GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches


{kind=link}
Abstract
1. Introduction
2. Pathogenetic Mutations of the GBA Gene
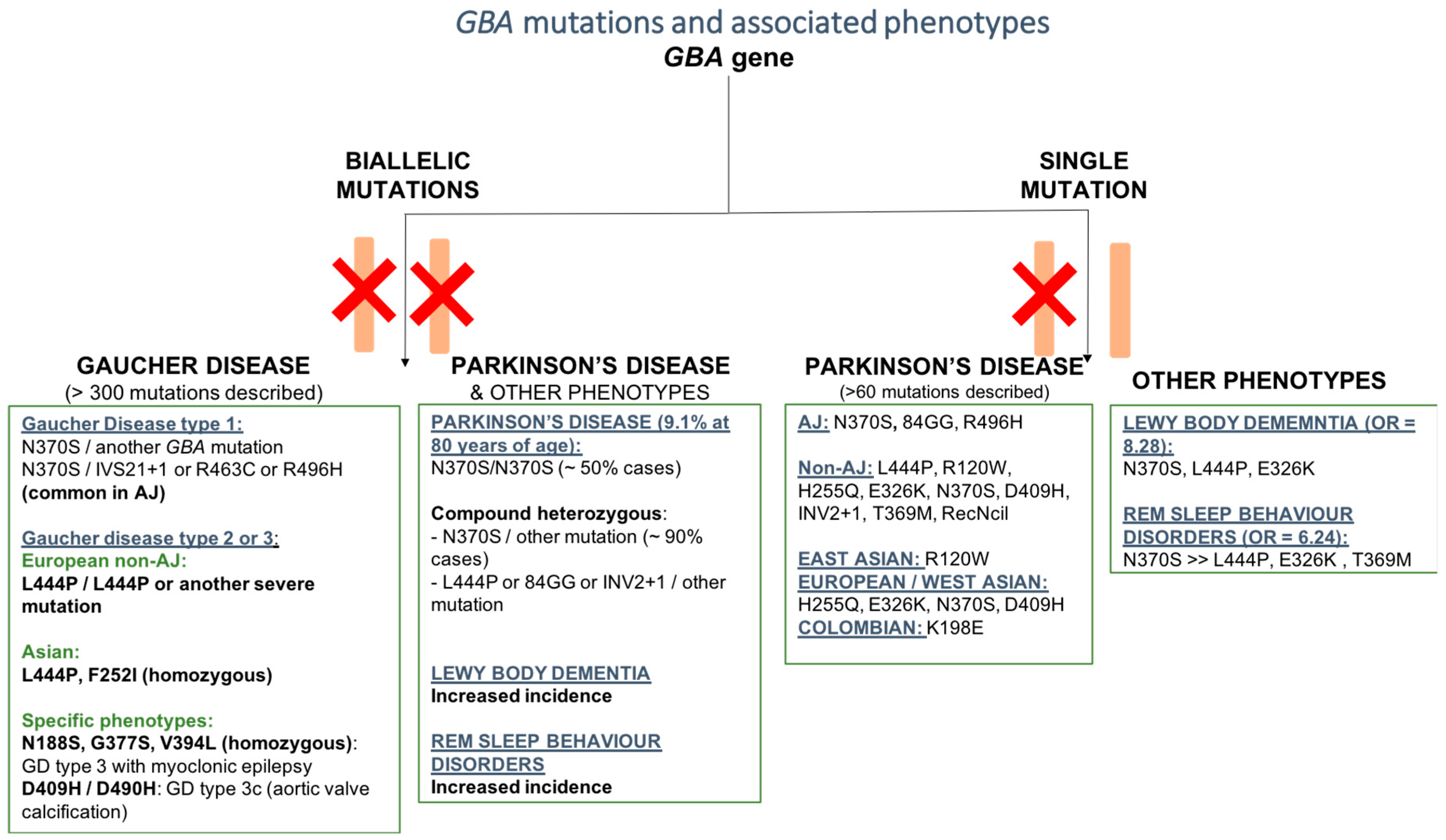
2.1. GBA Mutation and Gaucher’s Disease (GD)
Different Pathogenic Mutations of GBA Associated with Gaucher’s Disease (GD) Subtypes
2.2. GBA Mutation and Parkinson’s Disease (PD)
2.2.1. Pathogenic Mutations of GBA Associated with PD
2.2.2. GBA Mutations and Parkinson’s Disease Phenotype
2.2.3. GBA Mutations and Other Phenotypes
GBA and Dementia with Lewy Bodies
GBA and REM Sleep Behavior Disorders
3. New Targeted Treatments for GBA–PD Patients
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef]
- Eblan, M.J.; Walker, J.M.; Sidransky, E. The glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2005, 352, 728–731. [Google Scholar] [CrossRef]
- Sidransky, E. Heterozygosity for a Mendelian disorder as a risk factor for complex disease. Clin. Genet. 2006, 70, 275–282. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef]
- Klein, A.D.; Mazzulli, J.R. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Cilia, R.; Cerri, S.; Pezzoli, G.; Schapira, A.H.V.; Mullin, S.; Lanciego, J.L. Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Roca-Espiau, M.; Weinreb, N.J.; Bembi, B. Skeletal aspects of Gaucher disease: A review. Br. J. Radiol. 2002, 75 (Suppl. 1), A2–A12. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Abrahamov, A.; Dweck, A.; Hadas-Halpern, I.; Zimran, A. Gaucher disease: Pediatric concerns. Paediatr. Drugs 2002, 4, 417–426. [Google Scholar] [CrossRef]
- Andersson, H.; Kaplan, P.; Kacena, K.; Yee, J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008, 122, 1182–1190. [Google Scholar] [CrossRef]
- Kauli, R.; Zaizov, R.; Lazar, L.; Pertzelan, A.; Laron, Z.; Galatzer, A.; Phillip, M.; Yaniv, Y.; Cohen, I.J. Delayed growth and puberty in patients with Gaucher disease type 1: Natural history and effect of splenectomy and/or enzyme replacement therapy. ISR Med. Assoc. J. 2000, 2, 158–163. [Google Scholar] [PubMed]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Elstein, D. Gaucher disease and related Lysosomal Storage Diseases. In Williams’ Hematology; Lichtman, M.A., Kaushansky, K., Prchal, J.T., Levi, M., Burns, L.J., Press, O.W., Caligiuri, M.A., Eds.; McGraw-Hill: New York, NY, USA, 2016; Volume 72, p. 1121. [Google Scholar]
- Gupta, P.; Pastores, G. Pharmacological treatment of pediatric Gaucher disease. Expert Rev. Clin. Pharm. 2018, 11, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Grabowski, G. Gaucher disease. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Beaudet, A.L., Scriver, C.R., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill International Book Co.: New York, NY, USA, 2001; pp. 3635–3668. [Google Scholar]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.; Hughes, D. Gaucher Disease. In GeneReviews® [Internet]; Ardinger, H.H., Adam, M.P., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2000; (updated 2018 June 21). [Google Scholar]
