Chronic Hepatitis C Virus Infection Impairs M1 Macrophage Differentiation and Contributes to CD8+ T-Cell Dysfunction

 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Study Groups
2.2. Isolation of Monocytes and Generation of Macrophage Subsets
2.3. Macrophage Subset Phenotyping by Flow Cytometry
2.4. Quantification of Secreted Cytokines
2.5. Macrophage:CD8+ T-Cell Co-Culture
2.6. CD8+ T-Cell Functions
Quantification of IFN-γ+ and Perforin+ Cells and Detection of Degranulating (CD107a+) Cells
2.7. Statistical Analysis
3. Results
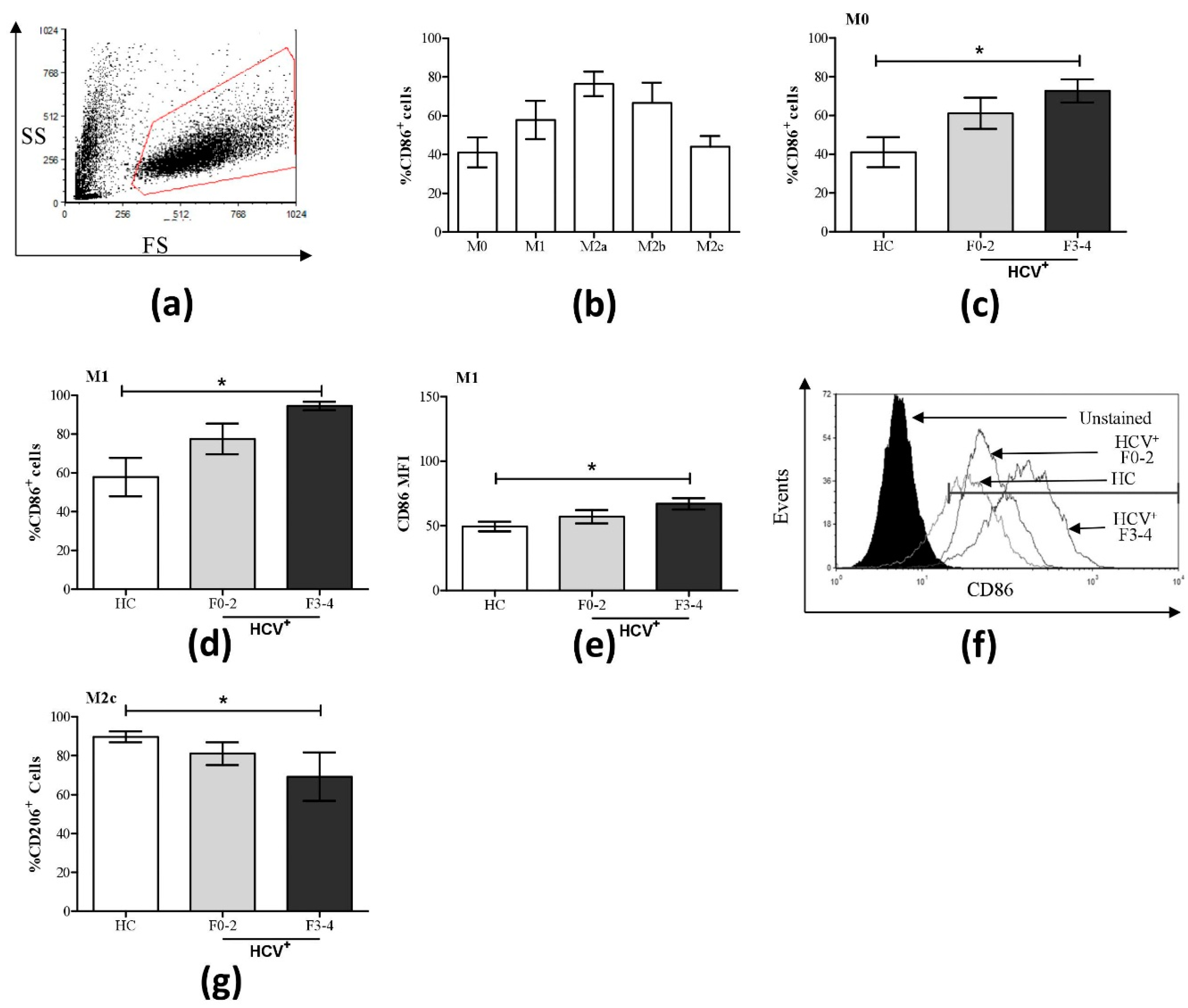
3.1. Altered Phenotypic Surface Marker Expression on Macrophage Subsets from Chronic HCV-Infected Patients
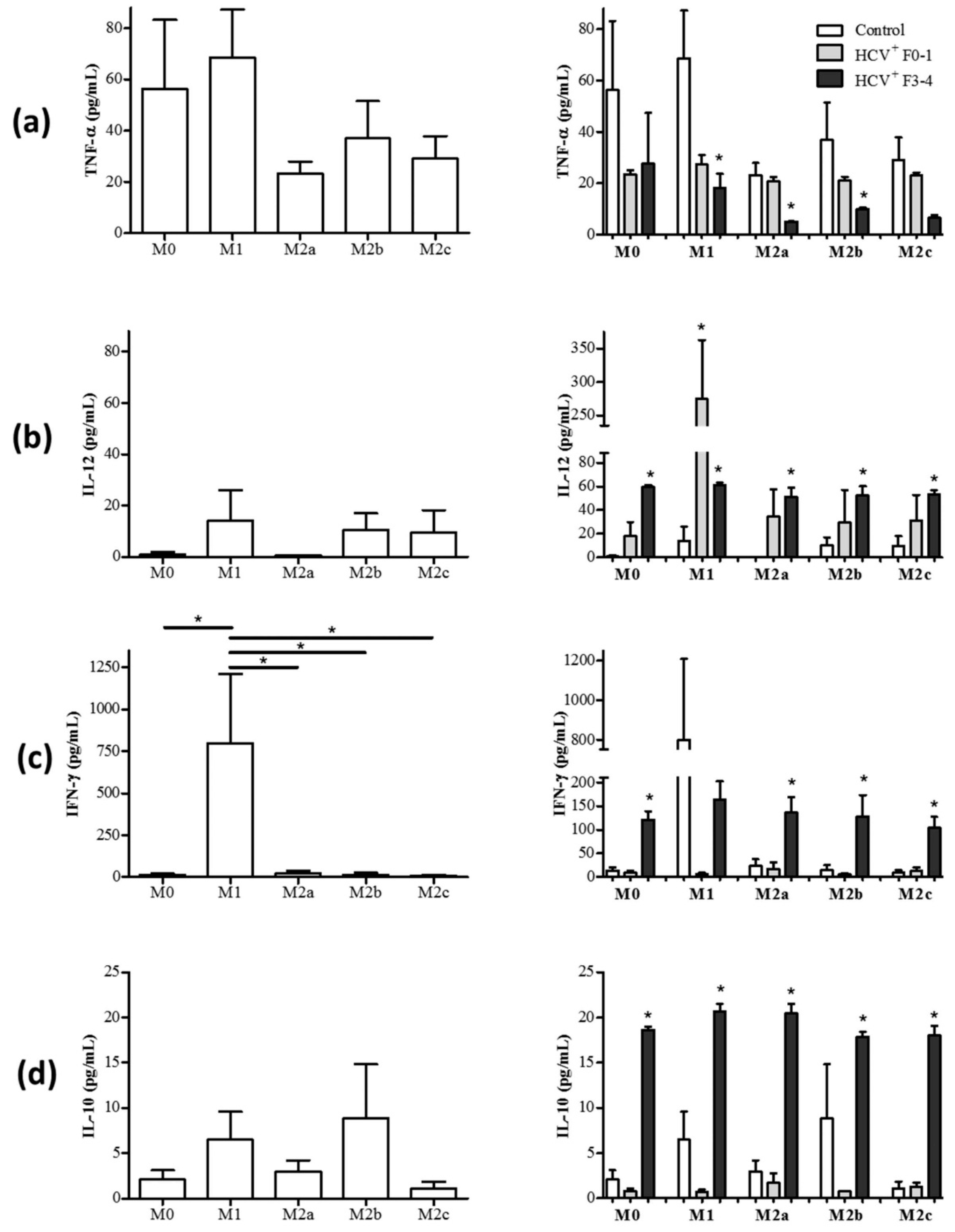
3.2. Altered Pro- and Anti-Inflammatory Cytokine Expression on Macrophage Subsets from HCV-Infected Patients
3.2.1. M1 Cells Acquire Cytokine Secretion Features of M2 Cells in HCV+(F3-4) Individuals
3.2.2. M2 Cells Acquire Cytokine Secretion Features of M1 Cells in HCV+(F3-4) Individuals
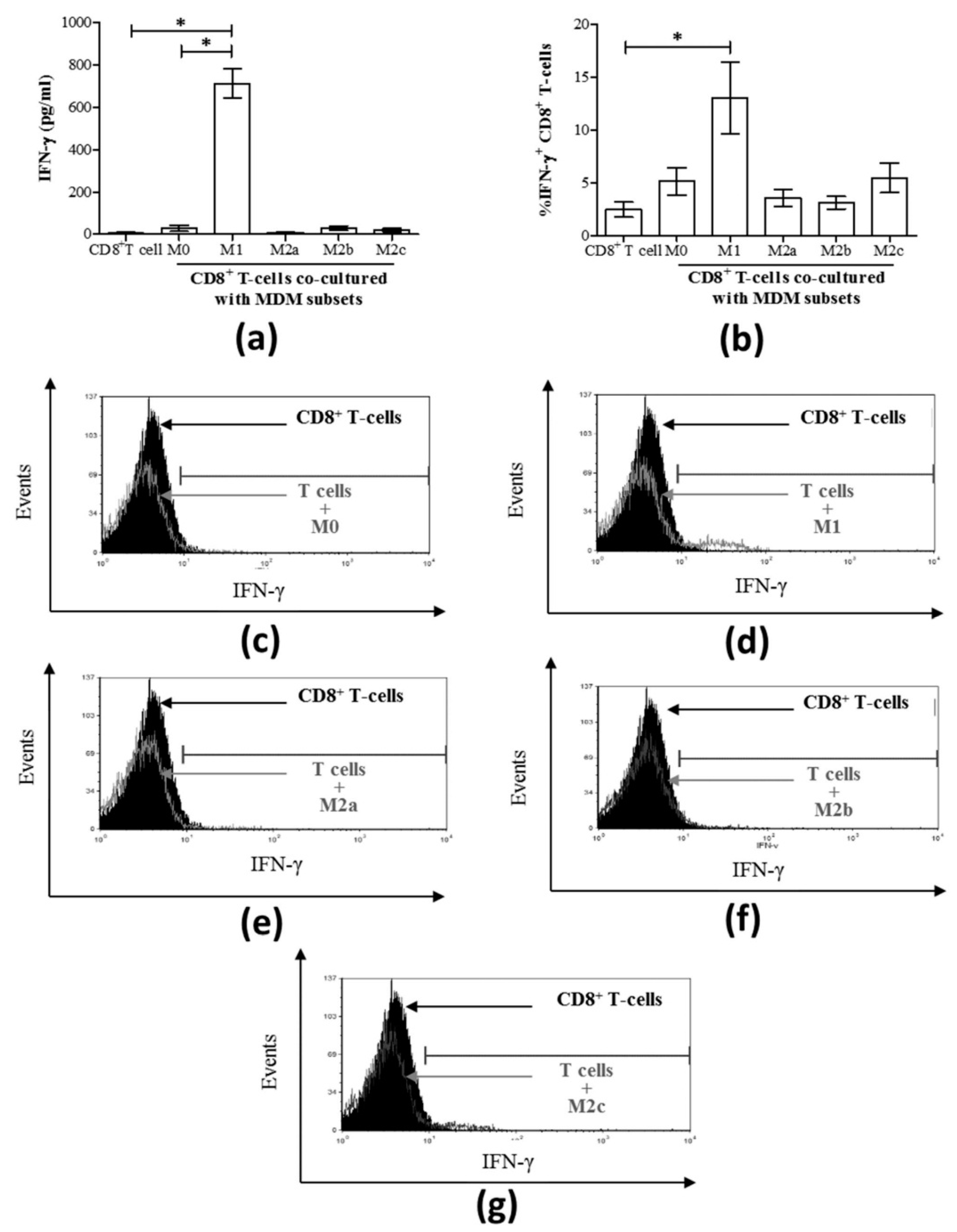
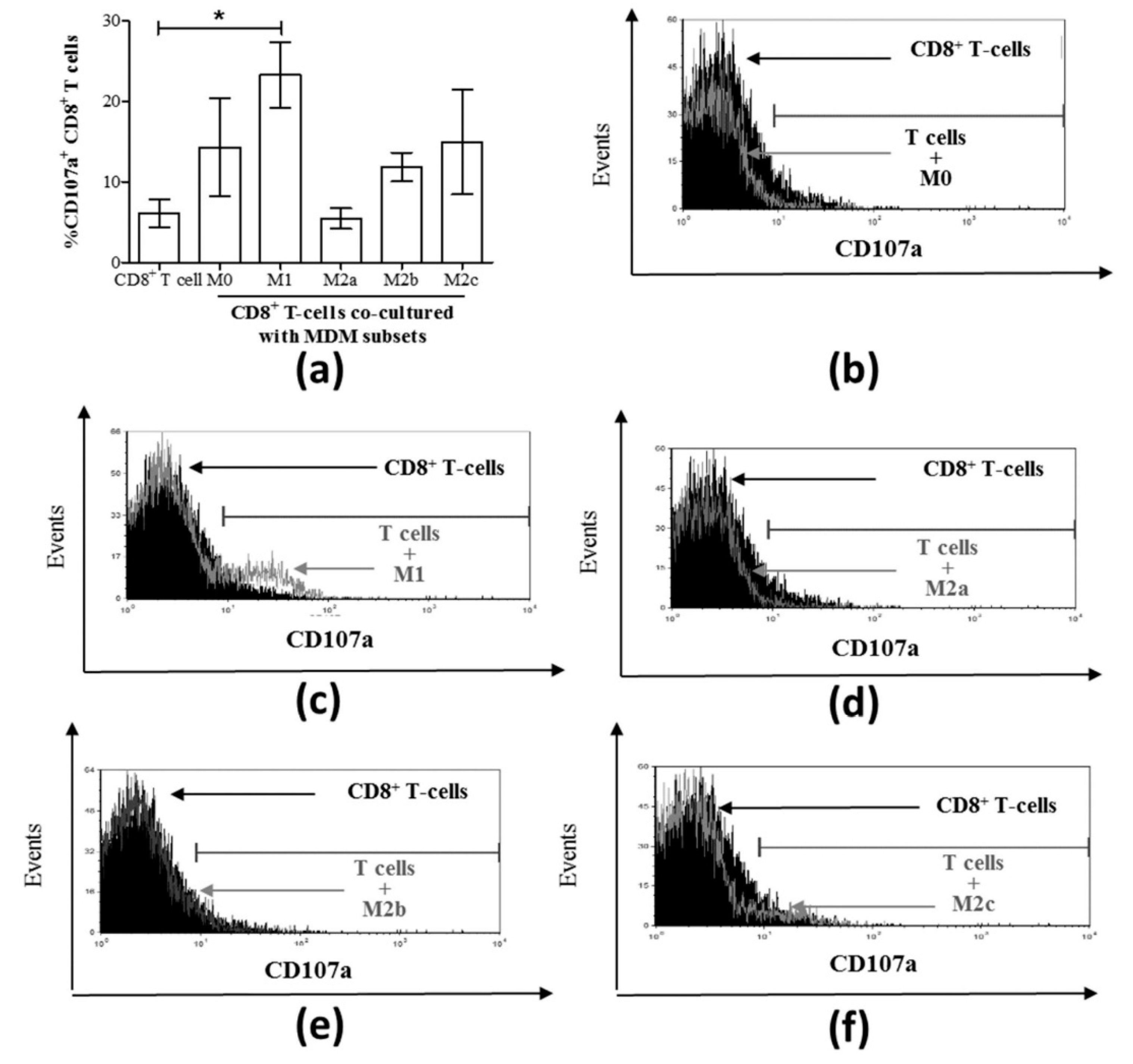
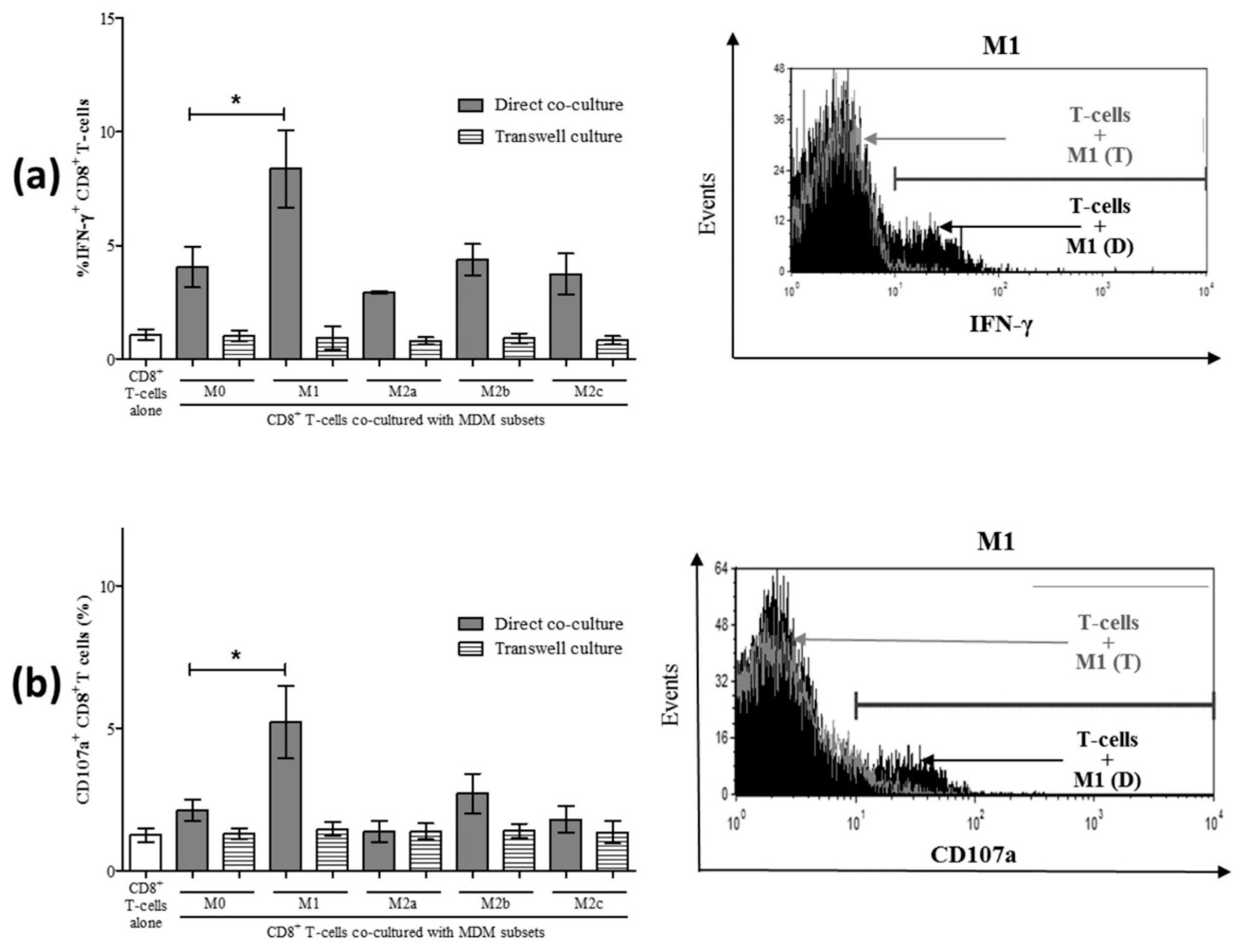
3.3. M1 Macrophages Enhance CD8+ T-Cell Functions in Normal Healthy Individuals via Contact-Dependent Mechanisms
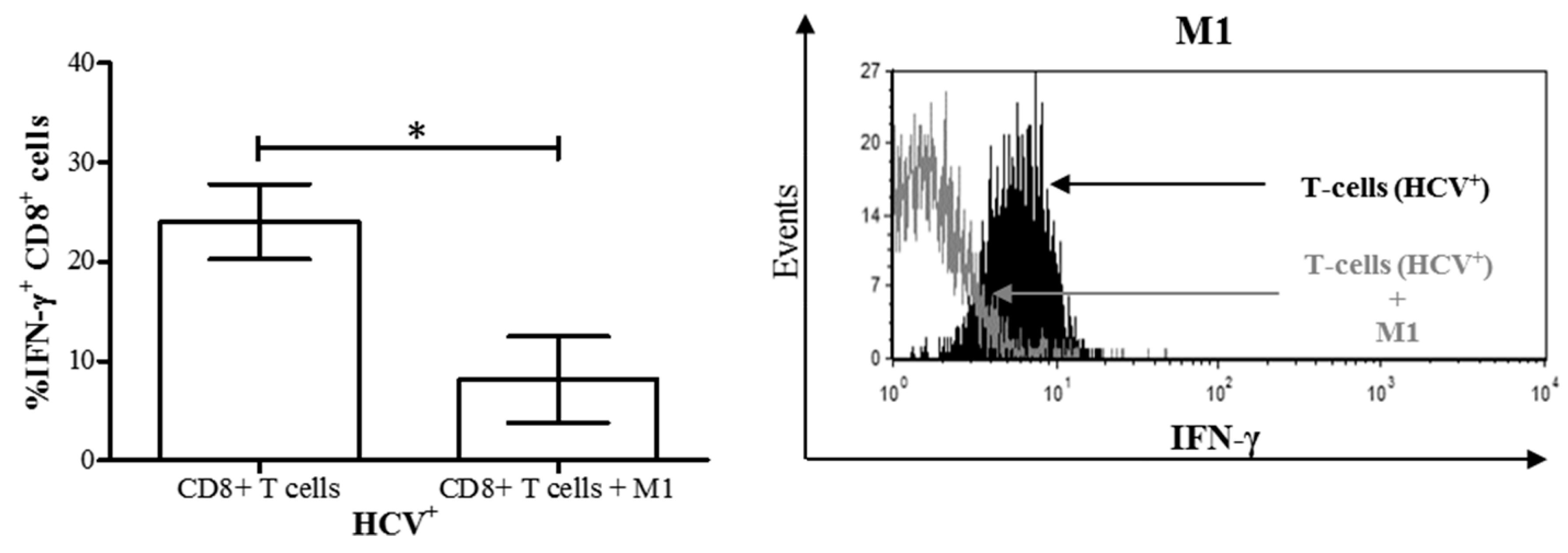
3.4. M1 Macrophages Inhibit IFN-γ Expression on CD8+ T-Cells in Chronic HCV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, A.; Tsien, C.; Mansour, S.; Cooper, C. Remaining clinical issues in hepatitis C treatment. Can. Liver J. 2018, 1, 66–77. [Google Scholar] [CrossRef]
- Serti, E.; Werner, J.M.; Chattergoon, M.; Cox, A.L.; Lohmann, V.; Rehermann, B. Monocytes activate natural killer cells via inflammasome-induced interleukin 18 in response to hepatitis C virus replication. Gastroenterology 2014, 147, 209–220. [Google Scholar] [CrossRef]
