitamura, N.; Shinogi, D.; Yoshida, M.; Goda, O.; Murai, R.; Kamino, H.; Arakawa, H. BNIP3 and NIX mediate Mieap-induced accumulation of lysosomal proteins within mitochondria. PLoS ONE 2012, 7, e30767. [Google Scholar] [CrossRef]
- Gugnoni, M.; Sancisi, V.; Gandolfi, G.; Manzotti, G.; Ragazzi, M.; Giordano, D.; Tamagnini, I.; Tigano, M.; Frasoldati, A.; Piana, S.; et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene 2017, 36, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnaune-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Wang, C.; Li, F.; Peng, J.; Wen, B.; Gong, Q.; Shi, Y.; Tang, Y. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 2017, 8, 25–38. [Google Scholar] [CrossRef]
- Wu, X.; Wu, F.H.; Wu, Q.; Zhang, S.; Chen, S.; Sima, M. Phylogenetic and Molecular Evolutionary Analysis of Mitophagy Receptors under Hypoxic Conditions. Front. Physiol. 2017, 8, 539. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef]
- Sekine, S.; Kanamaru, Y.; Koike, M.; Nishihara, A.; Okada, M.; Kinoshita, H.; Kamiyama, M.; Maruyama, J.; Uchiyama, Y.; Ishihara, N.; et al. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J. Biol. Chem. 2012, 287, 34635–34645. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Sugo, M.; Kimura, H.; Arasaki, K.; Amemiya, T.; Hirota, N.; Dohmae, N.; Imai, Y.; Inoshita, T.; Shiba-Fukushima, K.; Hattori, N.; et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Chen, Z.; Siraj, S.; Liu, L.; Chen, Q. MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 2017, 13, 1244–1245. [Google Scholar] [CrossRef]
- Chu, C.T.; Bayir, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef]
- Anton, Z.; Landajuela, A.; Hervas, J.H.; Montes, L.R.; Hernandez-Tiedra, S.; Velasco, G.; Goni, F.M.; Alonso, A. Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 2016, 12, 2386–2403. [Google Scholar] [CrossRef]
- Kagan, V.E.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Desbourdes, C.; Cottet-Rousselle, C.; Dar, H.H.; Verma, M.; Tyurin, V.A.; Kapralov, A.A.; et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016, 23, 1140–1151. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Di Rita, A.; D’Acunzo, P.; Simula, L.; Campello, S.; Strappazzon, F.; Cecconi, F. AMBRA1-Mediated Mitophagy Counteracts Oxidative Stress and Apoptosis Induced by Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Front. Cell. Neurosci. 2018, 12, 92. [Google Scholar] [CrossRef]
- Di Rita, A.; Peschiaroli, A.; Pasquale, D.; Strobbe, D.; Hu, Z.; Gruber, J.; Nygaard, M.; Lambrughi, M.; Melino, G.; Papaleo, E.; et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat. Commun. 2018, 9, 3755. [Google Scholar] [CrossRef]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [PubMed]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, A.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 1–17. [Google Scholar] [CrossRef]
- Ju, L.; Chen, S.; Alimujiang, M.; Bai, N.; Yan, H.; Fang, Q.; Han, J.; Ma, X.; Yang, Y.; Jia, W. A novel role for Bcl2l13 in promoting beige adipocyte biogenesis. Biochem. Biophys. Res. Commun. 2018, 506, 485–491. [Google Scholar] [CrossRef]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjottem, E.; Brenne, H.B.; Overvatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Saita, S.; Shirane, M.; Nakayama, K.I. Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 2013, 4, 1410. [Google Scholar] [CrossRef]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Munch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. eLife 2018, 7. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Lyu, Z.; Chen, Y.; Ji, X.; Cao, H.; Jin, M.; Zhu, J.; Yang, J.; Ling, R.; et al. Positive feedback loop between mitochondrial fission and Notch signaling promotes survivin-mediated survival of TNBC cells. Cell Death Dis. 2018, 9, 1050. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Philley, J.V.; Kannan, A.; Qin, W.; Sauter, E.R.; Ikebe, M.; Hertweck, K.L.; Troyer, D.A.; Semmes, O.J.; Dasgupta, S. Complex-I Alteration and Enhanced Mitochondrial Fusion Are Associated With Prostate Cancer Progression. J. Cell Physiol. 2016, 231, 1364–1374. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Murphy, B.; Mustafi, S.B.; Dey, A.; Xiong, X.; Rao, G.; Naz, S.; Zhang, M.; Yang, D.; Dhanasekaran, D.N.; et al. Cystathionine beta-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018, 32, 4145–4157. [Google Scholar] [CrossRef]
- Soares, C.D.; Morais, T.M.L.; Carlos, R.; de Almeida, O.P.; Mariano, F.V.; Altemani, A.; de Carvalho, M.G.F.; Correa, M.B.; Dos Reis, R.R.D.; Amorim, L.S.; et al. Prognostic importance of mitochondrial markers in mucosal and cutaneous head and neck melanomas. Hum. Pathol. 2019, 85, 279–289. [Google Scholar] [CrossRef]
- Fang, C.L.; Sun, D.P.; Chen, H.K.; Lin, C.C.; Hung, S.T.; Uen, Y.H.; Lin, K.Y. Overexpression of Mitochondrial GTPase MFN2 Represents a Negative Prognostic Marker in Human Gastric Cancer and Its Inhibition Exerts Anti-Cancer Effects. J. Cancer 2017, 8, 1153–1161. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Sun, J.; Gong, W.; Sun, P.; Kong, X.; Yang, M.; Zhang, W. Mitofusin-2 acts as biomarker for predicting poor prognosis in hepatitis B virus related hepatocellular carcinoma. Infect. Agent Cancer 2018, 13, 36. [Google Scholar] [CrossRef]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, D.; Xu, X.; Zhao, X.; Huang, P.; Zhou, X.; Song, W.; Guo, H.; Wang, W.; Zheng, S. Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma. World J. Surg. Oncol. 2016, 14, 179. [Google Scholar] [CrossRef]
- Cheng, C.T.; Kuo, C.Y.; Ouyang, C.; Li, C.F.; Chung, Y.; Chan, D.C.; Kung, H.J.; Ann, D.K. Metabolic Stress-Induced Phosphorylation of KAP1 Ser473 Blocks Mitochondrial Fusion in Breast Cancer Cells. Cancer Res. 2016, 76, 5006–5018. [Google Scholar] [CrossRef]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef]
- Kannan, A.; Wells, R.B.; Sivakumar, S.; Komatsu, S.; Singh, K.P.; Samten, B.; Philley, J.V.; Sauter, E.R.; Ikebe, M.; Idell, S.; et al. Mitochondrial Reprogramming Regulates Breast Cancer Progression. Clin. Cancer Res. 2016, 22, 3348–3360. [Google Scholar] [CrossRef]