- Bennett, L.L.; Mohan, D. Gaucher disease and its treatment options. Ann. Pharm. 2013, 47, 1182–1193. [Google Scholar] [CrossRef]
- Dreborg, S.; Erikson, A.; Hagberg, B. Gaucher disease—Norrbottnian type. I. General clinical description. Eur. J. Pediatrics 1980, 133, 107–118. [Google Scholar] [CrossRef]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Horowitz, M. Gaucher’s disease: Molecular, genetic and enzymological aspects. Baillieres Clin. Haematol. 1997, 10, 635–656. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Hruska, K.S.; Orvisky, E.; Kishnani, P.S.; Stubblefield, B.K.; Schiffmann, R.; Sidransky, E. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J. Med. Genet. 2005, 42, e37. [Google Scholar] [CrossRef]
- Cindik, N.; Ozcay, F.; Suren, D.; Akkoyun, I.; Gokdemir, M.; Varan, B.; Alehan, F.; Ozbek, N.; Tokel, K. Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin. Cardiol 2010, 33, E26–E30. [Google Scholar] [CrossRef] [PubMed]
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef]
- Kowarz, L.; Goker-Alpan, O.; Banerjee-Basu, S.; LaMarca, M.E.; Kinlaw, L.; Schiffmann, R.; Baxevanis, A.D.; Sidransky, E. Gaucher mutation N188S is associated with myoclonic epilepsy. Hum. Mutat. 2005, 26, 271–273. [Google Scholar] [CrossRef]
- Park, J.K.; Orvisky, E.; Tayebi, N.; Kaneski, C.; Lamarca, M.E.; Stubblefield, B.K.; Martin, B.M.; Schiffmann, R.; Sidransky, E. Myoclonic epilepsy in Gaucher disease: Genotype-phenotype insights from a rare patient subgroup. Pediatr. Res. 2003, 53, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Tayebi, N.; Stubblefield, B.K.; LaMarca, M.E.; MacKenzie, J.J.; Stone, D.L.; Sidransky, E. The E326K mutation and Gaucher disease: Mutation or polymorphism? Clin. Genet. 2002, 61, 32–34. [Google Scholar] [CrossRef]
- Horowitz, M.; Pasmanik-Chor, M.; Ron, I.; Kolodny, E.H. The enigma of the E326K mutation in acid beta-glucocerebrosidase. Mol. Genet. Metab. 2011, 104, 35–38. [Google Scholar] [CrossRef]
- Chabas, A.; Gort, L.; Diaz-Font, A.; Montfort, M.; Santamaria, R.; Cidras, M.; Grinberg, D.; Vilageliu, L. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: Effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol. Dis. 2005, 35, 253–258. [Google Scholar] [CrossRef]
- Liou, B.; Grabowski, G.A. Is E326K glucocerebrosidase a polymorphic or pathological variant? Mol. Genet. Metab. 2012, 105, 528–529. [Google Scholar] [CrossRef]
- Neudorfer, O.; Giladi, N.; Elstein, D.; Abrahamov, A.; Turezkite, T.; Aghai, E.; Reches, A.; Bembi, B.; Zimran, A. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996, 89, 691–694. [Google Scholar] [CrossRef]
- Machaczka, M.; Rucinska, M.; Skotnicki, A.B.; Jurczak, W. Parkinson’s syndrome preceding clinical manifestation of Gaucher’s disease. Am. J. Hematol. 1999, 61, 216–217. [Google Scholar] [CrossRef]
- Perez-Calvo, J.; Bernal, M.; Giraldo, P.; Torralba, M.A.; Civeira, F.; Giralt, M.; Pocovi, M. Co-morbidity in Gaucher’s disease results of a nationwide enquiry in Spain. Eur. J. Med. Res. 2000, 5, 231–235. [Google Scholar]