- Villacres, M.C.; Literat, O.; DeGiacomo, M.; Du, W.; Frederick, T.; Kovacs, A. Defective response to Toll-like receptor 3 and 4 ligands by activated monocytes in chronic hepatitis C virus infection. J. Viral Hepat. 2008, 15, 137–144. [Google Scholar] [CrossRef]
- Woitas, R.P.; Petersen, U.; Moshage, D.; Brackmann, H.H.; Matz, B.; Sauerbruch, T.; Spengler, U. HCV-specific cytokine induction in monocytes of patients with different outcomes of hepatitis C. World J. Gastroenterol.: WJG 2002, 8, 562–566. [Google Scholar] [CrossRef]
- Orito, E.; Mizokami, M.; Tanaka, T.; Lau, J.Y.; Suzuki, K.; Yamauchi, M.; Ohta, Y.; Hasegawa, A.; Tanaka, S.; Kohara, M. Quantification of serum hepatitis C virus core protein level in patients chronically infected with different hepatitis C virus genotypes. Gut 1996, 39, 876–880. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Q.; Wang, Y.; Zhai, N.; Song, H.; Li, H.; Yang, Y.; Li, T.; Guo, X.; Chi, B.; Niu, J.; et al. HCV core protein inhibits polarization and activity of both M1 and M2 macrophages through the TLR2 signaling pathway. Sci. Rep. 2016, 6, 36160. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Labonte, A.C.; Tosello-Trampont, A.C.; Hahn, Y.S. The role of macrophage polarization in infectious and inflammatory diseases. Mol. Cells 2014, 37, 275–285. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Bility, M.T.; Nio, K.; Li, F.; McGivern, D.R.; Lemon, S.M.; Feeney, E.R.; Chung, R.T.; Su, L. Chronic hepatitis C infection-induced liver fibrogenesis is associated with M2 macrophage activation. Sci. Rep. 2016, 6, 39520. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Louvet, A.; Teixeira-Clerc, F.; Chobert, M.N.; Deveaux, V.; Pavoine, C.; Zimmer, A.; Pecker, F.; Mallat, A.; Lotersztajn, S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011, 54, 1217–1226. [Google Scholar] [CrossRef]