- Cruz, M.D.; Ledbetter, S.; Chowdhury, S.; Tiwari, A.K.; Momi, N.; Wali, R.K.; Bliss, C.; Huang, C.; Lichtenstein, D.; Bhattacharya, S.; et al. Metabolic reprogramming of the premalignant colonic mucosa is an early event in carcinogenesis. Oncotarget 2017, 8, 20543–20557. [Google Scholar] [CrossRef]
- Fang, H.Y.; Chen, C.Y.; Chiou, S.H.; Wang, Y.T.; Lin, T.Y.; Chang, H.W.; Chiang, I.P.; Lan, K.J.; Chow, K.C. Overexpression of optic atrophy 1 protein increases cisplatin resistance via inactivation of caspase-dependent apoptosis in lung adenocarcinoma cells. Hum. Pathol. 2012, 43, 105–114. [Google Scholar] [CrossRef]
- Kang, J.U.; Koo, S.H.; Kwon, K.C.; Park, J.W.; Kim, J.M. Identification of novel candidate target genes, including EPHB3, MASP1 and SST at 3q26.2-q29 in squamous cell carcinoma of the lung. BMC Cancer 2009, 9, 237. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, C.; Puszyk, W.M.; Ogunwobi, O.O.; Cao, M.; Wang, T.; Cabrera, R.; Nelson, D.R.; Liu, C. OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab. Investig. 2013, 93, 8–19. [Google Scholar] [CrossRef]
- Tanwar, D.K.; Parker, D.J.; Gupta, P.; Spurlock, B.; Alvarez, R.D.; Basu, M.K.; Mitra, K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget 2016, 7, 60021–60037. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Yun, S.H.; Yun, J. Downregulation of Drp1, a fission regulator, is associated with human lung and colon cancers. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 209–215. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Chen, W.T.; Ke, T.W.; Chen, T.W.; You, Y.S.; Lin, C.Y.; Chao, K.S.C.; Huang, C.Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Wieder, S.Y.; Serasinghe, M.N.; Sung, J.C.; Choi, D.C.; Birge, M.B.; Yao, J.L.; Bernstein, E.; Celebi, J.T.; Chipuk, J.E. Activation of the Mitochondrial Fragmentation Protein DRP1 Correlates with BRAF(V600E) Melanoma. J. Investig. Dermatol. 2015, 135, 2544–2547. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef]
- Fan, S.; Liu, B.; Sun, L.; Lv, X.B.; Lin, Z.; Chen, W.; Chen, W.; Tang, Q.; Wang, Y.; Su, Y.; et al. Mitochondrial fission determines cisplatin sensitivity in tongue squamous cell carcinoma through the BRCA1-miR-593-5p-MFF axis. Oncotarget 2015, 6, 14885–14904. [Google Scholar] [CrossRef]
- Abo Elwafa, R.; Gamaleldin, M.; Ghallab, O. The clinical and prognostic significance of FIS1, SPI1, PDCD7 and Ang2 expression levels in acute myeloid leukemia. Cancer Genet. 2018. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: Over-expression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef]
- Hsiao, C.P.; Wang, D.; Kaushal, A.; Saligan, L. Mitochondria-related gene expression changes are associated with fatigue in patients with nonmetastatic prostate cancer receiving external beam radiation therapy. Cancer Nurs. 2013, 36, 189–197. [Google Scholar] [CrossRef]
- Fan, S.; Chen, W.X.; Lv, X.B.; Tang, Q.L.; Sun, L.J.; Liu, B.D.; Zhong, J.L.; Lin, Z.Y.; Wang, Y.Y.; Li, Q.X.; et al. miR-483-5p determines mitochondrial fission and cisplatin sensitivity in tongue squamous cell carcinoma by targeting FIS1. Cancer Lett. 2015, 362, 183–191. [Google Scholar] [CrossRef]
- Chang, G.; Zhang, W.; Ma, Y.; Wen, Q. PINK1 Expression Is Associated with Poor Prognosis in Lung Adenocarcinoma. Tohoku J. Exp Med. 2018, 245, 115–121. [Google Scholar] [CrossRef]
- Yamashita, K.; Miyata, H.; Makino, T.; Masuike, Y.; Furukawa, H.; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. High Expression of the Mitophagy-Related Protein Pink1 is Associated with a Poor Response to Chemotherapy and a Poor Prognosis for Patients Treated with Neoadjuvant Chemotherapy for Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2017, 24, 4025–4032. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar] [CrossRef]
- Bhat, Z.I.; Kumar, B.; Bansal, S.; Naseem, A.; Tiwari, R.R.; Wahabi, K.; Sharma, G.D.; Alam Rizvi, M.M. Association of PARK2 promoter polymorphisms and methylation with colorectal cancer in North Indian population. Gene 2019, 682, 25–32. [Google Scholar] [CrossRef]
- da Silva-Camargo, C.C.V.; Svoboda Baldin, R.K.; Costacurta Polli, N.L.; Agostinho, A.P.; Olandosk, M.; de Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Kannengiesser, C.; Lesage, S.; Andre, J.; Mourah, S.; Michel, L.; Descamps, V.; Basset-Seguin, N.; Bagot, M.; Bensussan, A.; et al. PARKIN Inactivation Links Parkinson’s Disease to Melanoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Gao, G.; Kasperbauer, J.L.; Tombers, N.M.; Wang, V.; Mayer, K.; Smith, D.I. A selected group of large common fragile site genes have decreased expression in oropharyngeal squamous cell carcinomas. Genes Chromosom. Cancer 2014, 53, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.I.; Wuttig, D.; Kaiser, S.; Herr, A.; Weber, T.; Zastrow, S.; Koch, R.; Meinhardt, M.; Baretton, G.B.; Wirth, M.P.; et al. PARK2 and PACRG are commonly downregulated in clear-cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes Chromosom. Cancer 2013, 52, 265–273. [Google Scholar] [CrossRef]
- Mehdi, S.J.; Ali, A.; Rizvi, M.M. Parkin gene alterations in ovarian carcinoma from northern Indian population. Pathol. Oncol. Res. 2011, 17, 579–586. [Google Scholar] [CrossRef]
- Agirre, X.; Roman-Gomez, J.; Vazquez, I.; Jimenez-Velasco, A.; Garate, L.; Montiel-Duarte, C.; Artieda, P.; Cordeu, L.; Lahortiga, I.; Calasanz, M.J.; et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. Int. J. Cancer 2006, 118, 1945–1953. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455. [Google Scholar] [CrossRef]
- Jiang, Z.; Yu, F.; Li, M. Upregulation of BCL2 19 kD Protein-Interacting Protein 3 (BNIP3) is Predictive of Unfavorable Prognosis in Uveal Melanoma. Med. Sci. Monit. 2018, 24, 4711–4717. [Google Scholar] [CrossRef]