- Varkonyi, J.; Simon, Z.; Soos, K.; Poros, A. Gaucher disease type I complicated with Parkinson’s syndrome. Haematologia 2002, 32, 271–275. [Google Scholar] [CrossRef]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; LaMarca, M.E.; Wong, K.; Rosenbaum, H.; Schiffmann, R.; Bembi, B.; Sidransky, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Bembi, B.; Zambito Marsala, S.; Sidransky, E.; Ciana, G.; Carrozzi, M.; Zorzon, M.; Martini, C.; Gioulis, M.; Pittis, M.G.; Capus, L. Gaucher’s disease with Parkinson’s disease: Clinical and pathological aspects. Neurology 2003, 61, 99–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Shu, L.; Zhou, X.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. A Meta-Analysis of GBA-Related Clinical Symptoms in Parkinson’s Disease. Parkinsons Dis. 2018, 2018, 3136415. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Lorenzo-Betancor, O.; Jimenez-Del-Rio, M.; Moreno, S.; Lopera, F.; Cornejo-Olivas, M.; Torres, L.; Inca-Martinez, M.; Mazzetti, P.; Cosentino, C.; et al. The distribution and risk effect of GBA variants in a large cohort of PD patients from Colombia and Peru. Parkinsonism Relat. Disord. 2019. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.C.; Honore, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Boot, B.; Locascio, J.J.; Jansen, I.E.; Winder-Rhodes, S.; Eberly, S.; Elbaz, A.; Brice, A.; Ravina, B.; van Hilten, J.J.; et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016, 80, 674–685. [Google Scholar] [CrossRef]
- Thaler, A.; Gurevich, T.; Bar Shira, A.; Gana Weisz, M.; Ash, E.; Shiner, T.; Orr-Urtreger, A.; Giladi, N.; Mirelman, A. A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat. Disord. 2017, 36, 47–51. [Google Scholar] [CrossRef]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaiti, B.; Bonaiti-Pellie, C.; Brice, A. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef]
- McNeill, A.; Duran, R.; Proukakis, C.; Bras, J.; Hughes, D.; Mehta, A.; Hardy, J.; Wood, N.W.; Schapira, A.H. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 526–532. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal Dysfunction and alpha-Synuclein Aggregation in Parkinson’s Disease: Diagnostic Links. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C., Jr.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of acid beta-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Sidransky, E.; Lee, J.C. Saposin C protects glucocerebrosidase against alpha-synuclein inhibition. Biochemistry 2013, 52, 7161–7163. [Google Scholar] [CrossRef] [PubMed]
- Ouled Amar Bencheikh, B.; Leveille, E.; Ruskey, J.A.; Spiegelman, D.; Liong, C.; Fon, E.A.; Rouleau, G.A.; Dauvilliers, Y.; Dupre, N.; Alcalay, R.N.; et al. Sequencing of the GBA coactivator, Saposin C, in Parkinson disease. Neurobiol. Aging 2018, 72, 187.e1–187.e3. [Google Scholar] [CrossRef] [PubMed]
- Offman, M.N.; Krol, M.; Silman, I.; Sussman, J.L.; Futerman, A.H. Molecular basis of reduced glucosylceramidase activity in the most common Gaucher disease mutant, N370S. J. Biol. Chem. 2010, 285, 42105–42114. [Google Scholar] [CrossRef]