- Klein, I.; Cornejo, J.C.; Polakos, N.K.; John, B.; Wuensch, S.A.; Topham, D.J.; Pierce, R.H.; Crispe, I.N. Kupffer cell heterogeneity: Functional properties of bone marrow derived and sessile hepatic macrophages. Blood 2007, 110, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Wohlleber, D.; Knolle, P.A. The role of liver sinusoidal cells in local hepatic immune surveillance. Clin. Transl. Immunol. 2016, 5, e117. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Knolle, P.A.; Stabenow, D. Mechanisms balancing tolerance and immunity in the liver. Dig. Dis. 2011, 29, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Zimmerer, J.M.; Horne, P.H.; Fisher, M.G.; Pham, T.A.; Lunsford, K.E.; Ringwald, B.A.; Avila, C.L.; Bumgardner, G.L. Unique CD8+ T Cell-Mediated Immune Responses Primed in the Liver. Transplantation 2016, 100, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Burke Schinkel, S.C.; Carrasco-Medina, L.; Cooper, C.L.; Crawley, A.M. Generalized Liver- and Blood-Derived CD8+ T-Cell Impairment in Response to Cytokines in Chronic Hepatitis C Virus Infection. PLoS ONE 2016, 11, e0157055. [Google Scholar] [CrossRef]
- Vranjkovic, A.D.F.; Kaka, S.; Cooper, C.L.; Crawley, A.M. Dysfunction of circulating CD8+ T-cells in chronic HCV infection is associated with severity of liver disease and is unresolved after HCV cure. Front. Immunol. 2019. submitted. [Google Scholar]
- Classen, A.; Lloberas, J.; Celada, A. Macrophage activation: Classical versus alternative. Methods Mol. Biol. 2009, 531, 29–43. [Google Scholar] [PubMed]