- Schulten, H.J.; Bangash, M.; Karim, S.; Dallol, A.; Hussein, D.; Merdad, A.; Al-Thoubaity, F.K.; Al-Maghrabi, J.; Jamal, A.; Al-Ghamdi, F.; et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J. Transl. Med. 2017, 15, 269. [Google Scholar] [CrossRef]
- Xu, Q.; Junttila, S.; Scherer, A.; Giri, K.R.; Kivela, O.; Skovorodkin, I.; Roning, J.; Quaggin, S.E.; Marti, H.P.; Shan, J.; et al. Renal carcinoma/kidney progenitor cell chimera organoid as a novel tumorigenesis gene discovery model. Dis. Model. Mech. 2017, 10, 1503–1515. [Google Scholar] [CrossRef]
- Chen, J.L.; David, J.; Cook-Spaeth, D.; Casey, S.; Cohen, D.; Selvendiran, K.; Bekaii-Saab, T.; Hays, J.L. Autophagy Induction Results in Enhanced Anoikis Resistance in Models of Peritoneal Disease. Mol. Cancer Res. 2017, 15, 26–34. [Google Scholar] [CrossRef]
- Fujimoto, T.; Ohtsuka, T.; Date, K.; Kimura, H.; Matsunaga, T.; Mori, Y.; Miyasaka, Y.; Mochidome, N.; Oda, Y.; Nakamura, M. Expression of Bcl-2 19-kDa interacting protein 3 predicts prognosis after ampullary carcinoma resection. J. Hepatobiliary Pancreat Sci. 2016, 23, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.S.; Kim, J.W.; Yoon, J.S. Expression of Autophagy and Reactive Oxygen Species-Related Proteins in Lacrimal Gland Adenoid Cystic Carcinoma. Yonsei Med. J. 2016, 57, 482–489. [Google Scholar] [CrossRef]
- Petrova, V.; Mancini, M.; Agostini, M.; Knight, R.A.; Annicchiarico-Petruzzelli, M.; Barlev, N.A.; Melino, G.; Amelio, I. TAp73 transcriptionally represses BNIP3 expression. Cell Cycle 2015, 14, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Yongvanit, P.; Loilome, W.; Seubwai, W.; Phunicom, K.; Tassaneeyakul, W.; Pairojkul, C.; Promkotra, W.; Techasen, A.; Namwat, N. High expression of HIF-1alpha, BNIP3 and PI3KC3: Hypoxia-induced autophagy predicts cholangiocarcinoma survival and metastasis. Asian Pac. J. Cancer Prev. 2014, 15, 5873–5878. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, C.; Dachs, G.U.; Munn, D.; Currie, M.J.; Robinson, B.A.; Pearson, J.F.; Vissers, M.C. Increased Tumor Ascorbate is Associated with Extended Disease-Free Survival and Decreased Hypoxia-Inducible Factor-1 Activation in Human Colorectal Cancer. Front. Oncol. 2014, 4, 10. [Google Scholar] [CrossRef]
- Karpathiou, G.; Sivridis, E.; Koukourakis, M.; Mikroulis, D.; Bouros, D.; Froudarakis, M.; Bougioukas, G.; Maltezos, E.; Giatromanolaki, A. Autophagy and Bcl-2/BNIP3 death regulatory pathway in non-small cell lung carcinomas. APMIS 2013, 121, 592–604. [Google Scholar] [CrossRef]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Petry, I.B.; Fieber, E.; Schmidt, M.; Gehrmann, M.; Gebhard, S.; Hermes, M.; Schormann, W.; Selinski, S.; Freis, E.; Schwender, H.; et al. ERBB2 induces an antiapoptotic expression pattern of Bcl-2 family members in node-negative breast cancer. Clin. Cancer Res. 2010, 16, 451–460. [Google Scholar] [CrossRef]
- Lukashova-v Zangen, I.; Kneitz, S.; Monoranu, C.M.; Rutkowski, S.; Hinkes, B.; Vince, G.H.; Huang, B.; Roggendorf, W. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. 2007, 113, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.; Horn, L.C.; Hockel, M. Hypoxia and expression of the proapoptotic regulator BNIP3 in cervical cancer. Int. J. Gynecol. Cancer 2006, 16, 1314–1320. [Google Scholar] [CrossRef]
- An, J.S.; Huang, M.N.; Song, Y.M.; Li, N.; Wu, L.Y.; Zhan, Q.M. A preliminary study of genes related to concomitant chemoradiotherapy resistance in advanced uterine cervical squamous cell carcinoma. Chin. Med. J. (Engl.) 2013, 126, 4109–4115. [Google Scholar] [PubMed]
- Macher-Goeppinger, S.; Keith, M.; Hatiboglu, G.; Hohenfellner, M.; Schirmacher, P.; Roth, W.; Tagscherer, K.E. Expression and Functional Characterization of the BNIP3 Protein in Renal Cell Carcinomas. Transl. Oncol. 2017, 10, 869–875. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.F.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Pei, L.; Jiang, H.; Yang, W.; Chen, J.; Liang, H. Chemoresistance of colorectal cancer to 5-fluorouracil is associated with silencing of the BNIP3 gene through aberrant methylation. J. Cancer 2017, 8, 1187–1196. [Google Scholar] [CrossRef]
- Sambuudash, O.; Kim, H.S.; Cho, M.Y. Lack of Aberrant Methylation in an Adjacent Area of Left-Sided Colorectal Cancer. Yonsei Med. J. 2017, 58, 749–755. [Google Scholar] [CrossRef]
- Peng, X.; Xue, H.; Lu, L.; Shi, P.; Wang, J.; Wang, J. Accumulated promoter methylation as a potential biomarker for esophageal cancer. Oncotarget 2017, 8, 679–691. [Google Scholar] [CrossRef]
- Kamino, H.; Nakamura, Y.; Tsuneki, M.; Sano, H.; Miyamoto, Y.; Kitamura, N.; Futamura, M.; Kanai, Y.; Taniguchi, H.; Shida, D.; et al. Mieap-regulated mitochondrial quality control is frequently inactivated in human colorectal cancer. Oncogenesis 2016, 4, e181. [Google Scholar] [CrossRef] [PubMed]
- Simsek, B.C.; Turk, B.A.; Ozen, F.; Tuzcu, M.; Kanter, M. Investigation of telomerase activity and apoptosis on invasive ductal carcinoma of the breast using immunohistochemical and Western blot methods. Eur. Rev. Med. Pharm. Sci. 2015, 19, 3089–3099. [Google Scholar]
- Liu, Z.; Zhang, J.; Gao, Y.; Pei, L.; Zhou, J.; Gu, L.; Zhang, L.; Zhu, B.; Hattori, N.; Ji, J.; et al. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin. Cancer Res. 2014, 20, 4598–4612. [Google Scholar] [CrossRef]
- Garcia-Baquero, R.; Puerta, P.; Beltran, M.; Alvarez-Mujica, M.; Alvarez-Ossorio, J.L.; Sanchez-Carbayo, M. Methylation of tumor suppressor genes in a novel panel predicts clinical outcome in paraffin-embedded bladder tumors. Tumour Biol.: J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 5777–5786. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Baquero, R.; Puerta, P.; Beltran, M.; Alvarez, M.; Sacristan, R.; Alvarez-Ossorio, J.L.; Sanchez-Carbayo, M. Methylation of a novel panel of tumor suppressor genes in urine moves forward noninvasive diagnosis and prognosis of bladder cancer: A 2-center prospective study. J. Urol. 2013, 190, 723–730. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, C.M.; Chen, N.; Li, L.; Wang, X.D.; Zhang, W.; Bi, F.; Tang, Q.L.; Li, Z.P.; Wang, W. Chemotherapy and radiotherapy downregulate the activity and expression of DNA methyltransferase and enhance Bcl-2/E1B-19-kDa interacting protein-3-induced apoptosis in human colorectal cancer cells. Chemotherapy 2012, 58, 445–453. [Google Scholar] [CrossRef]