- Wei, R.R.; Hughes, H.; Boucher, S.; Bird, J.J.; Guziewicz, N.; Van Patten, S.M.; Qiu, H.; Pan, C.Q.; Edmunds, T. X-ray and biochemical analysis of N370S mutant human acid beta-glucosidase. J. Biol. Chem. 2011, 286, 299–308. [Google Scholar] [CrossRef]
- Horowitz, M.; Wilder, S.; Horowitz, Z.; Reiner, O.; Gelbart, T.; Beutler, E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 1989, 4, 87–96. [Google Scholar] [CrossRef]
- Imai, K.; Nakamura, M.; Yamada, M.; Asano, A.; Yokoyama, S.; Tsuji, S.; Ginns, E.I. A novel transcript from a pseudogene for human glucocerebrosidase in non-Gaucher disease cells. Gene 1993, 136, 365–368. [Google Scholar]
- Leija-Salazar, M.; Sedlazeck, F.J.; Toffoli, M.; Mullin, S.; Mokretar, K.; Athanasopoulou, M.; Donald, A.; Sharma, R.; Hughes, D.; Schapira, A.H.V.; et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genom. Med. 2019, e564. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs. mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef]
- Jesus, S.; Huertas, I.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Caceres-Redondo, M.T.; Vargas-Gonzalez, L.; Gomez-Llamas, M.; Carrillo, F.; Calderon, E.; Carballo, M.; et al. GBA Variants Influence Motor and Non-Motor Features of Parkinson’s Disease. PLoS ONE 2016, 11, e0167749. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Kim, J.; Wiggs, E.; Cintron, D.; Groden, C.; Tayebi, N.; Mistry, P.K.; Pastores, G.M.; Zimran, A.; Goker-Alpan, O.; et al. Clinical course and prognosis in patients with Gaucher disease and parkinsonism. Neurol Genet. 2016, 2, e57. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Tong, J.; Fook-Chong, S.; Yih, Y.; Wong, M.C.; Pavanni, R.; Zhao, Y. Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch. Neurol. 2007, 64, 1056–1058. [Google Scholar] [CrossRef]
- Eblan, M.J.; Nguyen, J.; Ziegler, S.G.; Lwin, A.; Hanson, M.; Gallardo, M.; Weiser, R.; De Lucca, M.; Singleton, A.; Sidransky, E. Glucocerebrosidase mutations are also found in subjects with early-onset parkinsonism from Venezuela. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Chen, C.M.; Chao, C.Y.; Ro, L.S.; Lyu, R.K.; Chang, K.H.; Lee-Chen, G.J. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J. Neurol. Neurosurg. Psychiatry 2007, 78, 977–979. [Google Scholar] [CrossRef]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef]
- Li, Y.; Sekine, T.; Funayama, M.; Li, L.; Yoshino, H.; Nishioka, K.; Tomiyama, H.; Hattori, N. Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol. Aging 2014, 35. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Hauser, A.K.; Schulte, C.; Csoti, I.; Gasser, T.; Berg, D. GBA-associated PD presents with nonmotor characteristics. Neurology 2011, 77, 276–280. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 407–411. [Google Scholar] [CrossRef]
- Lythe, V.; Athauda, D.; Foley, J.; Mencacci, N.E.; Jahanshahi, M.; Cipolotti, L.; Hyam, J.; Zrinzo, L.; Hariz, M.; Hardy, J.; et al. GBA-Associated Parkinson’s Disease: Progression in a Deep Brain Stimulation Cohort. J. Parkinsons Dis. 2017, 7, 635–644. [Google Scholar] [CrossRef]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013, 70, 727–735. [Google Scholar] [CrossRef]