- Biswas, S.K.; Chittezhath, M.; Shalova, I.N.; Lim, J.Y. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012, 53, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Suttles, J. Functional plasticity of macrophages: Reversible adaptation to changing microenvironments. J. Leukoc. Biol. 2004, 76, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Watkins, S.K.; Suttles, J. Functional plasticity of macrophages: In situ reprogramming of tumor-associated macrophages. J. Leukoc. Biol. 2009, 86, 1105–1109. [Google Scholar] [CrossRef]
- Busca, A.; Saxena, M.; Kumar, A. Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J. Biol. Chem. 2012, 287, 15118–15133. [Google Scholar] [CrossRef]
- Busca, A.; Konarski, Y.; Gajanayaka, N.; O’Hara, S.; Angel, J.; Kozlowski, M.; Kumar, A. cIAP1/2-TRAF2-SHP-1-Src-MyD88 Complex Regulates Lipopolysaccharide-Induced IL-27 Production through NF-kappaB Activation in Human Macrophages. J. Immunol. 2018, 200, 1593–1606. [Google Scholar]
- Iqbal, S.; Kumar, A. Characterization of in vitro generated human polarized macrophages. J. Clin. Cell. Immunol. 2015, 6, 380. [Google Scholar] [CrossRef]
- Novelli, F.; Casanova, J.L. The role of IL-12, IL-23 and IFN-gamma in immunity to viruses. Cytokine Growth Factor Rev. 2004, 15, 367–377. [Google Scholar] [CrossRef]
- Said, E.A.; Al-Reesi, I.; Al-Riyami, M.; Al-Naamani, K.; Al-Sinawi, S.; Al-Balushi, M.S.; Koh, C.Y.; Al-Busaidi, J.Z.; Idris, M.A.; Al-Jabri, A.A. Increased CD86 but Not CD80 and PD-L1 Expression on Liver CD68+ Cells during Chronic HBV Infection. PLoS ONE 2016, 11, e0158265. [Google Scholar] [CrossRef]