- Jin, T.; Lin, H.X.; Lin, H.; Guo, L.B.; Ge, N.; Cai, X.Y.; Sun, R.; Chen, W.K.; Li, Q.L.; Hu, W.H. Expression TGM2 and BNIP3 have prognostic significance in laryngeal cancer patients receiving surgery and postoperative radiotherapy: A retrospective study. J. Transl. Med. 2012, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Sugita, H.; Iida, S.; Inokuchi, M.; Kato, K.; Ishiguro, M.; Ishikawa, T.; Takagi, Y.; Enjoji, M.; Yamada, H.; Uetake, H.; et al. Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer. Oncol. Rep. 2011, 25, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Koga, Y.; Tanaka, T.; Nakamura, J.; Hashiguchi, K.; Noshiro, H.; Miyazaki, K. Aberrant gene methylation is a biomarker for the detection of cancer cells in peritoneal wash samples from advanced gastric cancer patients. Ann. Surg. Oncol. 2011, 18, 3013–3019. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Iida, S.; Ishiguro, M.; Uetake, H.; Ishikawa, T.; Takagi, Y.; Kobayashi, H.; Higuchi, T.; Enomoto, M.; Mogushi, K.; et al. Methylated BNIP3 gene in colorectal cancer prognosis. Oncol. Lett. 2010, 1, 865–872. [Google Scholar] [CrossRef]
- Hiraki, M.; Kitajima, Y.; Nakafusa, Y.; Nakamura, J.; Hashiguchi, K.; Sumi, K.; Noshiro, H.; Miyazaki, K. CpG island methylation of BNIP3 predicts resistance against S-1/CPT-11 combined therapy in colorectal cancer patients. Oncol. Rep. 2010, 23, 191–197. [Google Scholar]
- Jourdan, M.; Reme, T.; Goldschmidt, H.; Fiol, G.; Pantesco, V.; De Vos, J.; Rossi, J.F.; Hose, D.; Klein, B. Gene expression of anti- and pro-apoptotic proteins in malignant and normal plasma cells. Br. J. Haematol. 2009, 145, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Schmidt, W.M.; Ziegler, B.; Holzer, S.; Mullauer, L.; Bilban, M.; Zielinski, C.C.; Drach, J.; Zochbauer-Muller, S. Genome-wide transcriptional response to 5-aza-2’-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008, 68, 44–54. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Buchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef] [PubMed]
- Akada, M.; Crnogorac-Jurcevic, T.; Lattimore, S.; Mahon, P.; Lopes, R.; Sunamura, M.; Matsuno, S.; Lemoine, N.R. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin. Cancer Res. 2005, 11, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.; Grau, L.; Puerta, P.; Gimenez, L.; Venditti, J.; Quadrelli, S.; Sanchez-Carbayo, M. Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J. Transl. Med. 2010, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, J.W.; Wang, Y.; Chen, T.; Ren, N.; Yang, L.; Xu, W.; He, H.; Jiang, Y.; Chen, X.; et al. Abnormal miRNA-30e Expression is Associated with Breast Cancer Progression. Clin. Lab. 2016, 62, 121–128. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, L.; He, M.; Huang, W.; Li, H.; Wang, Y.; Kong, J.; Qi, S.; Ouyang, J.; Qiu, X. Nix protein positively regulates NF-kappaB activation in gliomas. PLoS ONE 2012, 7, e44559. [Google Scholar] [CrossRef]
- Liu, W.; Xie, C.C.; Zhu, Y.; Li, T.; Sun, J.; Cheng, Y.; Ewing, C.M.; Dalrymple, S.; Turner, A.R.; Sun, J.; et al. Homozygous deletions and recurrent amplifications implicate new genes involved in prostate cancer. Neoplasia 2008, 10, 897–907. [Google Scholar] [CrossRef]
- Eisele, L.; Klein-Hitpass, L.; Chatzimanolis, N.; Opalka, B.; Boes, T.; Seeber, S.; Moritz, T.; Flasshove, M. Differential expression of drug-resistance-related genes between sensitive and resistant blasts in acute myeloid leukemia. Acta Haematol. 2007, 117, 8–15. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, D.; Zhou, L.; Pei, Y.; Zhuang, Y.; Cui, W.; Chen, J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 2019, 41, 384–394. [Google Scholar] [CrossRef]
- Hui, L.; Wu, H.; Wang, T.W.; Yang, N.; Guo, X.; Jang, X.J. Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 2018. [Google Scholar] [CrossRef]
- Hou, H.; Er, P.; Cheng, J.; Chen, X.; Ding, X.; Wang, Y.; Chen, X.; Yuan, Z.; Pang, Q.; Wang, P.; et al. High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 2017, 6, 1871–1881. [Google Scholar] [CrossRef]
- Cheng, J.; Qian, D.; Ding, X.; Song, T.; Cai, M.; Dan, X.; Wang, Y.; Zhao, J.; Liu, Z.; Wu, Z.; et al. High PGAM5 expression induces chemoresistance by enhancing Bcl-xL-mediated anti-apoptotic signaling and predicts poor prognosis in hepatocellular carcinoma patients. Cell Death Dis. 2018, 9, 991. [Google Scholar] [CrossRef]
- Ng Kee Kwong, F.; Nicholson, A.G.; Pavlidis, S.; Adcock, I.M.; Chung, K.F. PGAM5 expression and macrophage signatures in non-small cell lung cancer associated with chronic obstructive pulmonary disease (COPD). BMC Cancer 2018, 18, 1238. [Google Scholar] [CrossRef]
- Randall, E.C.; Zadra, G.; Chetta, P.; Lopez, B.G.C.; Syamala, S.; Basu, S.S.; Agar, J.N.; Loda, M.; Tempany, C.M.; Fennessy, F.M.; et al. Molecular Characterization of Prostate Cancer with Associated Gleason Score Using Mass Spectrometry Imaging. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Sapandowski, A.; Stope, M.; Evert, K.; Evert, M.; Zimmermann, U.; Peter, D.; Page, I.; Burchardt, M.; Schild, L. Cardiolipin composition correlates with prostate cancer cell proliferation. Mol. Cell Biochem. 2015, 410, 175–185. [Google Scholar] [CrossRef]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: A novel link between oxidative stress and cancer. Free Radic. Biol. Med. 2017, 102, 67–76. [Google Scholar] [CrossRef]
- Cai, X.W.; Yu, W.W.; Yu, W.; Zhang, Q.; Feng, W.; Liu, M.N.; Sun, M.H.; Xiang, J.Q.; Zhang, Y.W.; Fu, X.L. Tissue-based quantitative proteomics to screen and identify the potential biomarkers for early recurrence/metastasis of esophageal squamous cell carcinoma. Cancer Med. 2018, 7, 2504–2517. [Google Scholar] [CrossRef]
