NFκB- and MAP-Kinase Signaling Contribute to the Activation of Murine Myeloid Dendritic Cells by a Flagellin A: Allergen Fusion Protein


Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Recombinant Proteins
2.2. Mice
2.3. In Vitro Generation of Mouse Bone Marrow-Derived Dendritic Cells, Stimulation, and Flow Cytometry
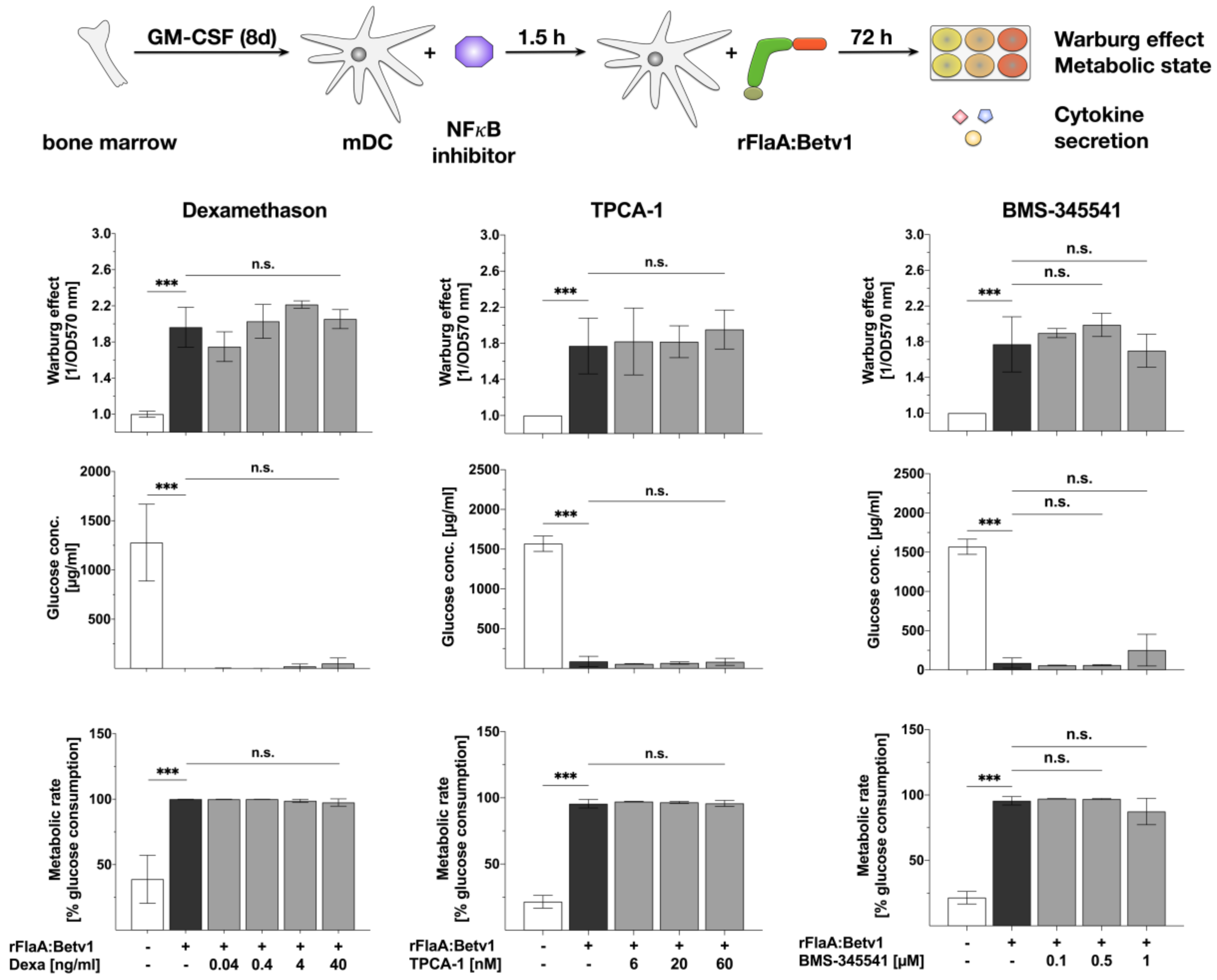
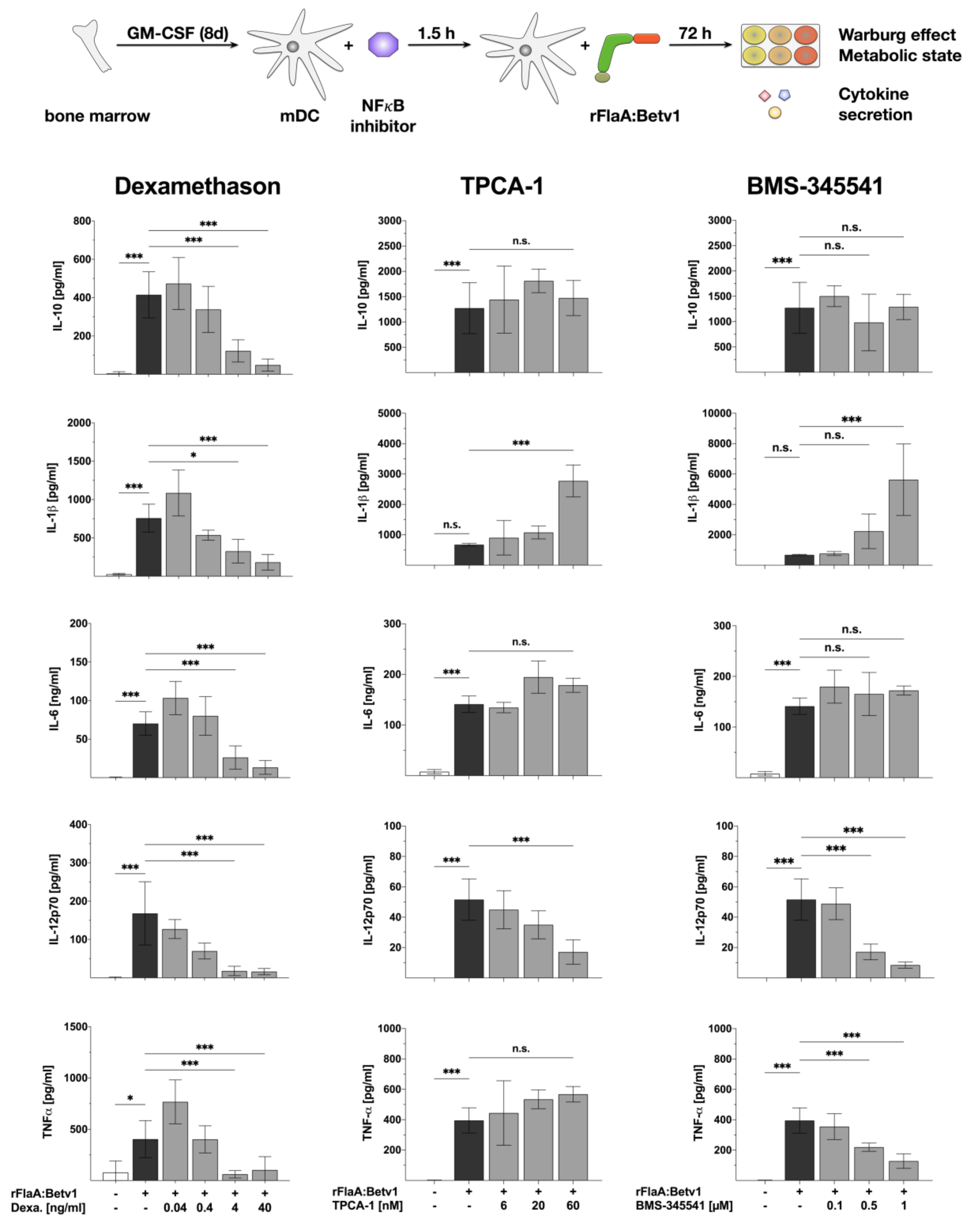
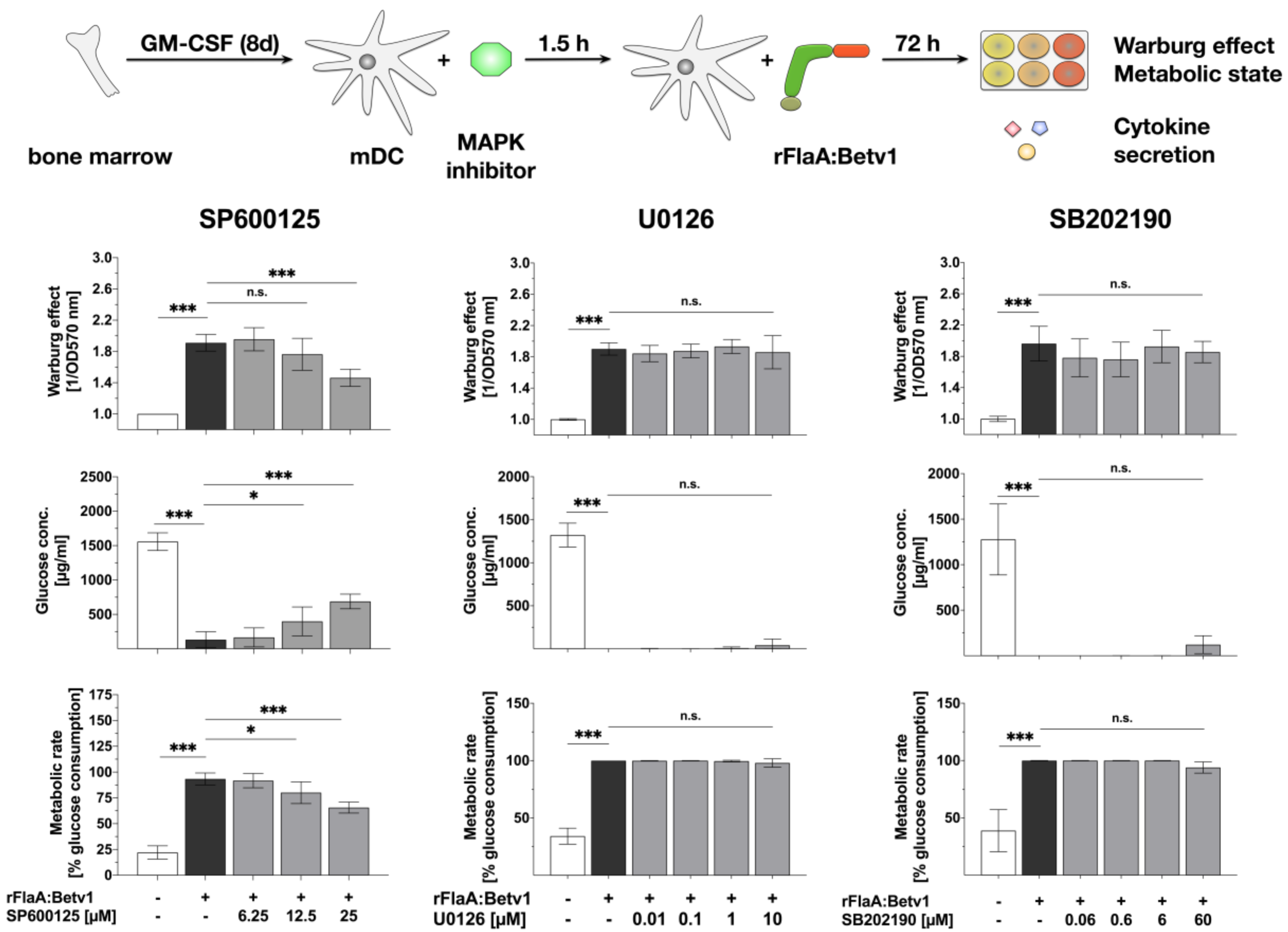
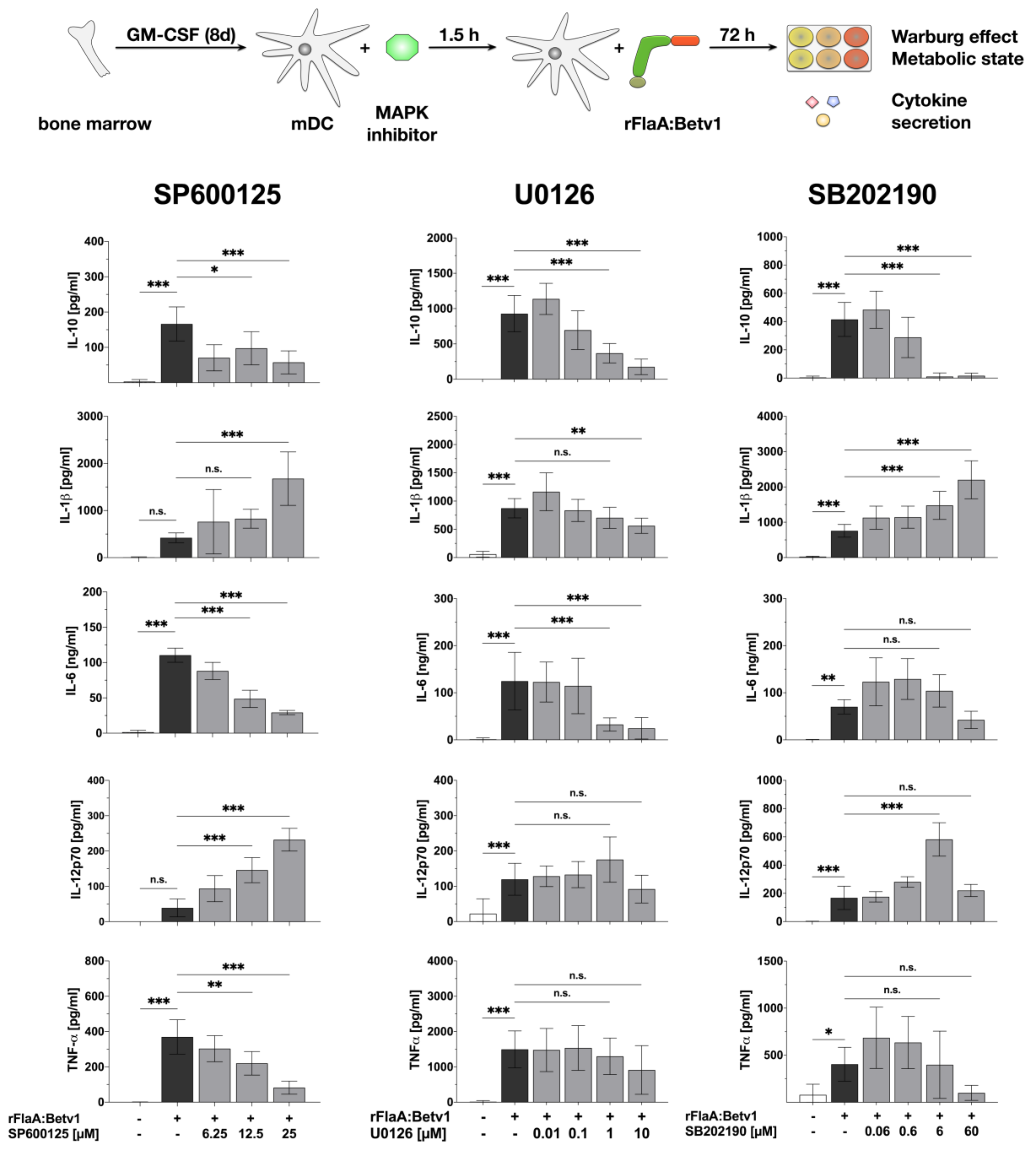
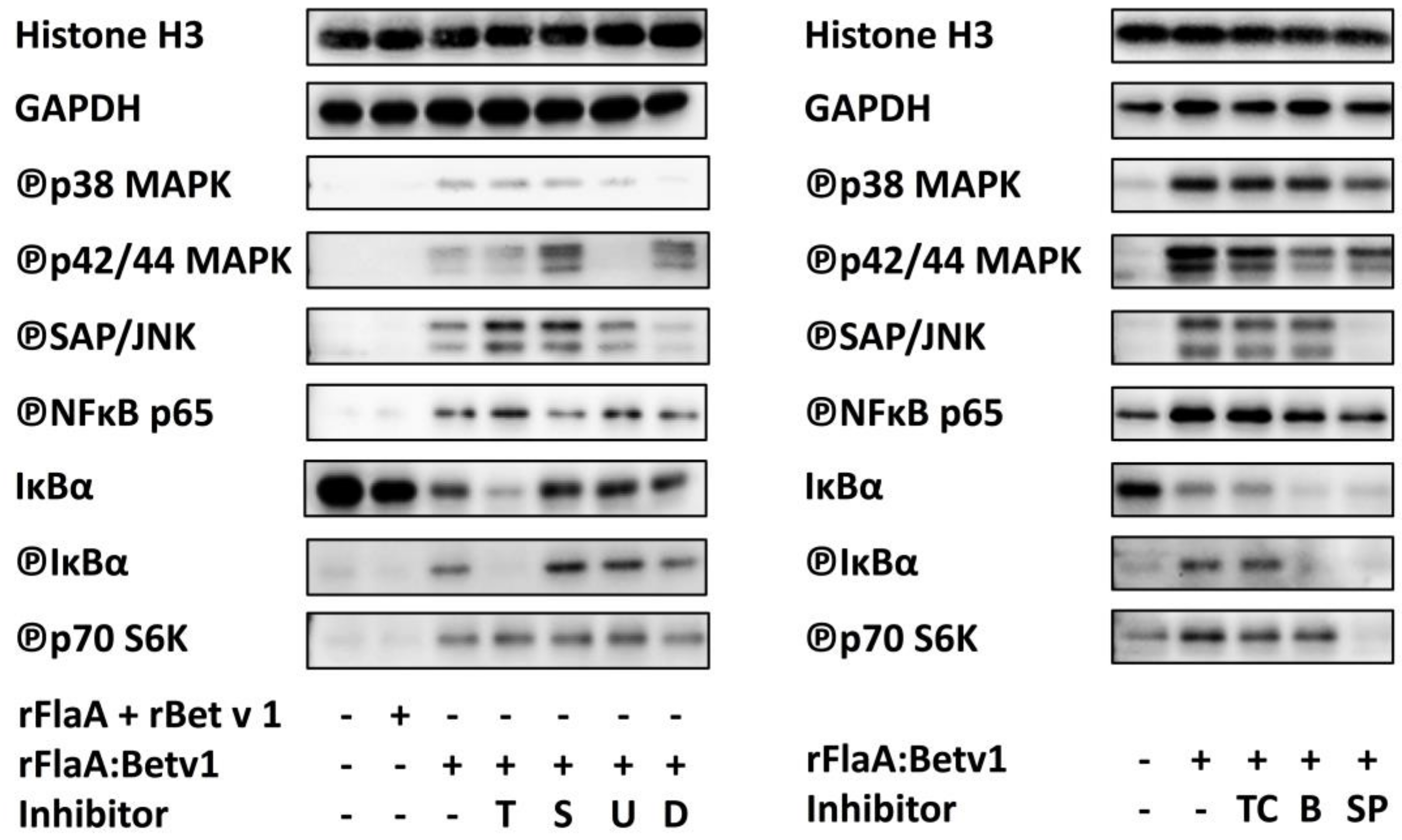
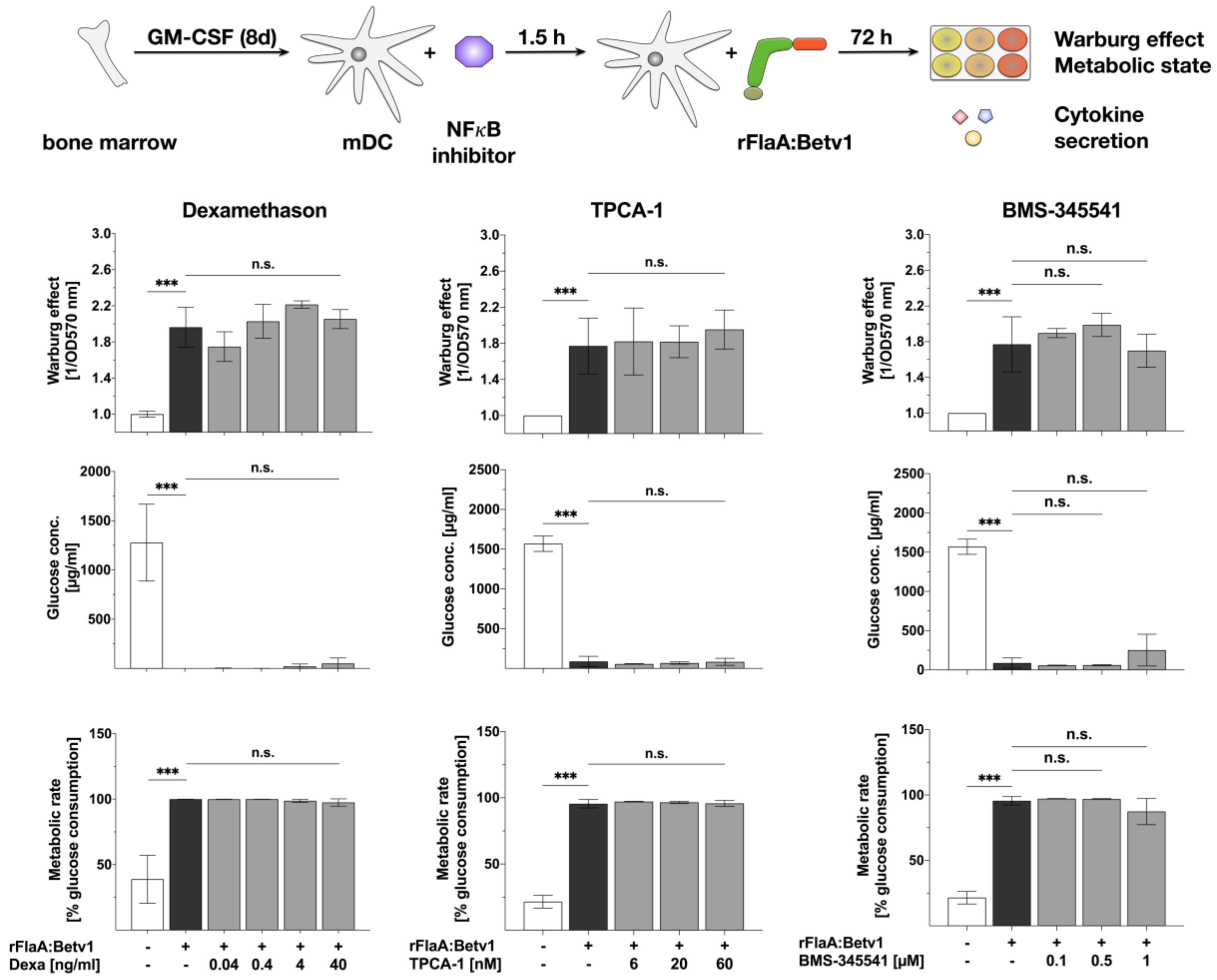
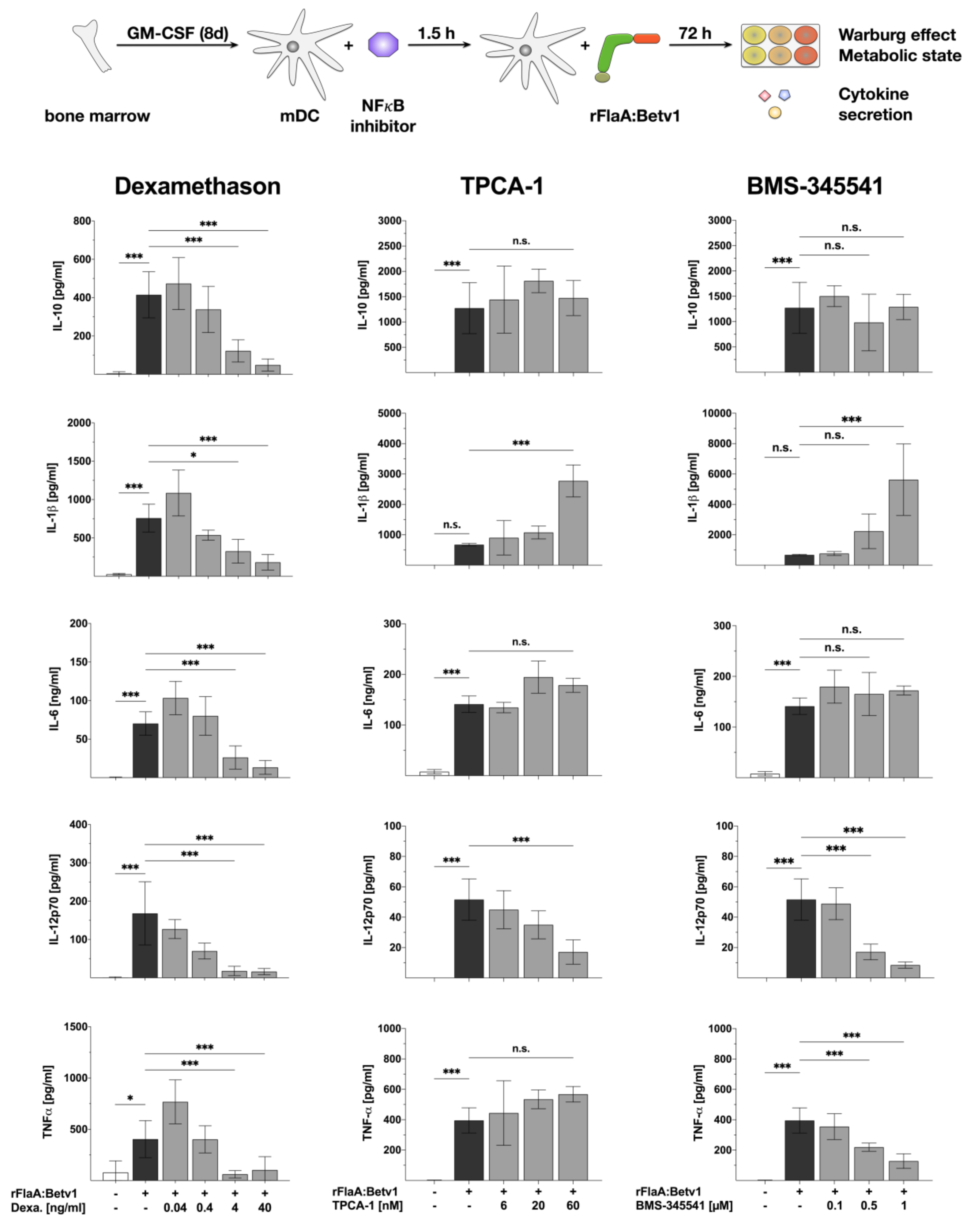
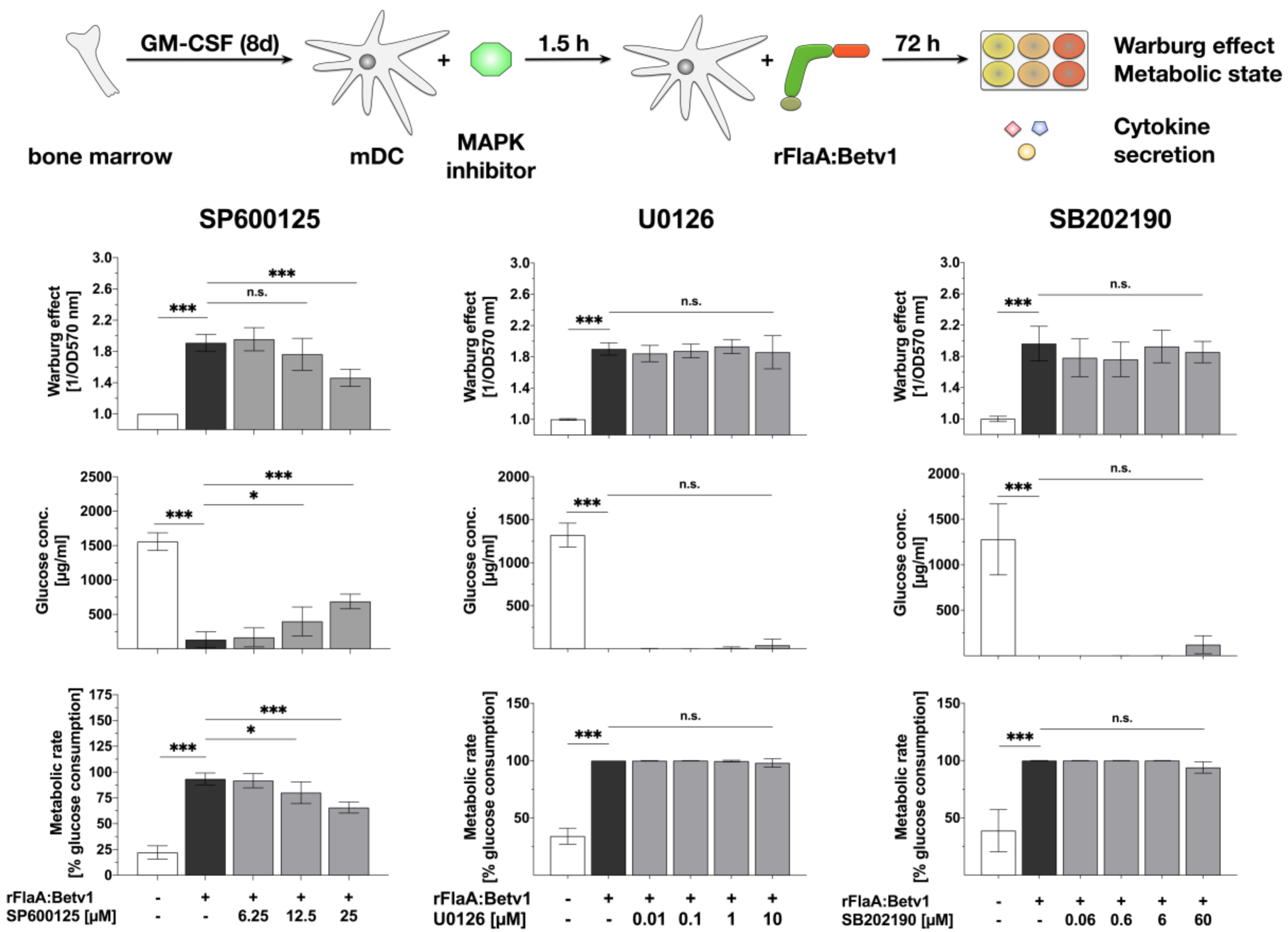
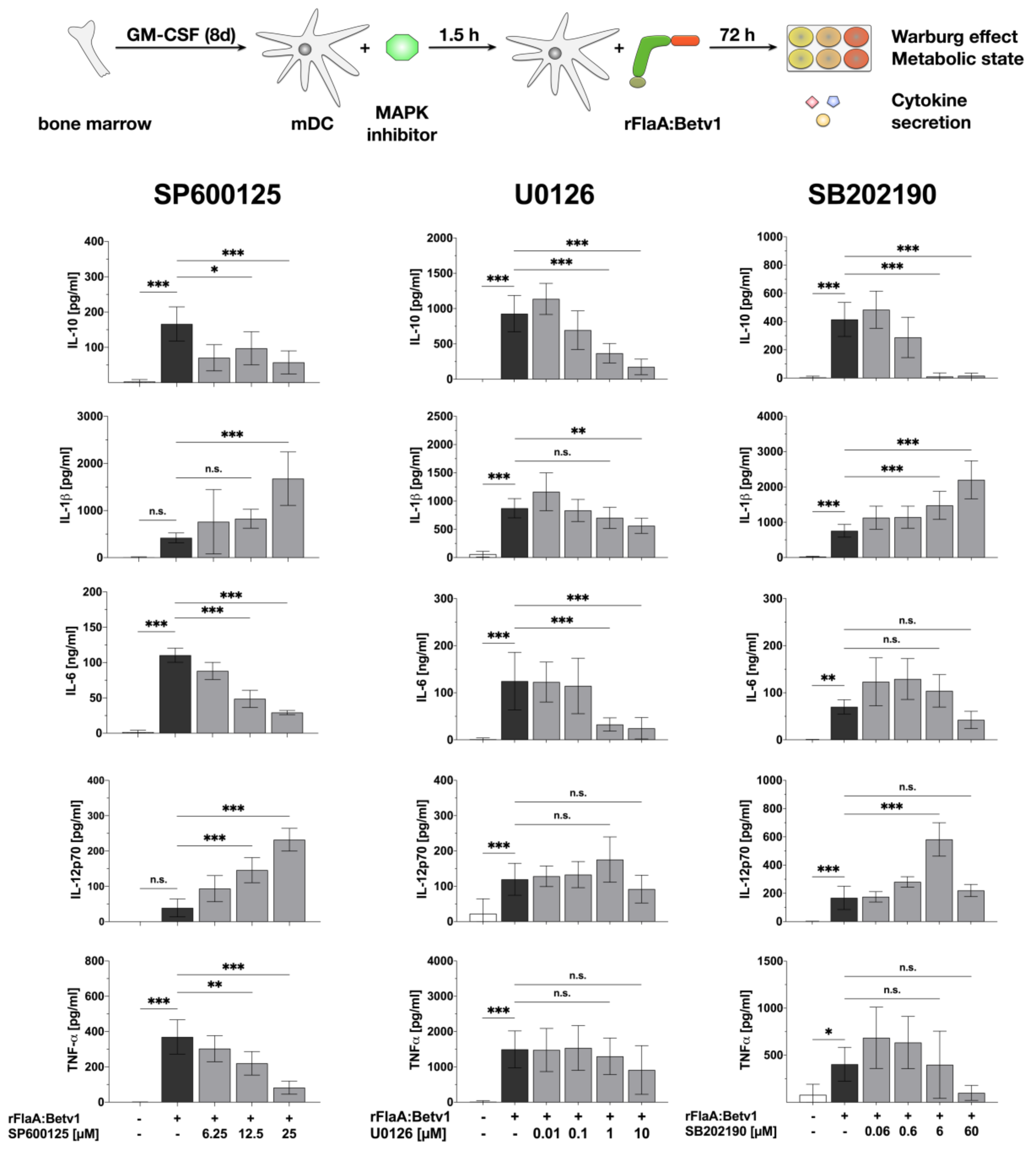
2.4. Inhibitors
2.5. SDS-PAGE and Western Blot
2.6. Analysis of Cell Metabolic State
2.7. Statistical Analysis
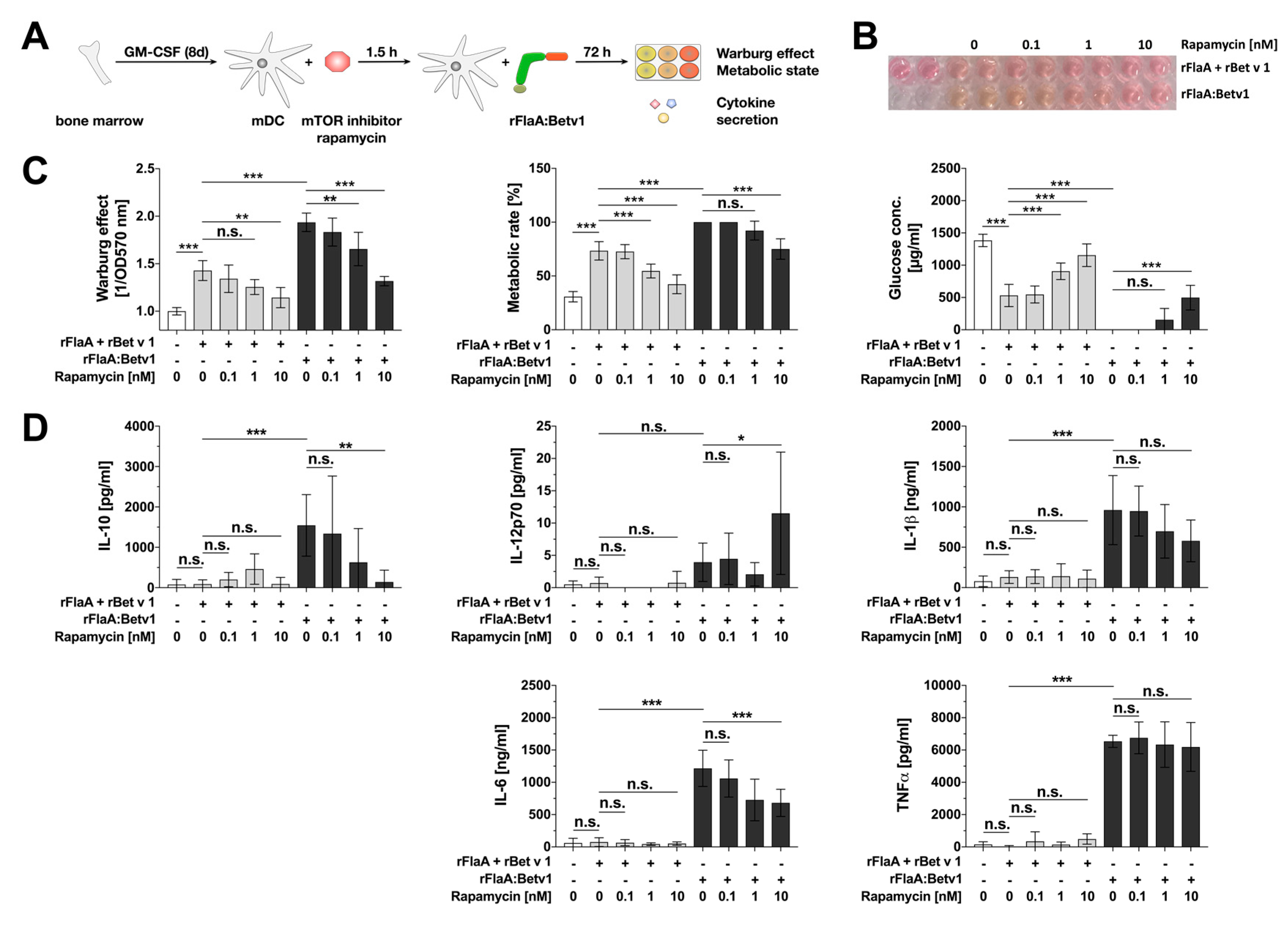
3. Results
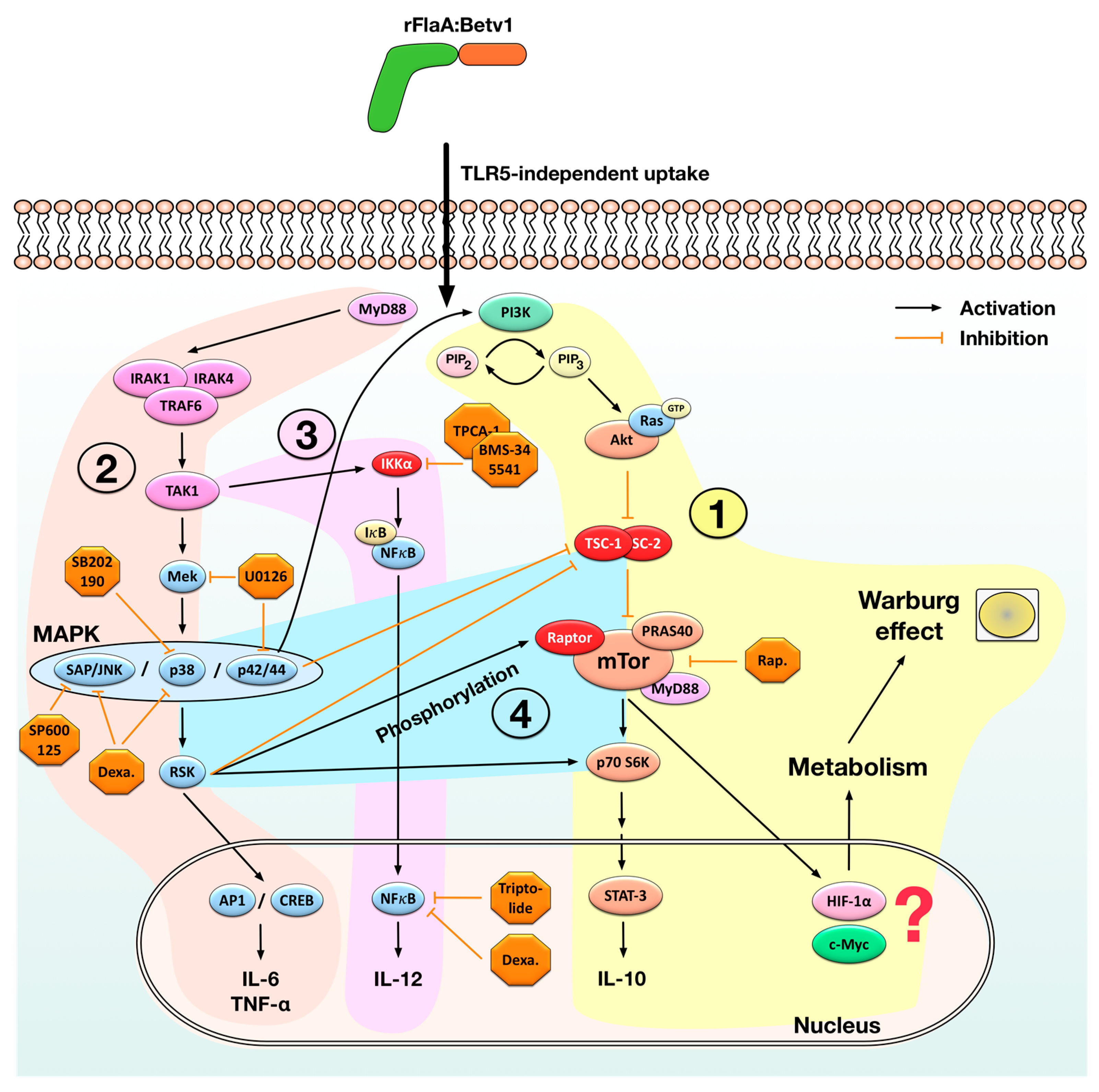
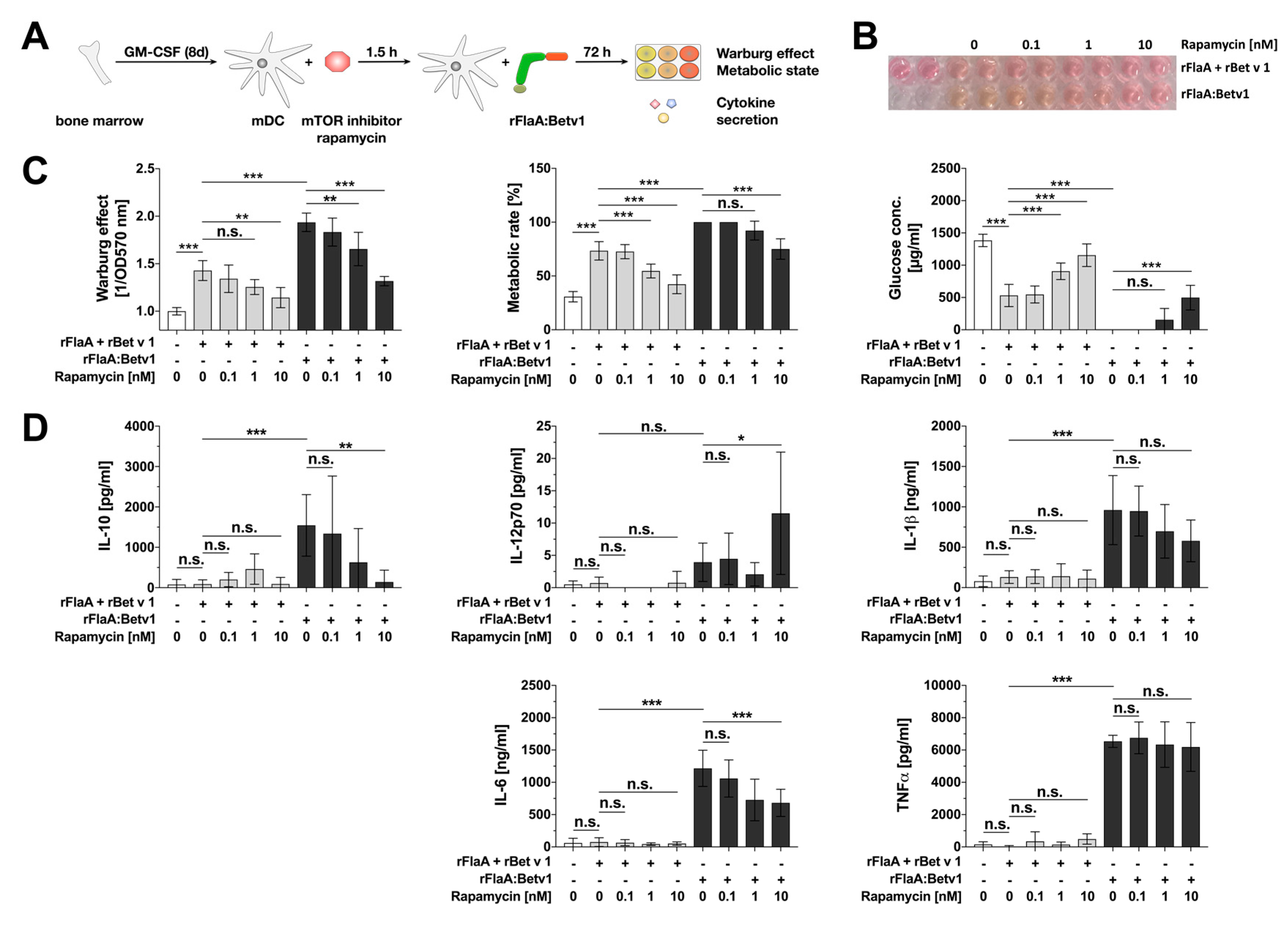
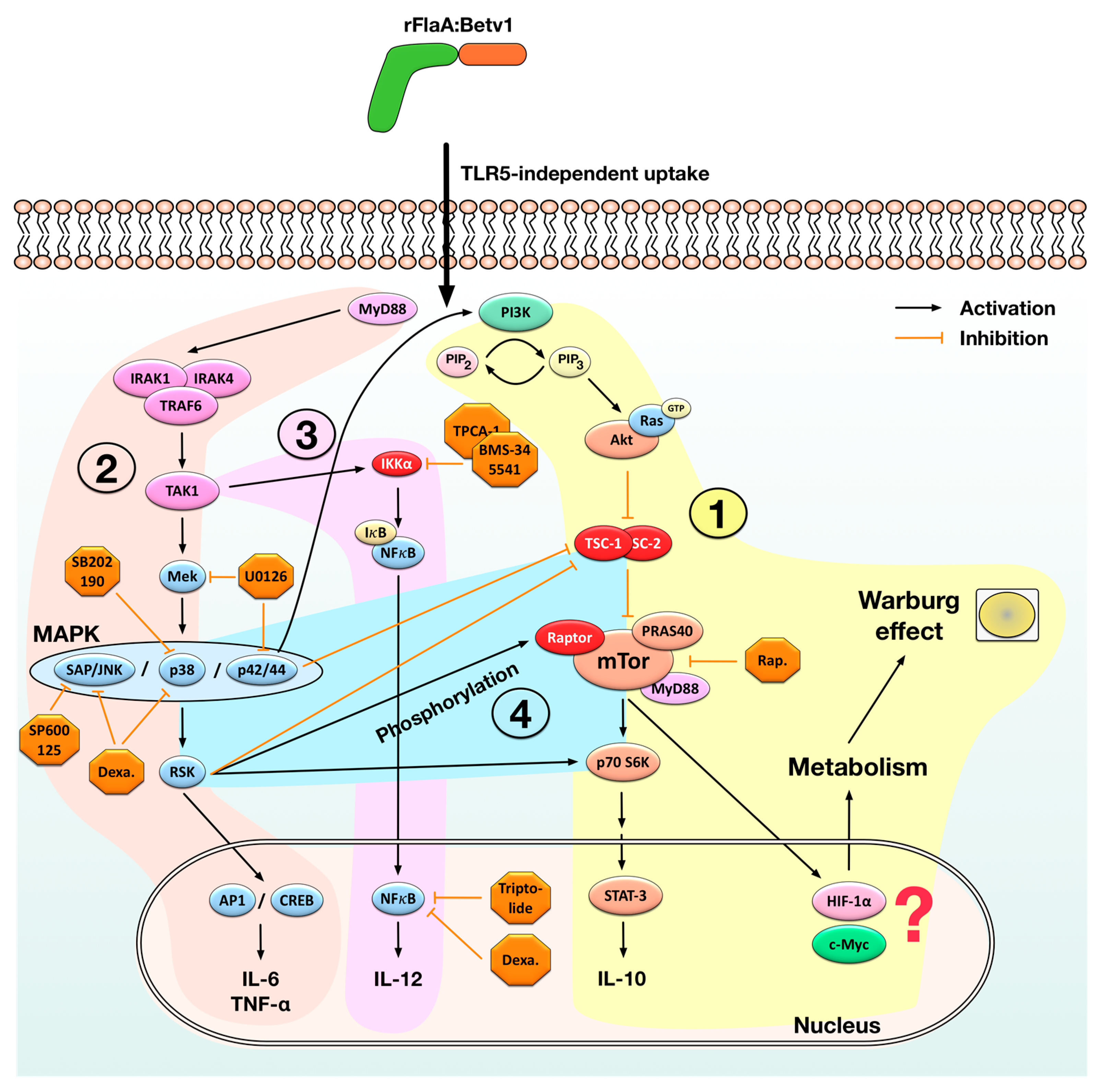
3.1. rFlaA:Betv1-Induced Activation of mDC Metabolism and IL-10 Secretion is Mediated by an Activation of the mTOR1 Complex, Whereas Pro-Inflammatory Cytokine Secretion Is Mostly mTOR-Independent
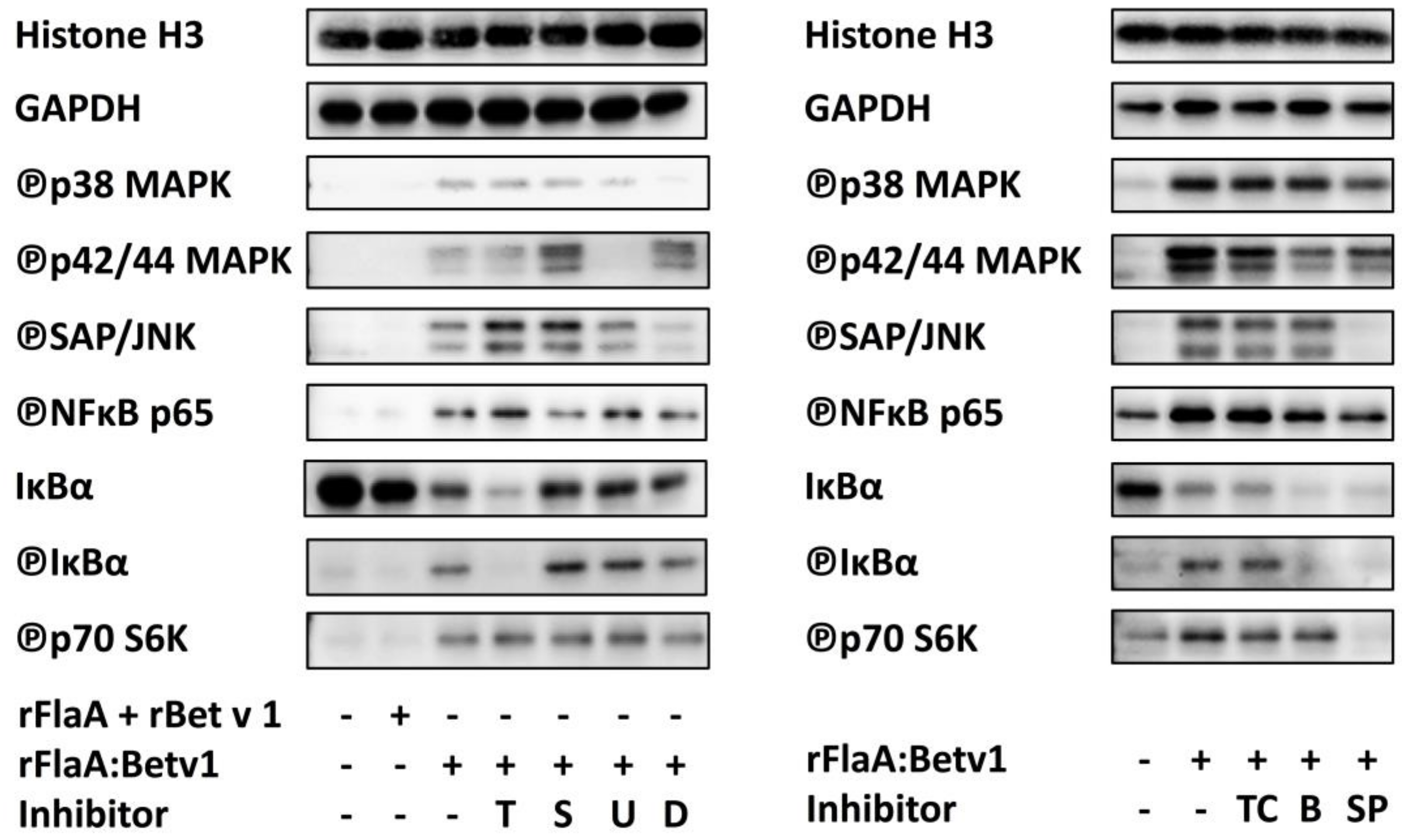
3.2. NFκB Inhibition Suppresses rFlaA:Betv1-Induced IL-12 Secretion while Enhancing IL-1β Secretion
3.3. MAPK Signalling Contributes to Both rFlaABetv1-Induced Pro- and Anti-Inflammatory Cytokine Secretion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP1 | Activator protein 1 |
| APC | Antigen presenting cell |
| Akt | Protein kinase B |
| c-Myc | Avian myelocytomatosis virus oncogene cellular homology |
| ConA | Concanavalin A |
| CREB | cAMP response element-binding protein |
| DC | Dendritic cell |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| IFN-γ | Interferon γ |
| Ig | Immunoglobulin |
| IκB | Inhibitor of kappa B |
| IL | Interleukin |
| IRAK | Interleukin-1 receptor-associated kinase |
| LPS | Lipopolysaccharide |
| Mal/TIRAP | MyD88 adapter-like/toll-interleukin 1 receptor domain containing adaptor protein |