- Yoshida, T.; Hachimura, S.; Ishimori, M.; Ise, W.; Totsuka, M.; Ametani, A.; Kaminogawa, S. Interleukin 12 and CD86 Regulate Th1 and Th2 Development Induced by a Range of Antigen Doses Presented by Peyer’s Patch and Spleen Cells. Cytotechnology 2003, 43, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F. B7-1 but not B7-2 efficiently costimulates CD8+ T lymphocytes in the P815 tumor system in vitro. J. Immunol. 1996, 156, 465–472. [Google Scholar] [PubMed]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van de Velde, L.A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef]
- Choi, K.M.; Kashyap, P.C.; Dutta, N.; Stoltz, G.J.; Ordog, T.; Shea Donohue, T.; Bauer, A.J.; Linden, D.R.; Szurszewski, J.H.; Gibbons, S.J.; et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 2010, 138, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.; Bishop, G.A.; McGuinness, P.H.; Painter, D.M.; McCaughan, G.W. Progressive liver injury in chronic hepatitis C infection correlates with increased intrahepatic expression of Th1-associated cytokines. Hepatology 1996, 24, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, C.J.; Ni, L.; Zhang, C.L.; Wu, X.Y.; Kumaraguru, U.; Li, C.F.; Moorman, J.P.; Yao, Z.Q. Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J. Immunol. 2011, 186, 3093–3103. [Google Scholar] [CrossRef] [PubMed]
- Eisen-Vandervelde, A.L.; Waggoner, S.N.; Yao, Z.Q.; Cale, E.M.; Hahn, C.S.; Hahn, Y.S. Hepatitis C virus core selectively suppresses interleukin-12 synthesis in human macrophages by interfering with AP-1 activation. J. Biol. Chem. 2004, 279, 43479–43486. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Kodys, K.; Kopasz, A.; Marshall, C.; Do, T.; Romics, L., Jr.; Mandrekar, P.; Zapp, M.; Szabo, G. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J. Immunol. 2003, 170, 5615–5624. [Google Scholar] [CrossRef]