- Gamez-Valero, A.; Prada-Dacasa, P.; Santos, C.; Adame-Castillo, C.; Campdelacreu, J.; Rene, R.; Gascon-Bayarri, J.; Ispierto, L.; Alvarez, R.; Ariza, A.; et al. GBA Mutations Are Associated With Earlier Onset and Male Sex in Dementia With Lewy Bodies. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 1066–1070. [Google Scholar] [CrossRef]
- Guerreiro, R.; Ross, O.A.; Kun-Rodrigues, C.; Hernandez, D.G.; Orme, T.; Eicher, J.D.; Shepherd, C.E.; Parkkinen, L.; Darwent, L.; Heckman, M.G.; et al. Investigating the genetic architecture of dementia with Lewy bodies: A two-stage genome-wide association study. Lancet Neurol. 2018, 17, 64–74. [Google Scholar] [CrossRef]
- Asselta, R.; Rimoldi, V.; Siri, C.; Cilia, R.; Guella, I.; Tesei, S.; Solda, G.; Pezzoli, G.; Duga, S.; Goldwurm, S. Glucocerebrosidase mutations in primary parkinsonism. Parkinsonism Relat. Disord. 2014, 20, 1215–1220. [Google Scholar] [CrossRef]
- Shiner, T.; Mirelman, A.; Gana Weisz, M.; Bar-Shira, A.; Ash, E.; Cialic, R.; Nevler, N.; Gurevich, T.; Bregman, N.; Orr-Urtreger, A.; et al. High Frequency of GBA Gene Mutations in Dementia With Lewy Bodies Among Ashkenazi Jews. JAMA Neurol. 2016, 73, 1448–1453. [Google Scholar] [CrossRef]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Factor, S.A.; Wood-Siverio, C.; et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 95–102. [Google Scholar] [CrossRef]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef]
- Perez-Roca, L.; Adame-Castillo, C.; Campdelacreu, J.; Ispierto, L.; Vilas, D.; Rene, R.; Alvarez, R.; Gascon-Bayarri, J.; Serrano-Munoz, M.A.; Ariza, A.; et al. Glucocerebrosidase mRNA is Diminished in Brain of Lewy Body Diseases and Changes with Disease Progression in Blood. Aging Dis. 2018, 9, 208–219. [Google Scholar] [CrossRef]
- Clark, L.N.; Kartsaklis, L.A.; Wolf Gilbert, R.; Dorado, B.; Ross, B.M.; Kisselev, S.; Verbitsky, M.; Mejia-Santana, H.; Cote, L.J.; Andrews, H.; et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch. Neurol. 2009, 66, 578–583. [Google Scholar] [CrossRef]
- Boeve, B.F.; Silber, M.H.; Ferman, T.J.; Lin, S.C.; Benarroch, E.E.; Schmeichel, A.M.; Ahlskog, J.E.; Caselli, R.J.; Jacobson, S.; Sabbagh, M.; et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013, 14, 754–762. [Google Scholar] [CrossRef]
- Galbiati, A.; Verga, L.; Giora, E.; Zucconi, M.; Ferini-Strambi, L. The risk of neurodegeneration in REM sleep behavior disorder: A systematic review and meta-analysis of longitudinal studies. Sleep Med. Rev. 2019, 43, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ferini-Strambi, L.; Marelli, S.; Galbiati, A.; Rinaldi, F.; Giora, E. REM Sleep Behavior Disorder (RBD) as a marker of neurodegenerative disorders. Arch. Ital. Biol. 2014, 152, 129–146. [Google Scholar] [CrossRef]