- Ross, J.A.; Robles-Escajeda, E.; Oaxaca, D.M.; Padilla, D.L.; Kirken, R.A. The prohibitin protein complex promotes mitochondrial stabilization and cell survival in hematologic malignancies. Oncotarget 2017, 8, 65445–65456. [Google Scholar] [CrossRef]
- Yoshimaru, T.; Ono, M.; Bando, Y.; Chen, Y.A.; Mizuguchi, K.; Shima, H.; Komatsu, M.; Imoto, I.; Izumi, K.; Honda, J.; et al. A-kinase anchoring protein BIG3 coordinates oestrogen signalling in breast cancer cells. Nat. Commun. 2017, 8, 15427. [Google Scholar] [CrossRef]
- Zuo, X.; Chen, L.; Liu, L.; Zhang, Z.; Zhang, X.; Yu, Q.; Feng, L.; Zhao, X.; Qin, T. Identification of a panel of complex autoantigens (LGALS3, PHB2, MUC1, and GK2) in combination with CA15-3 for the diagnosis of early-stage breast cancer. Tumour Biol.: J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 1309–1317. [Google Scholar] [CrossRef]
- Mengwasser, J.; Piau, A.; Schlag, P.; Sleeman, J.P. Differential immunization identifies PHB1/PHB2 as blood-borne tumor antigens. Oncogene 2004, 23, 7430–7435. [Google Scholar] [CrossRef]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of ceramide synthase 1 increases C18-ceramide and leads to lethal autophagy in human glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef]
- Qu, B.; Yao, L.; Ma, H.L.; Chen, H.L.; Zhang, Z.; Xie, J. Prognostic significance of autophagy-related proteins expression in resected human gastric adenocarcinoma. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 37–43. [Google Scholar] [CrossRef]
- Nitta, T.; Sato, Y.; Ren, X.S.; Harada, K.; Sasaki, M.; Hirano, S.; Nakanuma, Y. Autophagy may promote carcinoma cell invasion and correlate with poor prognosis in cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4913–4921. [Google Scholar]
- Ko, Y.H.; Cho, Y.S.; Won, H.S.; Jeon, E.K.; An, H.J.; Hong, S.U.; Park, J.H.; Lee, M.A. Prognostic significance of autophagy-related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas 2013, 42, 829–835. [Google Scholar] [CrossRef]
- Falasca, L.; Torino, F.; Marconi, M.; Costantini, M.; Pompeo, V.; Sentinelli, S.; De Salvo, L.; Patrizio, M.; Padula, C.; Gallucci, M.; et al. AMBRA1 and SQSTM1 expression pattern in prostate cancer. Apoptosis 2015, 20, 1577–1586. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Yang, D.; Cheng, D.; Tu, Q.; Yang, H.; Sun, B.; Yan, L.; Dai, H.; Luo, J.; Mao, B.; Cao, Y.; et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018, 8, 3517–3529. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Z.; Gu, S.; Zheng, J.; Feng, D.; Qin, Q.; He, J. Global Analysis of miRNA-mRNA Interaction Network in Breast Cancer with Brain Metastasis. Anticancer Res. 2017, 37, 4455–4468. [Google Scholar] [CrossRef][Green Version]
- Kodama, T.; Newberg, J.Y.; Kodama, M.; Rangel, R.; Yoshihara, K.; Tien, J.C.; Parsons, P.H.; Wu, H.; Finegold, M.J.; Copeland, N.G.; et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, E3384–E3393. [Google Scholar] [CrossRef]
- Cheng, D.D.; Yu, T.; Hu, T.; Yao, M.; Fan, C.Y.; Yang, Q.C. MiR-542-5p is a negative prognostic factor and promotes osteosarcoma tumorigenesis by targeting HUWE1. Oncotarget 2015, 6, 42761–42772. [Google Scholar] [CrossRef]
- Yang, Y.L.; Lin, S.R.; Chen, J.S.; Lin, S.W.; Yu, S.L.; Chen, H.Y.; Yen, C.T.; Lin, C.Y.; Lin, J.F.; Lin, K.H.; et al. Expression and prognostic significance of the apoptotic genes BCL2L13, Livin, and CASP8AP2 in childhood acute lymphoblastic leukemia. Leuk Res. 2010, 34, 18–23. [Google Scholar] [CrossRef]
- Tahir, S.K.; Wass, J.; Joseph, M.K.; Devanarayan, V.; Hessler, P.; Zhang, H.; Elmore, S.W.; Kroeger, P.E.; Tse, C.; Rosenberg, S.H.; et al. Identification of expression signatures predictive of sensitivity to the Bcl-2 family member inhibitor ABT-263 in small cell lung carcinoma and leukemia/lymphoma cell lines. Mol. Cancer Ther. 2010, 9, 545–557. [Google Scholar] [CrossRef]
- Holleman, A.; den Boer, M.L.; de Menezes, R.X.; Cheok, M.H.; Cheng, C.; Kazemier, K.M.; Janka-Schaub, G.E.; Gobel, U.; Graubner, U.B.; Evans, W.E.; et al. The expression of 70 apoptosis genes in relation to lineage, genetic subtype, cellular drug resistance, and outcome in childhood acute lymphoblastic leukemia. Blood 2006, 107, 769–776. [Google Scholar] [CrossRef]
- Millino, C.; Maretto, I.; Pacchioni, B.; Digito, M.; De Paoli, A.; Canzonieri, V.; D’Angelo, E.; Agostini, M.; Rizzolio, F.; Giordano, A.; et al. Gene and MicroRNA Expression Are Predictive of Tumor Response in Rectal Adenocarcinoma Patients Treated With Preoperative Chemoradiotherapy. J. Cell Physiol. 2017, 232, 426–435. [Google Scholar] [CrossRef]
- Leon-Mateos, L.; Casas, H.; Abalo, A.; Vieito, M.; Abreu, M.; Anido, U.; Gomez-Tato, A.; Lopez, R.; Abal, M.; Muinelo-Romay, L. Improving circulating tumor cells enumeration and characterization to predict outcome in first line chemotherapy mCRPC patients. Oncotarget 2017, 8, 54708–54721. [Google Scholar] [CrossRef]
- da Silva, S.D.; Marchi, F.A.; Xu, B.; Bijian, K.; Alobaid, F.; Mlynarek, A.; Rogatto, S.R.; Hier, M.; Kowalski, L.P.; Alaoui-Jamali, M.A. Predominant Rab-GTPase amplicons contributing to oral squamous cell carcinoma progression to metastasis. Oncotarget 2015, 6, 21950–21963. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Et Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef]
- Chang, H.W.; Kim, M.R.; Lee, H.J.; Lee, H.M.; Kim, G.C.; Lee, Y.S.; Nam, H.Y.; Lee, M.; Jang, H.J.; Lee, K.E.; et al. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Maes, H.; Van Eygen, S.; Krysko, D.V.; Vandenabeele, P.; Nys, K.; Rillaerts, K.; Garg, A.D.; Verfaillie, T.; Agostinis, P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014, 5, e1127. [Google Scholar] [CrossRef]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, L.; Fei, X.C.; Jiang, X.F.; Zheng, Z.; Zhao, Y.; Wang, C.F.; Li, B.; Chen, S.J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1alpha in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef]