- Pozzi, L.A.; Maciaszek, J.W.; Rock, K.L. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J. Immunol. 2005, 175, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.E.; Hammarlund, E.; Raue, H.P.; Slifka, M.K. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 9971–9976. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.K.; Betts, M.R.; Price, D.A.; Gostick, E.; Horton, H.; Roederer, M.; De Rosa, S.C. The cytolytic enzymes granyzme A, granzyme B, and perforin: Expression patterns, cell distribution, and their relationship to cell maturity and bright CD57 expression. J. Leukoc. Biol. 2009, 85, 88–97. [Google Scholar] [CrossRef]
- Ge, Q.; Palliser, D.; Eisen, H.N.; Chen, J. Homeostatic T cell proliferation in a T cell-dendritic cell coculture system. Proc. Natl. Acad. Sci. USA 2002, 99, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Ogasawara, K.; Takeda, K.; Hashimoto, W.; Sakihara, H.; Kumagai, K.; Anzai, R.; Satoh, M.; Seki, S. LPS induces NK1.1+ alpha beta T cells with potent cytotoxicity in the liver of mice via production of IL-12 from Kupffer cells. J. Immunol. 1996, 156, 2436–2442. [Google Scholar]
- Seki, S.; Nakashima, H.; Nakashima, M.; Kinoshita, M. Antitumor immunity produced by the liver Kupffer cells, NK cells, NKT cells, and CD8 CD122 T cells. Clin.Dev. Immunol. 2011, 2011, 868345. [Google Scholar] [CrossRef]
- Mills, C.D.; Lenz, L.L.; Ley, K. Macrophages at the fork in the road to health or disease. Front. Immunol. 2015, 6, 59. [Google Scholar] [CrossRef]
- Porcheray, F.; Viaud, S.; Rimaniol, A.C.; Leone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage activation switching: An asset for the resolution of inflammation. Clin. Exp. Immunol. 2005, 142, 481–489. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Antignano, F.; von Rossum, A.; Boucher, J.L.; Bennewith, K.L.; Krystal, G. TLR agonists that induce IFN-beta abrogate resident macrophage suppression of T cells. J. Immunol. 2010, 185, 4545–4553. [Google Scholar] [CrossRef]