| MAP(K) | Mitogen-activated protein (kinase) |
| mDC | Myeloid dendritic cell |
| MEK1/2 | MAP kinase/ERK kinase-1/2 |
| mTOR(1-complex) | Mammalian target of rapamycin (complex 1) |
| MyD88 | Myeloid differentiation primary response 88 |
| NFAT | Nuclear factor of activated T cells |
| NFκB | Nuclear factor “kapa-light-enhancer” of activated B cells |
| OD | Optical density |
| PBMC | Peripheral blood mononuclear cells |
| PI3K | Phosphatidylinositol 3-kinase |
| PRR | Pattern recognition receptor |
| RAPTOR | Regulatory associated protein of mTOR |
| Ras-ERK | Ras-extracellular signal-regulated kinase |
| rBet v 1 | Betula verucosa allergen 1 |
| rFlaA | Recombinant flagellin A |
| rFlaA:Betv1 | Recombinant fusion protein consisting of FlaA and Bet v 1 |
| Rel(A) | Reticuloendotheliosis oncogene cellular homolog (A) |
| RSK | ERK/ribosomal S6 kinase |
| STAT-3 | Signal transducer and activator of transcription 3 |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
| TC | T cell |
| TNF-α | Tumor necrosis factor α |
| TH1/2 | T helper type 1/2 |
| TLR(5) | “Toll”-like receptor (5) |
| TRAF6 | TNF receptor associated factor 6 |
| TRAM/TICAM-2 | TRIF-related adaptor molecule/ TIR domain-containing adapter molecule 2 |
| TRIF/TICAM-1 | TIR-domain-containing adapter-inducing interferon-β/ TIR domain-containing adapter molecule 1 |
| TSC-1/2 | Tuberous sclerosis complex ½ |
References
- Gupta, R.; Sheikh, A.; Strachan, D.P.; Anderson, H.R. Burden of allergic disease in the UK: Secondary analyses of national databases. Clin. Exp. Allergy 2004, 34, 520–526. [Google Scholar] [PubMed]
- Sicherer, S.H.; Leung, D.Y.M. Advances in allergic skin disease, anaphylaxis, and hypersensitivity reactions to foods, drugs, and insects in 2014. J. Allergy Clin. Immunol. 2015, 135, 357–367. [Google Scholar]
- Justicia, J.L.; Cardona, V.; Guardia, P.; Ojeda, P.; Olaguíbel, J.M.; Vega, J.M.; Vidal, C.; Baró, E.; García, M.A. Validation of the first treatment-specific questionnaire for the assessment of patient satisfaction with allergen-specific immunotherapy in allergic patients: The ESPIA questionnaire. J. Allergy Clin. Immunol. 2013, 131, 1539–1546. [Google Scholar] [PubMed]