- Park, J.H.; Ko, J.; Park, Y.S.; Park, J.; Hwang, J.; Koh, H.C. Clearance of Damaged Mitochondria Through PINK1 Stabilization by JNK and ERK MAPK Signaling in Chlorpyrifos-Treated Neuroblastoma Cells. Mol. Neurobiol. 2017, 54, 1844–1857. [Google Scholar] [CrossRef]
- Bazil, J.N.; Beard, D.A.; Vinnakota, K.C. Catalytic Coupling of Oxidative Phosphorylation, ATP Demand, and Reactive Oxygen Species Generation. Biophys. J. 2016, 110, 962–971. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Maes, H.; Van Dingenen, S.; Agostinis, P. BNIP3 contributes to the glutamine-driven aggressive behavior of melanoma cells. Biol. Chem. 2019, 400, 187–193. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Du, Q.; Tan, Z.; Shi, F.; Tang, M.; Xie, L.; Zhao, L.; Li, Y.; Hu, J.; Zhou, M.; Bode, A.; et al. PGC1alpha/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer Sci. 2019. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1alpha buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706. [Google Scholar] [CrossRef]
- Inoue, S.; Hao, Z.; Elia, A.J.; Cescon, D.; Zhou, L.; Silvester, J.; Snow, B.; Harris, I.S.; Sasaki, M.; Li, W.Y.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef]
- Strappazzon, F.; Cecconi, F. AMBRA1-induced mitophagy: A new mechanism to cope with cancer? Mol. Cell Oncol. 2015, 2, e975647. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef]
- Gargini, R.; Garcia-Escudero, V.; Izquierdo, M.; Wandosell, F. Oncogene-mediated tumor transformation sensitizes cells to autophagy induction. Oncol. Rep. 2016, 35, 3689–3695. [Google Scholar] [CrossRef]
- Huang, X.Y.; Li, D.; Chen, Z.X.; Huang, Y.H.; Gao, W.Y.; Zheng, B.Y.; Wang, X.Z. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp. Cell Res. 2018, 368, 75–83. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef]
- Kang, R.; Xie, Y.; Zeh, H.J.; Klionsky, D.J.; Tang, D. Mitochondrial quality control mediated by PINK1 and PRKN: Links to iron metabolism and tumor immunity. Autophagy 2019, 15, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, O.; De Oliveira, T.; Diamanti, M.A.; Muller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef]
- Fessler, E.; Dijkgraaf, F.E.; De Sousa, E.M.F.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett. 2013, 341, 97–104. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292. [Google Scholar] [CrossRef]
- Lathia, J.D.; Liu, H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef]
- Whelan, K.A.; Chandramouleeswaran, P.M.; Tanaka, K.; Natsuizaka, M.; Guha, M.; Srinivasan, S.; Darling, D.S.; Kita, Y.; Natsugoe, S.; Winkler, J.D.; et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene 2017, 36, 4843–4858. [Google Scholar] [CrossRef]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.A.; Wang, C.Y.; Hsieh, Y.T.; Chen, Y.J.; Wei, Y.H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, R.; Shen, J.; Wu, J.; Lu, W.; Zhang, J.; Zhang, J.; Liu, K. Human Bone Marrow Mesenchymal Stem Cells Rescue Endothelial Cells Experiencing Chemotherapy Stress by Mitochondrial Transfer Via Tunneling Nanotubes. Stem Cells Dev. 2019. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [PubMed]
- Nzigou Mombo, B.; Gerbal-Chaloin, S.; Bokus, A.; Daujat-Chavanieu, M.; Jorgensen, C.; Hugnot, J.P.; Vignais, M.L. MitoCeption: Transferring Isolated Human MSC Mitochondria to Glioblastoma Stem Cells. J. Vis. Exp.: JoVE 2017. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.P.; Trevor, J.L.; Strenkowski, J.G.; Wang, Y.; Chacko, B.K.; Tousif, S.; Chanda, D.; Steele, C.; Antony, V.B.; Dokland, T.; et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 2018, 18, 54–64. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernandez-Delgado, I.; Latorre-Pellicer, A.; Acin-Perez, R.; Martin-Cofreces, N.B.; Jaso-Tamame, A.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Lou, E.; Zhai, E.; Sarkari, A.; Desir, S.; Wong, P.; Iizuka, Y.; Yang, J.; Subramanian, S.; McCarthy, J.; Bazzaro, M.; et al. Cellular and Molecular Networking Within the Ecosystem of Cancer Cell Communication via Tunneling Nanotubes. Front. Cell Dev. Biol. 2018, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Patheja, P.; Sahu, K. Macrophage conditioned medium induced cellular network formation in MCF-7 cells through enhanced tunneling nanotube formation and tunneling nanotube mediated release of viable cytoplasmic fragments. Exp. Cell Res. 2017, 355, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef] [PubMed]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.; Fitzgerald, K.A. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur. J. Immunol. 2014, 44, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Diamond, M.S.; Suthar, M.S. Negative regulators of the RIG-I-like receptor signaling pathway. Eur. J. Immunol. 2017, 47, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Park, E.; Kang, S.J. Stimulator of IFN genes-mediated DNA-sensing pathway is suppressed by NLRP3 agonists and regulated by mitofusin 1 and TBC1D15, mitochondrial dynamics mediators. FASEB J. 2017, 31, 4866–4878. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes As Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019. [Google Scholar] [CrossRef]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Gomez-Cabanas, L.; Lopez-Cotarelo, P.; Criado-Garcia, O.; Murphy, M.P.; Boya, P.; Rodriguez-Fernandez, J.L. Immunological Synapse Formation Induces Mitochondrial Clustering and Mitophagy in Dendritic Cells. J. Immunol. 2019, 202, 1715–1723. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef]
- Abdrakhmanov, A.; Kulikov, A.V.; Luchkina, E.A.; Zhivotovsky, B.; Gogvadze, V. Involvement of mitophagy in cisplatin-induced cell death regulation. Biol. Chem. 2019, 400, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Shao, L.J.; Jiang, Z.F.; Liu, R.Y. Gemcitabine-Induced Autophagy Protects Human Lung Cancer Cells from Apoptotic Death. Lung 2016, 194, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Mackeh, R.; Perdiz, D.; Lorin, S.; Codogno, P.; Pous, C. Autophagy and microtubules - new story, old players. J. Cell Sci. 2013, 126, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Cao, J.; Yao, S. Matrine promotes liver cancer cell apoptosis by inhibiting mitophagy and PINK1/Parkin pathways. Cell Stress Chaperones 2018, 23, 1295–1309. [Google Scholar] [CrossRef]