- Barber, T.R.; Lawton, M.; Rolinski, M.; Evetts, S.; Baig, F.; Ruffmann, C.; Gornall, A.; Klein, J.C.; Lo, C.; Dennis, G.; et al. Prodromal Parkinsonism and Neurodegenerative Risk Stratification in REM Sleep Behavior Disorder. Sleep 2017, 40. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015, 72, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gamez-Valero, A.; Iranzo, A.; Serradell, M.; Vilas, D.; Santamaria, J.; Gaig, C.; Alvarez, R.; Ariza, A.; Tolosa, E.; Beyer, K. Glucocerebrosidase gene variants are accumulated in idiopathic REM sleep behavior disorder. Parkinsonism Relat. Disord. 2018, 50, 94–98. [Google Scholar] [CrossRef]
- Fernandez-Santiago, R.; Iranzo, A.; Gaig, C.; Serradell, M.; Fernandez, M.; Tolosa, E.; Santamaria, J.; Ezquerra, M. Absence of LRRK2 mutations in a cohort of patients with idiopathic REM sleep behavior disorder. Neurology 2016, 86, 1072–1073. [Google Scholar] [CrossRef]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Ko, W.K.D.; Li, Q.; Bezard, E.; Schapira, A.H.V. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 2017, 71. [Google Scholar] [CrossRef]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease--Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef]
- Fernandes, H.J.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular alpha-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef]
- Bae, E.J.; Yang, N.Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Yang, C.; Swallows, C.L.; Zhang, C.; Lu, J.; Xiao, H.; Brady, R.O.; Zhuang, Z. Celastrol increases glucocerebrosidase activity in Gaucher disease by modulating molecular chaperones. Proc. Natl. Acad. Sci. USA 2014, 111, 249–254. [Google Scholar] [CrossRef]
- Yang, C.; Rahimpour, S.; Lu, J.; Pacak, K.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. USA 2013, 110, 966–971. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef]
- Sudhakar, V.; Richardson, R.M. Gene Therapy for Neurodegenerative Diseases. Neurother. J. Am. Soc. Exp. Neurother. 2019, 16, 166–175. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Parkinson’s disease genetics: Identifying novel risk loci, providing causal insights and improving estimates of heritable risk. BioRxiv 2018. [Google Scholar] [CrossRef]
- Ysselstein, D.; Shulman, J.M.; Krainc, D. Emerging links between pediatric lysosomal storage diseases and adult parkinsonism. Mov. Disord. Off. J. Mov. Disord. Soc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Zahs, K.; Wu, E.; Dakin, K.; Bras, J.; Guerreiro, R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018, 17, 721–730. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. https://doi.org/10.3390/cells8040364
Riboldi GM, Di Fonzo AB. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells. 2019; 8(4):364. https://doi.org/10.3390/cells8040364
Chicago/Turabian StyleRiboldi, Giulietta M., and Alessio B. Di Fonzo. 2019. "GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches" Cells 8, no. 4: 364. https://doi.org/10.3390/cells8040364
APA StyleRiboldi, G. M., & Di Fonzo, A. B. (2019). GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells, 8(4), 364. https://doi.org/10.3390/cells8040364