- Valenta, R. The future of antigen-specific immunotherapy of allergy. Nat. Rev. Immunol. 2002, 2, 446–453. [Google Scholar] [PubMed]
- Toussi, D.; Massari, P. Immune Adjuvant Effect of Molecularly-defined Toll-Like Receptor Ligands. Vaccines 2014, 2, 323–353. [Google Scholar] [PubMed]
- Hayashi, F.; Aderem, A.; Smith, K.D.; Smith, K.D.; Akira, S.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [PubMed]
- Turley, C.B.; Rupp, R.E.; Johnson, C.; Taylor, D.N.; Wolfson, J.; Tussey, L.; Kavita, U.; Stanberry, L.; Shaw, A. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine 2011, 29, 5145–5152. [Google Scholar] [PubMed]
- Treanor, J.J.; Taylor, D.N.; Tussey, L.; Hay, C.; Nolan, C.; Fitzgerald, T.; Liu, G.; Kavita, U.; Song, L.; Dark, I.; et al. Safety and immunogenicity of a recombinant hemagglutinin influenza-flagellin fusion vaccine (VAX125) in healthy young adults. Vaccine 2010, 28, 8268–8274. [Google Scholar]
- Lee, S.E.; Kim, S.Y.; Jeong, B.C.; Kim, Y.R.; Bae, S.J.; Ahn, O.S.; Lee, J.J.; Song, H.C.; Kim, J.M.; Choy, H.E.; et al. A bacterial flagellin, Vibrio vulnificus FlaB, has a strong mucosal adjuvant activity to induce protective immunity. Infect. Immun. 2006, 74, 694–702. [Google Scholar]
- Honko, A.N.; Sriranganathan, N.; Lees, C.J.; Mizel, S.B. Flagellin is an effective adjuvant for immunization against lethal respiratory challenge with Yersinia pestis. Infect. Immun. 2006, 74, 1113–1120. [Google Scholar]
- Huleatt, J.W.; Jacobs, A.R.; Tang, J.; Desai, P.; Kopp, E.B.; Huang, Y.; Song, L.; Nakaar, V.; Powell, T.J. Vaccination with recombinant fusion proteins incorporating Toll-like receptor ligands induces rapid cellular and humoral immunity. Vaccine 2007, 25, 763–775. [Google Scholar] [PubMed]
- Song, L.; Xiong, D.; Kang, X.; Yang, Y.; Wang, J.; Guo, Y.; Xu, H.; Chen, S.; Peng, D.; Pan, Z.; et al. An avian influenza A (H7N9) virus vaccine candidate based on the fusion protein of hemagglutinin globular head and Salmonella typhimurium flagellin. BMC Biotechnol. 2015, 15, 79. [Google Scholar]