- Zheng, R.; Yao, Q.; Xie, G.; Du, S.; Ren, C.; Wang, Y.; Yuan, Y. TAT-ODD-p53 enhances the radiosensitivity of hypoxic breast cancer cells by inhibiting Parkin-mediated mitophagy. Oncotarget 2015, 6, 17417–17429. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, Y.; Yan, L.; Yuan, N.; Zhang, S.; Xu, L.; Nie, M.; Zhang, X.; Wang, J. Erythroleukemia cells acquire an alternative mitophagy capability. Sci. Rep. 2016, 6, 24641. [Google Scholar] [CrossRef]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Guo, J.; Wang, L.; Liu, X. Ambra1 induces autophagy and desensitizes human prostate cancer cells to cisplatin. Biosci. Rep. 2017. [Google Scholar] [CrossRef]
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef]
- Qian, W.; Wang, J.; Roginskaya, V.; McDermott, L.A.; Edwards, R.P.; Stolz, D.B.; Llambi, F.; Green, D.R.; Van Houten, B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180–4194. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Wang, C.; Hu, N.; Li, Y.; Liu, M.; Lei, Y.; Chen, M.; Chen, L.; Chen, C.; Lan, P.; et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 2019, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Cristofani, R.; Montagnani Marelli, M.; Cicardi, M.E.; Fontana, F.; Marzagalli, M.; Limonta, P.; Poletti, A.; Moretti, R.M. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018, 9, 889. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.J.; Park, Y.J.; Park, S.Y.; Oh, B.M.; Park, Y.S.; Kim, B.Y.; Lee, Y.H.; Cho, H.J.; Yoon, S.R.; et al. Opa-interacting protein 5 modulates docetaxel-induced cell death via regulation of mitophagy in gastric cancer. Tumour Biol.: J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317733985. [Google Scholar] [CrossRef]
- Yan, C.; Luo, L.; Guo, C.Y.; Goto, S.; Urata, Y.; Shao, J.H.; Li, T.S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J. Biol. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Naik, P.P.; Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, C.K.; Mishra, R.; Patil, S.; Bhutia, S.K. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018, 51. [Google Scholar] [CrossRef]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.M.; Lan, K.L.; Huang, W.S.; Lee, Y.J.; Lee, T.W.; Chang, C.H.; Chuang, C.M. (188)Re-Liposome Can Induce Mitochondrial Autophagy and Reverse Drug Resistance for Ovarian Cancer: From Bench Evidence to Preliminary Clinical Proof-of-Concept. Int. J. Mol. Sci. 2017, 18, 903. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays: News Rev. Mol. Cell. Dev. Biol. 2015, 37, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. Vitr.: Int. J. Publ. Assoc. BIBRA 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: A mitochondrial division/mitophagy inhibitor. PLoS ONE 2013, 8, e77713. [Google Scholar] [CrossRef]
- Du, Q.; Zhu, B.; Zhai, Q.; Yu, B. Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am. J. Transl. Res. 2017, 9, 3360–3373. [Google Scholar]
- Warren, S. The immediate causes of death in cancer. Am. J. Med. Sci. 1932, 184, 610–615. [Google Scholar] [CrossRef]
- Martin, L.; Birdsell, L.; Macdonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, M.B.; Baracos, V.E. Cancer cachexia in the age of obesity: Skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Skipworth, R.J.; Stewart, G.D.; Dejong, C.H.; Preston, T.; Fearon, K.C. Pathophysiology of cancer cachexia: Much more than host-tumour interaction? Clin. Nutr. 2007, 26, 667–676. [Google Scholar] [CrossRef]
- von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. Cachexiasarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Caillet, P.; Liuu, E.; Raynaud Simon, A.; Bonnefoy, M.; Guerin, O.; Berrut, G.; Lesourd, B.; Jeandel, C.; Ferry, M.; Rolland, Y.; et al. Association between cachexia, chemotherapy and outcomes in older cancer patients: A systematic review. Clin. Nutr. 2017, 36, 1473–1482. [Google Scholar] [CrossRef]
- Op den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; de Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.; Schols, A.M. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef]
- Johns, N.; Hatakeyama, S.; Stephens, N.A.; Degen, M.; Degen, S.; Frieauff, W.; Lambert, C.; Ross, J.A.; Roubenoff, R.; Glass, D.J.; et al. Clinical classification of cancer cachexia: Phenotypic correlates in human skeletal muscle. PLoS ONE 2014, 9, e83618. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Rossi Fanelli, F.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Manne, N.D.; Lima, M.; Enos, R.T.; Wehner, P.; Carson, J.A.; Blough, E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int. J. Oncol. 2013, 42, 2134–2140. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Gallagher, I.J.; Rooyackers, O.; Skipworth, R.J.; Tan, B.H.; Marstrand, T.; Ross, J.A.; Guttridge, D.C.; Lundell, L.; Fearon, K.C.; et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med. 2010, 2, 1. [Google Scholar] [CrossRef]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 2010, 126, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Feather, C.E.; Lees, J.G.; Makker, P.G.S.; Goldstein, D.; Kwok, J.B.; Moalem-Taylor, G.; Polly, P. Oxaliplatin induces muscle loss and muscle-specific molecular changes in Mice. Muscle Nerve 2018, 57, 650–658. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef]
- Julienne, C.M.; Tardieu, M.; Chevalier, S.; Pinault, M.; Bougnoux, P.; Labarthe, F.; Couet, C.; Servais, S.; Dumas, J.F. Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer-induced cachexia without the involvement of adenine nucleotide translocase. Biochim. Et Biophys. Acta 2014, 1842, 726–733. [Google Scholar] [CrossRef]