- Saha, B.; Kodys, K.; Szabo, G. Hepatitis C Virus-Induced Monocyte Differentiation Into Polarized M2 Macrophages Promotes Stellate Cell Activation via TGF-beta. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 302–316. [Google Scholar] [CrossRef]
- Cabral-Piccin, M.P.; Guillermo, L.V.; Vellozo, N.S.; Filardy, A.A.; Pereira-Marques, S.T.; Rigoni, T.S.; Pereira-Manfro, W.F.; DosReis, G.A.; Lopes, M.F. Apoptotic CD8 T-lymphocytes disable macrophage-mediated immunity to Trypanosoma cruzi infection. Cell Death Dis. 2016, 7, e2232. [Google Scholar] [CrossRef]
- Gordon, S.; Pluddemann, A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front. Immunol. 2018, 9, 127. [Google Scholar] [CrossRef]
- Vranjkovic, A.; Crawley, A.M.; Patey, A.; Angel, J.B. IL-7-dependent STAT-5 activation and CD8+ T cell proliferation are impaired in HIV infection. J. Leukoc. Biol. 2011, 89, 499–506. [Google Scholar] [CrossRef]
- Tacke, R.S.; Tosello-Trampont, A.; Nguyen, V.; Mullins, D.W.; Hahn, Y.S. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J. Biol. Chem. 2011, 286, 10847–10855. [Google Scholar] [CrossRef]
- Brady, M.T.; MacDonald, A.J.; Rowan, A.G.; Mills, K.H. Hepatitis C virus non-structural protein 4 suppresses Th1 responses by stimulating IL-10 production from monocytes. Eur. J. Immunol. 2003, 33, 3448–3457. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, C. The Role of Innate Lymphoid Cells in Immune-Mediated Liver Diseases. Front. Immunol. 2017, 8, 695. [Google Scholar] [CrossRef]