- Stepanova, L.A.; Kotlyarov, R.Y.; Kovaleva, A.A.; Potapchuk, M.V.; Korotkov, A.V.; Sergeeva, M.V.; Kasianenko, M.A.; Kuprianov, V.V.; Ravin, N.V.; Tsybalova, L.M.; et al. Protection against multiple influenza A virus strains induced by candidate recombinant vaccine based on heterologous M2e peptides linked to flagellin. PLoS ONE 2015, 10, e0119520. [Google Scholar]
- Wang, B.-Z.; Gill, H.S.; He, C.; Ou, C.; Wang, L.; Wang, Y.-C.; Feng, H.; Zhang, H.; Prausnitz, M.R.; Compans, R.W. Microneedle delivery of an M2e-TLR5 ligand fusion protein to skin confers broadly cross-protective influenza immunity. J. Control. Release 2014, 178, 1–7. [Google Scholar] [PubMed]
- Delaney, K.N.; Phipps, J.P.; Johnson, J.B.; Mizel, S.B. A recombinant flagellin-poxvirus fusion protein vaccine elicits complement-dependent protection against respiratory challenge with vaccinia virus in mice. Viral Immunol. 2010, 23, 201–210. [Google Scholar] [PubMed]
- McDonald, W.F.; Huleatt, J.W.; Foellmer, H.G.; Hewitt, D.; Tang, J.; Desai, P.; Price, A.; Jacobs, A.; Takahashi, V.N.; Huang, Y.; et al. A West Nile virus recombinant protein vaccine that coactivates innate and adaptive immunity. J. Infect. Dis. 2007, 195, 1607–1617. [Google Scholar] [PubMed]
- Lee, S.E.; Nguyen, C.T.; Kim, S.Y.; Thi, T.N.; Rhee, J.H. Tetanus toxin fragment C fused to flagellin makes a potent mucosal vaccine. Clin. Exp. Vaccine Res. 2015, 4, 59–67. [Google Scholar]
- Weimer, E.T.; Ervin, S.E.; Wozniak, D.J.; Mizel, S.B. Immunization of young African green monkeys with OprF epitope 8-OprI-type A- and B-flagellin fusion proteins promotes the production of protective antibodies against nonmucoid Pseudomonas aeruginosa. Vaccine 2009, 27, 6762–6769. [Google Scholar]
- Kitzmüller, C.; Kalser, J.; Mutschlechner, S.; Hauser, M.; Zlabinger, G.J.; Ferreira, F.; Bohle, B. Fusion proteins of flagellin and the major birch pollen allergen Bet v 1 show enhanced immunogenicity, reduced allergenicity, and intrinsic adjuvanticity. J. Allergy Clin. Immunol. 2018, 141, 293–299. [Google Scholar]