- Dumas, J.F.; Goupille, C.; Julienne, C.M.; Pinault, M.; Chevalier, S.; Bougnoux, P.; Servais, S.; Couet, C. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J. Hepatol. 2011, 54, 320–327. [Google Scholar] [CrossRef]
- Tzika, A.A.; Fontes-Oliveira, C.C.; Shestov, A.A.; Constantinou, C.; Psychogios, N.; Righi, V.; Mintzopoulos, D.; Busquets, S.; Lopez-Soriano, F.J.; Milot, S.; et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 2013, 43, 886–894. [Google Scholar] [CrossRef]
- Bing, C.; Brown, M.; King, P.; Collins, P.; Tisdale, M.J.; Williams, G. Increased gene expression of brown fat uncoupling protein (UCP)1 and skeletal muscle UCP2 and UCP3 in MAC16-induced cancer cachexia. Cancer Res. 2000, 60, 2405–2410. [Google Scholar]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef] [PubMed]
- Lijie, G.; Yueyue, Z.; Nan, Z.; Ling, W.; Xuan, W.; Weijie, Y. Mitsugumin 53 promotes mitochondrial autophagy through regulating Ambra1 expression in C2C12 myoblast cells. Cell Biol. Int. 2019, 43, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Jang, G.M.; Huttenhain, R.; Gordon, D.E.; Du, D.; Newton, B.W.; Johnson, J.R.; Hiatt, J.; Hultquist, J.F.; Johnson, T.L.; et al. CRL4(AMBRA1) targets Elongin C for ubiquitination and degradation to modulate CRL5 signaling. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, Z.; Qi, J.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zeng, M.; Zhang, B.; Wang, N.; et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 2017, 8, 20988–21000. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef]




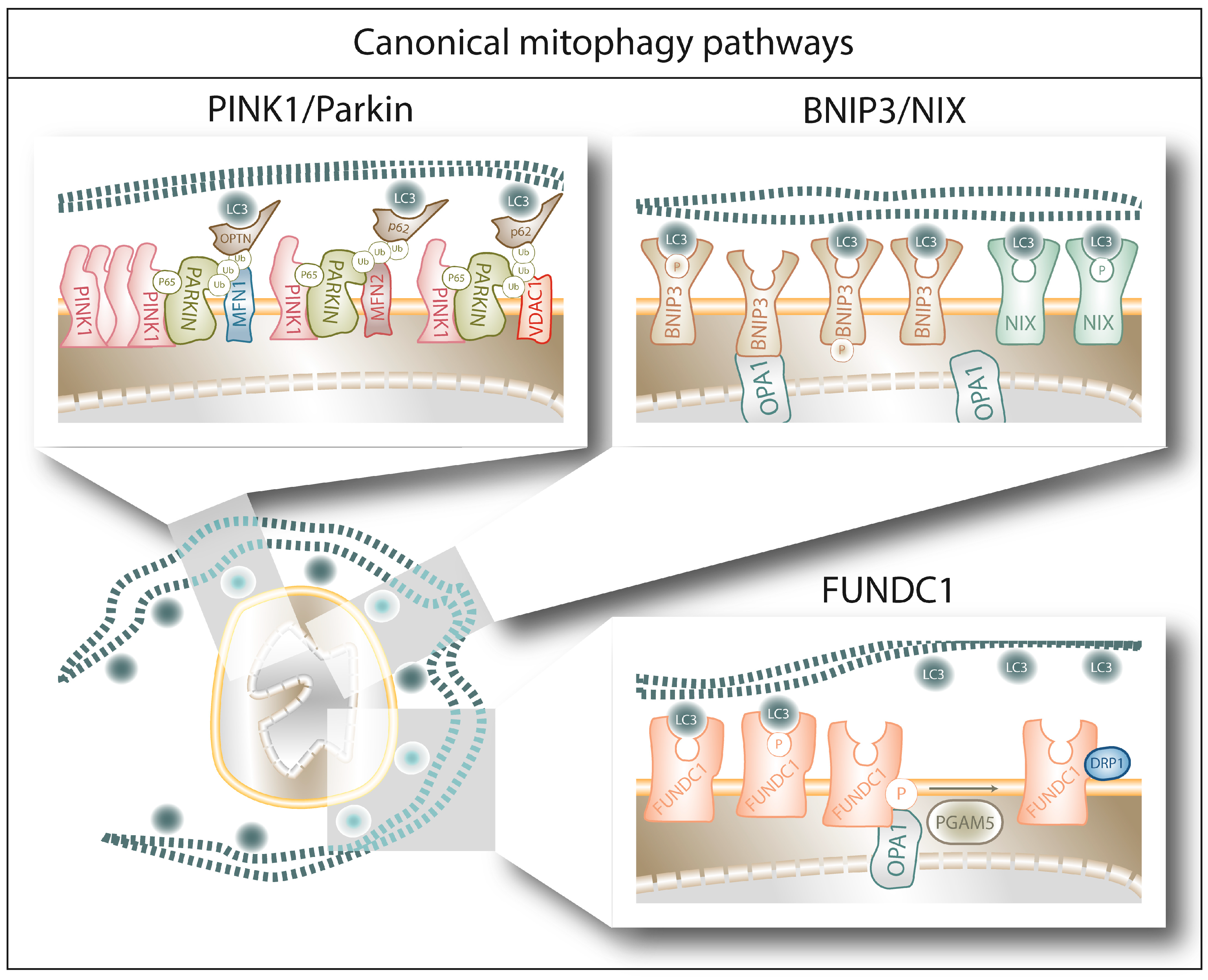{kind=link}
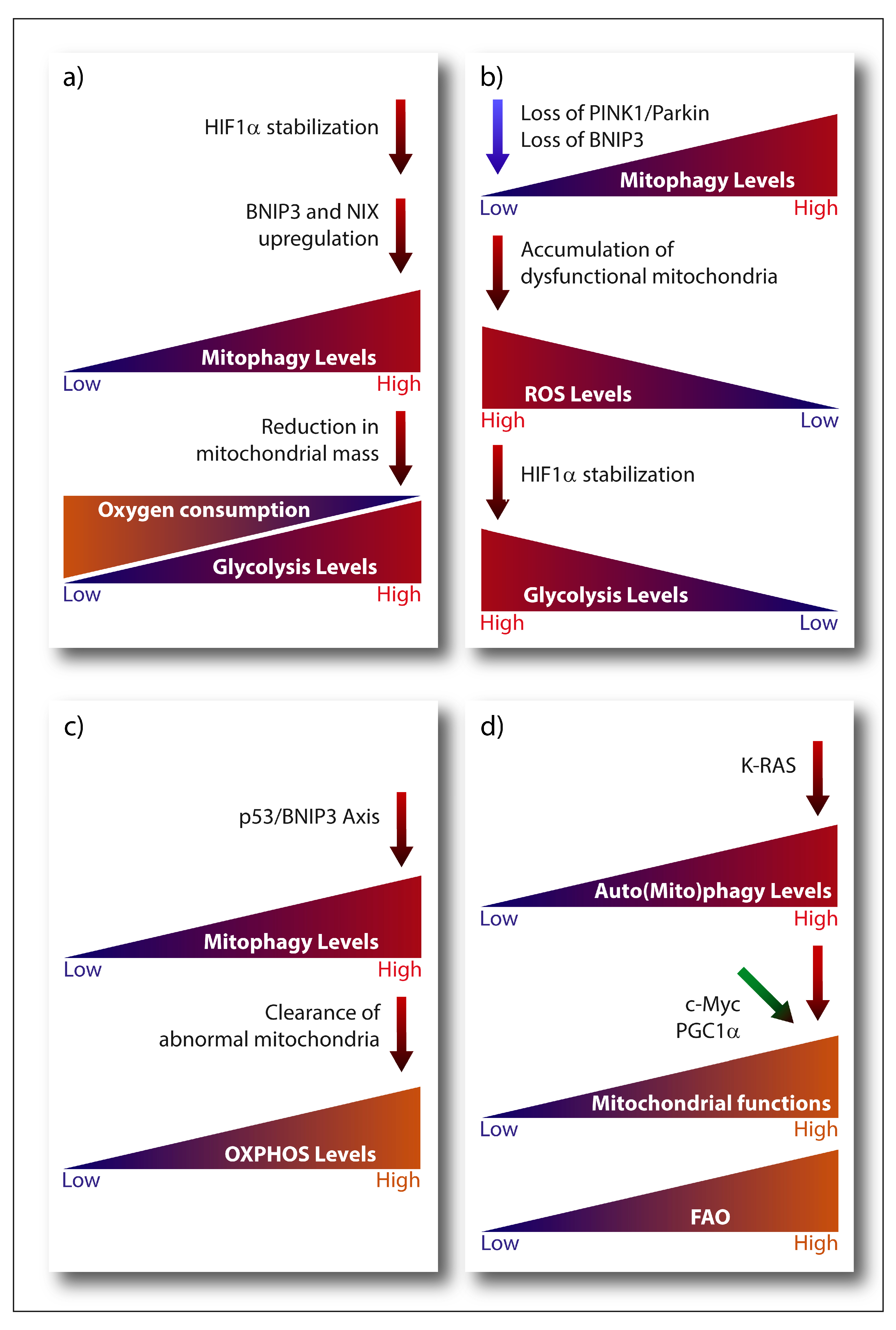{kind=link}
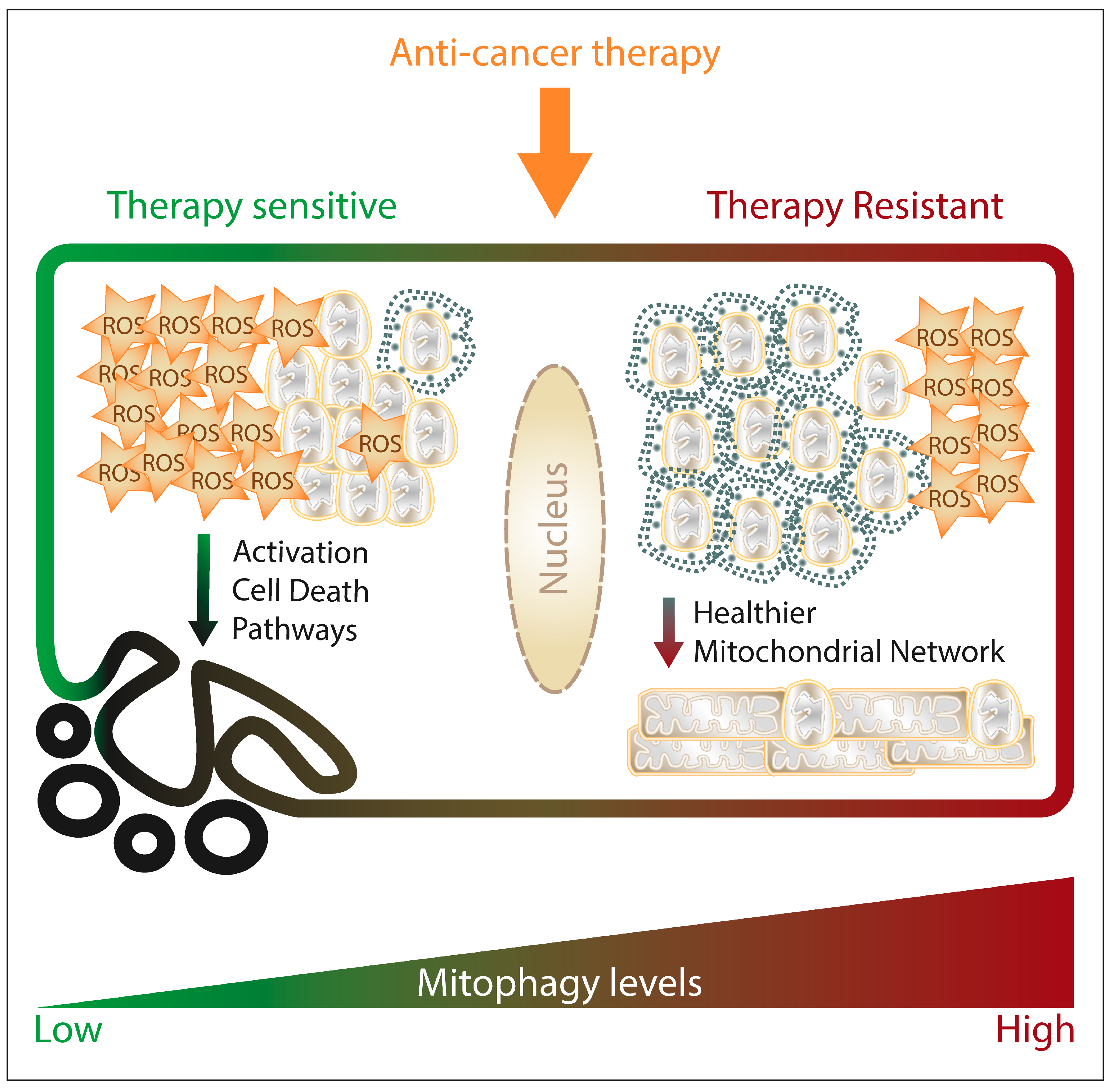{kind=link}
| Protein | Expression Levels in Patients | Cancer Type | Reference(s) |
|---|---|---|---|
| Mitochondrial Dynamics | |||
| MFN 1 | Downregulation | Triple Negative Breast Cancer 1,2, Hepatocellular Carcinoma 1,2 | [89,90] |
| MFN 2 | Upregulation | Cutaneous Melanoma 2, Gastric Cancer 2, Ovarian Cancer 1, Prostate Cancer 2 | [91,92,93,94] |
| MFN 2 | Downregulation | Breast Cancer 1,2, Hepatocellular Carcinoma 1,2, Non-Small Cell Lung Cancer 1 | [95,96,97,98,99,100] |
| Opa1 | Upregulation | Lung Cancer 2,3, Colorectal Cancer 1 | [101,102,103] |
| Opa1 | Downregulation | Hepatocellular Carcinoma 2 | [104] |
| DRP1 | Upregulation | Triple Negative Breast Cancer 1,2, Colorectal Cancer1, Hepatocellular Carcinoma 1,2, Ovarian Cancer 1,3 | [89,90,101,105] |
| DRP1 | Downregulation | Colorectal Cancer 2, Lung Cancer 2 | [106] |
| pDRP1 (Ser616) | Upregulation | Colorectal Cancer 2, Melanoma 2 | [107,108] |
| pDRP1 (Ser637) | Upregulation | Hepatocellular Carcinoma 2 | [109] |
| Mff | Upregulation | Hepatocellular Carcinoma 1,2 | [110] |
| Mff | Downregulation | Tongue Squamous Cell Carcinoma 2 | [111] |
| FIS1 | Upregulation | Acute Myeloid Leukemia 1,2, Oral Melanoma 2, Prostate Cancer 1 | [93,112,113,114] |
| FIS1 | Downregulation | Tongue Squamous Cell Carcinoma 2 | [115] |
| Canonical Mitophagy Pathways | |||
| PINK1 | Upregulation | Lung Cancer 2, Esophageal Squamous Cell Carcinoma 2 | [116,117] |
| PINK1 | Downregulation | Ovarian cancer 1 | [118] |
| Parkin | Downregulation | Acute Lymphoblastic Leukemia 4, Colorectal Cancer 1,2,4, Clear Cell Renal Cell Carcinoma 1,2, Melanoma 1,3, Oropharyngeal Squamous Cell Carcinoma 1, Ovarian Cancer 1,3, Pancreatic Cancer 1,2,3 | [119,120,121,122,123,124,125,126,127] |
| BNIP3 | Upregulation | Adenoid Cystic Carcinoma 2, Ampullary Carcinoma 2, Breast Cancer 1, Cervical Cancer 1,2, Cholangiocarcinoma 2, Colorectal Cancer 2, Ependydoma 1, Glioblastoma 2, Lung Cancer 1,2, Melanoma 2, Ovarian Cancer 1,2, Renal Carcinoma 1,2, Uterine-Cervical Squamous Cell Carcinoma 1 | [128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143] |
| BNIP3 | Downregulation | Bladder Cancer 4, Breast Cancer 1,2, Colorectal Cancer 2,4, Esophageal Cancer 4, Gastric Carcinoma 4, Laryngeal Squamous Cell Carcinoma 2, Lung Cancer 4, Multiple Myeloma 1,4, Pancreatic Cancer 1,2, | [142,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163] |
| NIX | Upregulation | Breast Cancer 1, Glioma 1,2 | [164,165] |
| NIX | Downregulation | Acute Myeloid Leukemia 1, Prostate Cancer 3 | [166,167] |
| FUNDC1 | Upregulation | Breast Cancer 1,2, Cervical Cancer 2, Laryngeal Cancer 2 | [168,169,170] |
| PGAM5 | Upregulation | Hepatocellular Carcinoma 2, Non-Small Cell Lung Cancer 2 | [171,172] |
| Non-Canonical Mitophagy Pathways | |||
| CL | Upregulation | Prostate Cancer | [173,174] |
| CL | Downregulation | Hepatocellular Carcinoma | [175] |
| PHB2 | Upregulation | Breast Cancer 1,2, Colorectal Cancer 2, Esophageal Squamous Cell carcinoma 1,2, Leukemia 2, Lymphoma 2 | [167,176,177,178,179,180] |
| C18-Ceramide | Downregulation | Glioblastoma, Glioma, Head and Neck Squamous Cell Carcinoma | [181,182,183] |
| AMBRA1 | Upregulation | Cholangiocarcinoma 2, Gastric Adenocarcinoma 2, Pancreatic Ductal Adenocarcinoma 2, Prostate Cancer 1,2 | [184,185,186,187] |
| HUWE1 | Upregulation | Lung Cancer 1,2, Multiple Myeloma 1 | [188,189] |
| HUWE1 | Downregulation | Breast Cancer 1, Hepatocellular Carcinoma 1, Osteosarcoma 1 | [190,191,192] |
| BCL2L13 (BCL-RAMBO) | Upregulation | Leukemia 1 | [193,194,195] |
| BCL2L13 (BCL-RAMBO) | Downregulation | Breast Cancer 1, Locally Advanced Rectal Cancer 1 | [139,196] |
| RAB7 | Upregulation | Oral Squamous Cell Carcinoma 2, Prostate Cancer 1, | [197,198] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. https://doi.org/10.3390/cells8050493
Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in Cancer: A Tale of Adaptation. Cells. 2019; 8(5):493. https://doi.org/10.3390/cells8050493
Chicago/Turabian StyleVara-Perez, Monica, Blanca Felipe-Abrio, and Patrizia Agostinis. 2019. "Mitophagy in Cancer: A Tale of Adaptation" Cells 8, no. 5: 493. https://doi.org/10.3390/cells8050493
APA StyleVara-Perez, M., Felipe-Abrio, B., & Agostinis, P. (2019). Mitophagy in Cancer: A Tale of Adaptation. Cells, 8(5), 493. https://doi.org/10.3390/cells8050493