- Krueger, P.D.; Narayanan, S.; Surette, F.A.; Brown, M.G.; Sung, S.J.; Hahn, Y.S. Murine liver-resident group 1 innate lymphoid cells regulate optimal priming of anti-viral CD8+ T cells. J. Leukoc. Biol. 2017, 101, 329–338. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Controls | HCV+ (F0-2)1 | HCV+ (F3-4)2 | |
|---|---|---|---|
| Number | 13 | 9 | 4 |
| Age, avg. years (SD3) | 31.2 (8.4) | 50 (14.9) | 56.5 (4.1) |
| Race | Caucasian | Caucasian (7), East Indian (1), African (1) | Caucasian (3), Native (1) |
| HCV-RNA (IU/mL) | 3.1 × 106 (2.2 × 106) | 1.6 × 106 (2.2 × 106) | |
| HCV genotype4 (n) | 1 (7: 1a n = 3, 1b n = 1), 2b (1) | 1a (3), 3 (1) | |
| Fibroscan, avg. kPa5 (SD) | 6.1 (1.9) | 19.3 (7.1) | |
| AST6 (U/L) | 25.4 (12.9) | 47.3 (37.3) | |
| ALT7 (U/L) | 47.3 (23.6) | 75.0 (42.0) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, F.; Ibrahim, A.; Cooper, C.L.; Kumar, A.; Crawley, A.M. Chronic Hepatitis C Virus Infection Impairs M1 Macrophage Differentiation and Contributes to CD8+ T-Cell Dysfunction. Cells 2019, 8, 374. https://doi.org/10.3390/cells8040374
Ahmed F, Ibrahim A, Cooper CL, Kumar A, Crawley AM. Chronic Hepatitis C Virus Infection Impairs M1 Macrophage Differentiation and Contributes to CD8+ T-Cell Dysfunction. Cells. 2019; 8(4):374. https://doi.org/10.3390/cells8040374
Chicago/Turabian StyleAhmed, Faria, Andrea Ibrahim, Curtis L. Cooper, Ashok Kumar, and Angela M. Crawley. 2019. "Chronic Hepatitis C Virus Infection Impairs M1 Macrophage Differentiation and Contributes to CD8+ T-Cell Dysfunction" Cells 8, no. 4: 374. https://doi.org/10.3390/cells8040374
APA StyleAhmed, F., Ibrahim, A., Cooper, C. L., Kumar, A., & Crawley, A. M. (2019). Chronic Hepatitis C Virus Infection Impairs M1 Macrophage Differentiation and Contributes to CD8+ T-Cell Dysfunction. Cells, 8(4), 374. https://doi.org/10.3390/cells8040374