- Schülke, S.; Burggraf, M.; Waibler, Z.; Wangorsch, A.; Wolfheimer, S.; Kalinke, U.; Vieths, S.; Toda, M.; Scheurer, S. A fusion protein of flagellin and ovalbumin suppresses the TH2 response and prevents murine intestinal allergy. J. Allergy Clin. Immunol. 2011, 128, 1340–1348. [Google Scholar]
- Schülke, S.; Kuttich, K.; Wolfheimer, S.; Duschek, N.; Wangorsch, A.; Reuter, A.; Briza, P.; Pablos, I.; Gadermaier, G.; Ferreira, F.; et al. Author Correction: Conjugation of wildtype and hypoallergenic mugwort allergen Art v 1 to flagellin induces IL-10-DC and suppresses allergen-specific TH2-responses in vivo. Sci. Rep. 2018, 8, 2745. [Google Scholar]
- Schülke, S.; Fiedler, A.-H.; Junker, A.-C.; Flaczyk, A.; Wolfheimer, S.; Wangorsch, A.; Heinz, A.; Beckert, H.; Nagl, B.; Bohle, B.; et al. Critical role of mammalian target of rapamycin for IL-10 dendritic cell induction by a flagellin A conjugate in preventing allergic sensitization. J. Allergy Clin. Immunol. 2018, 141, 1786–1798. [Google Scholar] [PubMed]
- Zhu, S.; Zhang, C.; Wang, J.; Wei, L.; Quan, R.; Yang, J.; Yan, X.; Li, Z.; She, R.; Hu, F.; et al. Immunity Elicited by an Experimental Vaccine Based on Recombinant Flagellin-Porcine Circovirus Type 2 Cap Fusion Protein in Piglets. PLoS ONE 2016, 11, e0147432. [Google Scholar]
- Huleatt, J.W.; Nakaar, V.; Desai, P.; Huang, Y.; Hewitt, D.; Jacobs, A.; Tang, J.; McDonald, W.; Song, L.; Evans, R.K.; Umlauf, S.; Tussey, L.; Powell, T.J. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine 2008, 26, 201–214. [Google Scholar]
- Narayanan, K.; Erathodiyil, N.; Gopalan, B.; Chong, S.; Wan, A.C.A.; Ying, J.Y. Targeting Warburg Effect in Cancers with PEGylated Glucose. Adv. Healthc. Mater. 2016, 5, 696–701. [Google Scholar]
- Soares-Silva, M.; Diniz, F.F.; Gomes, G.N.; Bahia, D. The Mitogen-Activated Protein Kinase (MAPK) Pathway: Role in Immune Evasion by Trypanosomatids. Front. Microbiol. 2016, 7, 183. [Google Scholar]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [PubMed]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [PubMed]
- Moynagh, P.N. The NF-κB pathway. J. Cell. Sci. 2005, 118, 4589–4592. [Google Scholar] [PubMed]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [PubMed]
- Ruland, J.; Mak, T.W. From antigen to activation: Specific signal transduction pathways linking antigen receptors to NF-κB. Semin. Immunol. 2003, 15, 177–183. [Google Scholar] [PubMed]
- Schülke, S.; Waibler, Z.; Mende, M.-S.; Zoccatelli, G.; Vieths, S.; Toda, M.; Scheurer, S. Fusion protein of TLR5-ligand and allergen potentiates activation and IL-10 secretion in murine myeloid DC. Mol. Immunol. 2010, 48, 341–350. [Google Scholar] [PubMed]
- Siebeneicher, S.; Reuter, S.; Krause, M.; Wangorsch, A.; Maxeiner, J.; Wolfheimer, S.; Schülke, S.; Naito, S.; Heinz, A.; Taube, C.; et al. Epicutaneous immune modulation with Bet v 1 plus R848 suppresses allergic asthma in a murine model. Allergy 2014, 69, 328–337. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [PubMed]
- Bhattacharyya, S.; Ratajczak, C.K.; Vogt, S.K.; Kelley, C.; Colonna, M.; Schreiber, R.D.; Muglia, L.J. TAK1 targeting by glucocorticoids determines JNK and IkappaB regulation in Toll-like receptor-stimulated macrophages. Blood 2010, 115, 1921–1931. [Google Scholar]
- Qiu, D.; Zhao, G.; Aoki, Y.; Shi, L.; Uyei, A.; Nazarian, S.; Ng, J.C.; Kao, P.N. Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-κB transcriptional activation. J. Biol. Chem. 1999, 274, 13443–13450. [Google Scholar]
- Chang, W.T.; Kang, J.J.; Lee, K.Y.; Wei, K.; Anderson, E.; Gotmare, S.; Ross, J.A.; Rosen, G.D. Triptolide and chemotherapy cooperate in tumor cell apoptosis. A role for the p53 pathway. J. Biol. Chem. 2001, 276, 2221–2227. [Google Scholar]
- Westerheide, S.D.; Kawahara, T.L.A.; Orton, K.; Morimoto, R.I. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 2006, 281, 9616–9622. [Google Scholar]
- Stanborough, T.; Niederhauser, J.; Koch, B.; Bergler, H.; Pertschy, B. Ribosomal protein S3 interacts with the NF-κB inhibitor IκBα. FEBS Lett. 2014, 588, 659–664. [Google Scholar]
- Russo, A.; Saide, A.; Cagliani, R.; Cantile, M.; Botti, G.; Russo, G. rpL3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating CBS and NFκB upon 5-FU treatment. Sci. Rep. 2016, 6, 38369. [Google Scholar]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar]
- Kodaki, T.; Woscholski, R.; Hallberg, B.; Rodriguez-Viciana, P.; Downward, J.; Parker, P.J. The activation of phosphatidylinositol 3-kinase by Ras. Curr. Biol. 1994, 4, 798–806. [Google Scholar]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar]
- Suire, S.; Hawkins, P.; Stephens, L. Activation of phosphoinositide 3-kinase gamma by Ras. Curr. Biol. 2002, 12, 1068–1075. [Google Scholar]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar]
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577. [Google Scholar]
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J. Biol. Chem. 2010, 285, 80–94. [Google Scholar]
- Menon, M.B.; Kotlyarov, A.; Gaestel, M. SB202190-induced cell type-specific vacuole formation and defective autophagy do not depend on p38 MAP kinase inhibition. PLoS ONE 2011, 6, e23054. [Google Scholar]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar]
- Kim, J.-H.; Yoon, M.-S.; Chen, J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J. Biol. Chem. 2009, 284, 35425–35432. [Google Scholar]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histon H3 | GAPDH | ℗p38 | ℗p42/44 | ℗SAP/JNK | ℗NFκBp65 | IκB-α | ℗IκB-α | ℗p70 S6K | |
|---|---|---|---|---|---|---|---|---|---|
| rFlaA + rBet v 1 | 1.06 ± 0.02 | 0.95 ± 0.16 | 1.07 ± 0.21 | 0.83 ± 0.16 | 1.68 ± 1.35 | 5.01 ± 5.45 | 0.79 ± 0.07 | 0.71 ± 0.15 | 0.71 ± 0.31 |
| rFlaA:Betv1 | 0.99 ± 0.07 | 1.09 ± 0.22 | 2.64 ± 1.19 | 3.57 ± 0.38 | 2.81 ± 0.22 | 16.79 ± 6.81 | 0.44 ± 0.02 | 1.52 ± 0.17 | 2.07 ± 0.85 |
| rFlaA:Betv1 + dexamethason | 1.17 ± 0.30 | 0.83 ± 0.16 | 0.65 ± 0.74 | 9.51 ± 6.49 | 1.97 ± 0.27 | 34.02 ± 32.76 | 0.37 ± 0.05 | 2.11 ± 0.40 | 3.64 ± 1.43 |
| rFlaA:Betv1 + TPCA-1 | 0.92 ± 0.04 | 1.05 ± 0.11 | 2.62 ± 1.01. | 2.71 ± 0.86 | 2.82 ± 0.52 | 2.30 ± 0.70 | 0.43 ± 0.05 | 1.46 ± 0.03 | 1.37 ± 0.15 |
| rFlaA:Betv1 + BMS-345541 | 0.88 ± 0.06 | 1.04 ± 0.23 | 2.57 ± 0.94 | 1.86 ± 0.57 | 2.97 ± 0.68 | 1.98 ± 0.69 | 0.29 ± 0.10 | 0.78 ± 0.16 | 1.57 ± 0.33 |
| rFlaA:Betv1 + triptolide | 1.17 ± 0.08 | 1.05 ± 0.24 | 3.87 ± 2.38 | 11.42 ± 12.43 | 5.79 ± 2.38 | 32.03 ± 20.29 | 0.18 ± 0.03 | 1.03 ± 0.18 | 4.99 ± 2.52 |
| rFlaA:Betv1 + Sp600125 | 0.92 ± 0.10 | 1.07 ± 0.12 | 1.96 ± 0.78 | 2.65 ± 1.20 | 1.14 ± 0.34 | 1.93 ± 0.60 | 0.31 ± 0.06 | 0.67 ± 0.25 | 0.74 ± 0.32 |
| rFlaA:Betv1 + U0126 | 1.16 ± 0.22 | 0.96 ± 0.29 | 2.66 ± 0.47 | 1.31 ± 0.83 | 4.28 ± 0.22 | 32.28 ± 23.97 | 0.42 ± 0.15 | 2.47 ± 0.08 | 4.55 ± 1.65 |
| rFlaA:Betv1 + SB202190 | 1.10 ± 0.11 | 0.93 ± 0.29 | 3.40 ± 1.54 | 15.80 ± 14.57 | 5.07 ± 2.71 | 38.11 ± 29.42 | 0.43 ± 0.15 | 2.46 ± 0.40 | 5.18 ± 2.81 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moeller, T.; Wolfheimer, S.; Goretzki, A.; Scheurer, S.; Schülke, S. NFκB- and MAP-Kinase Signaling Contribute to the Activation of Murine Myeloid Dendritic Cells by a Flagellin A: Allergen Fusion Protein. Cells 2019, 8, 355. https://doi.org/10.3390/cells8040355
Moeller T, Wolfheimer S, Goretzki A, Scheurer S, Schülke S. NFκB- and MAP-Kinase Signaling Contribute to the Activation of Murine Myeloid Dendritic Cells by a Flagellin A: Allergen Fusion Protein. Cells. 2019; 8(4):355. https://doi.org/10.3390/cells8040355
Chicago/Turabian StyleMoeller, Tobias, Sonja Wolfheimer, Alexandra Goretzki, Stephan Scheurer, and Stefan Schülke. 2019. "NFκB- and MAP-Kinase Signaling Contribute to the Activation of Murine Myeloid Dendritic Cells by a Flagellin A: Allergen Fusion Protein" Cells 8, no. 4: 355. https://doi.org/10.3390/cells8040355
APA StyleMoeller, T., Wolfheimer, S., Goretzki, A., Scheurer, S., & Schülke, S. (2019). NFκB- and MAP-Kinase Signaling Contribute to the Activation of Murine Myeloid Dendritic Cells by a Flagellin A: Allergen Fusion Protein. Cells, 8(4), 355. https://doi.org/10.3390/cells8040355