Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy

 , , ,
, , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. CRISPR/Cas9 Plasmid Construction and Transfection
2.2. Analysis of CRISPR/Cas9-Induced Mutations in GLA Gene
2.3. hESC Culture and Differentiation to Cardiomyocytes
2.4. Alkaline Phosphatase Staining
2.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.6. Immunofluorescence Staining
2.7. Measurement of Cardiomyocyte Size
2.8. Western Blotting
2.9. Lipid Extraction
2.10. Quantification of Gb3 by Thin Layer Chromatography (TLC)
2.11. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis
2.12. Transmission Electron Microscopy
2.13. Array-Based Comparative Genomic Hybridization (CGH-Array
2.14. Exosome Isolation and Characterization
2.15. Quantification of Isolated Exosomes
2.16. Mitochondrial Superoxide Stress Quantification
2.17. Annexin V Staining
2.18. GLA Enzyme Activity Assay
2.19. Statistical Analysis
3. Results
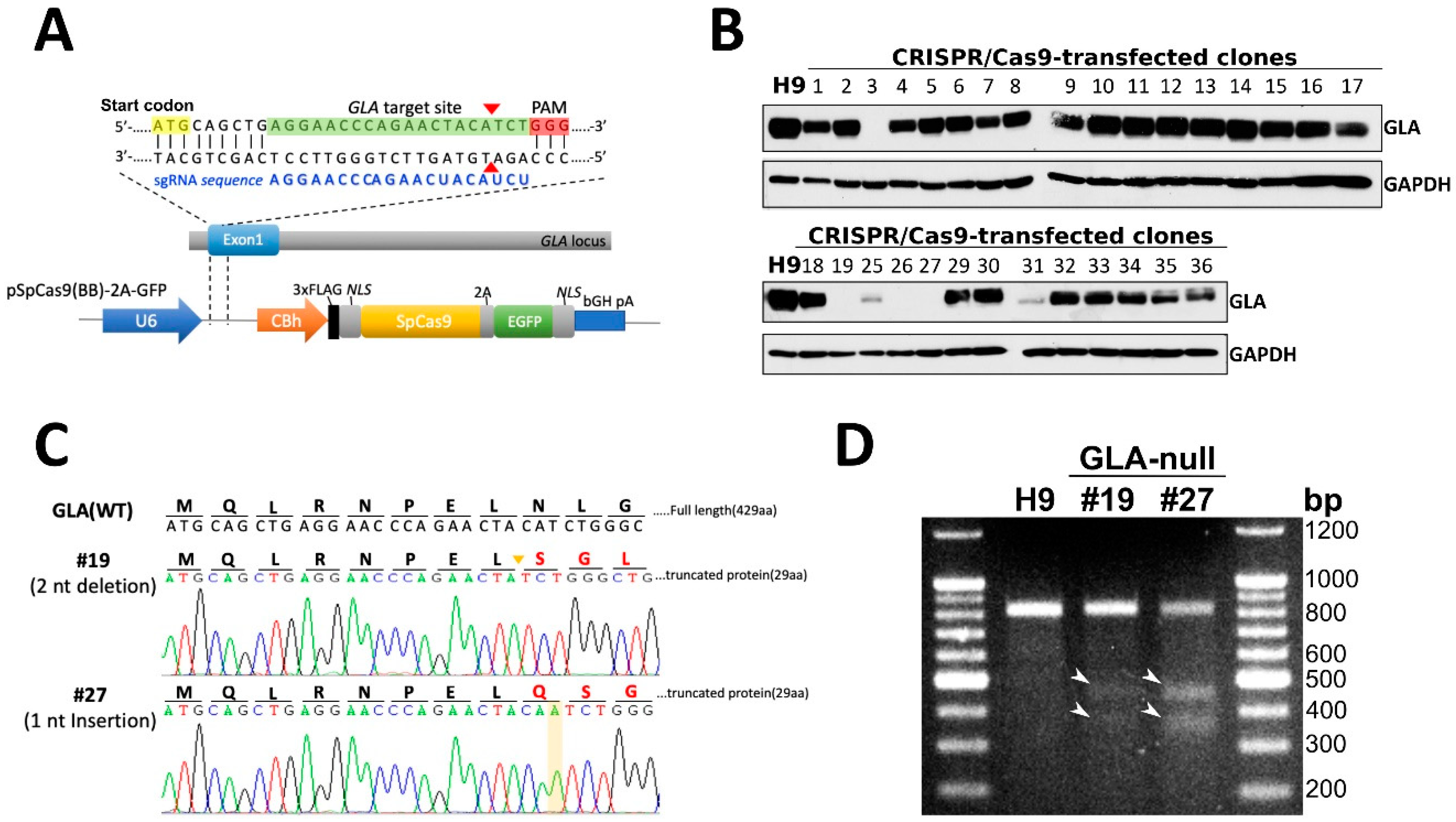
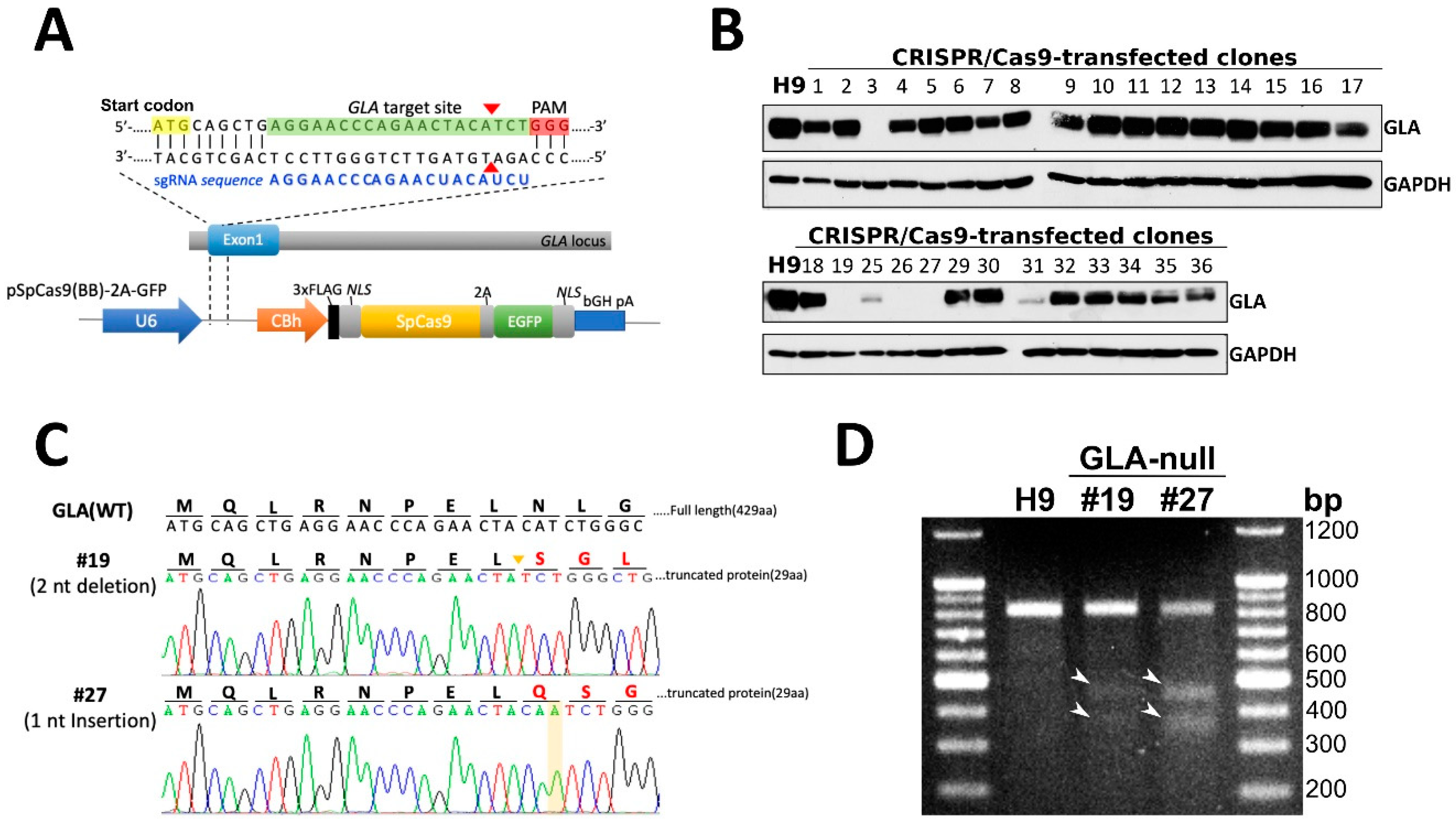
3.1. CRISPR/Cas9-Mediated Knockout of Expression of GLA in hESCs
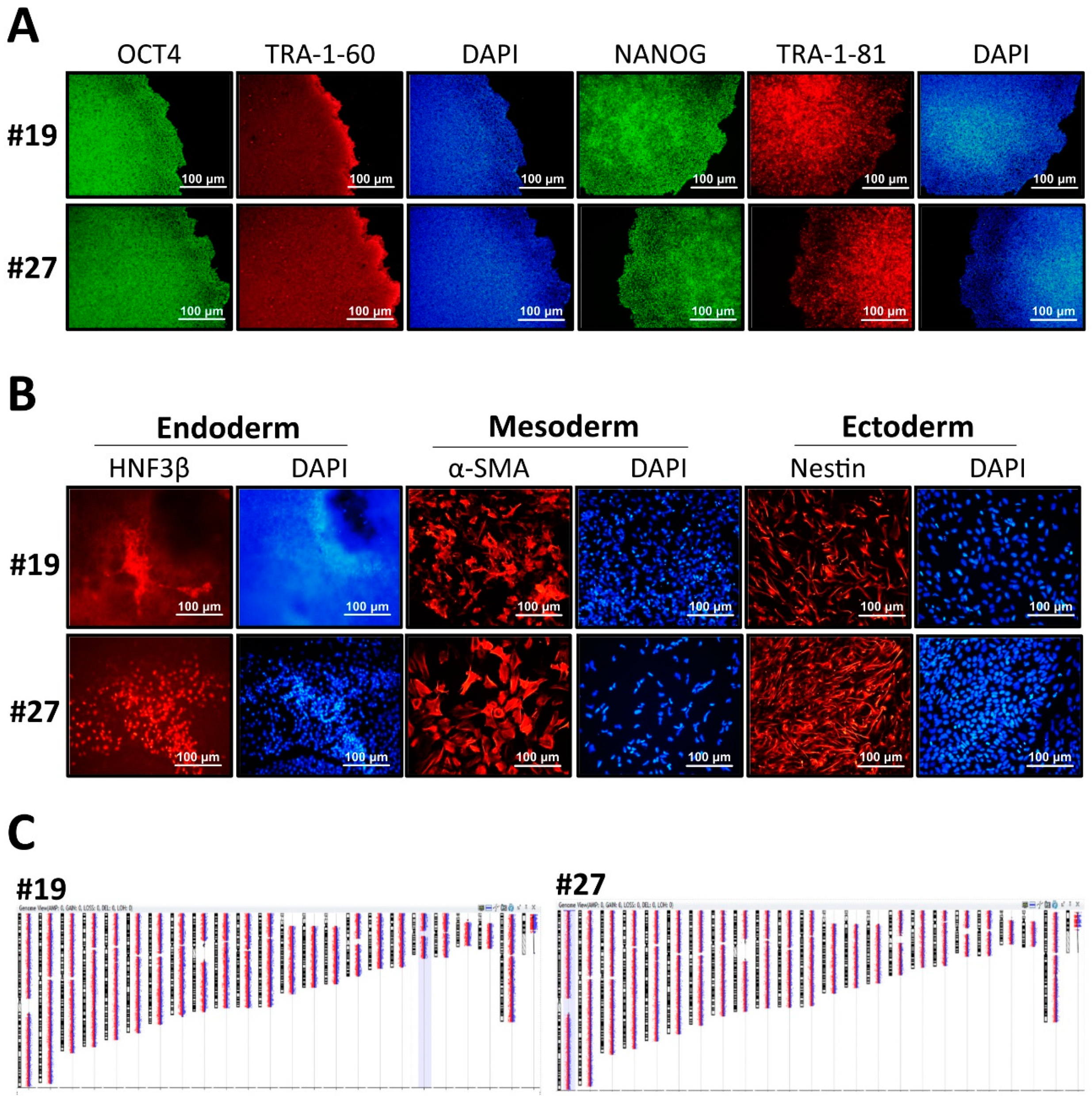
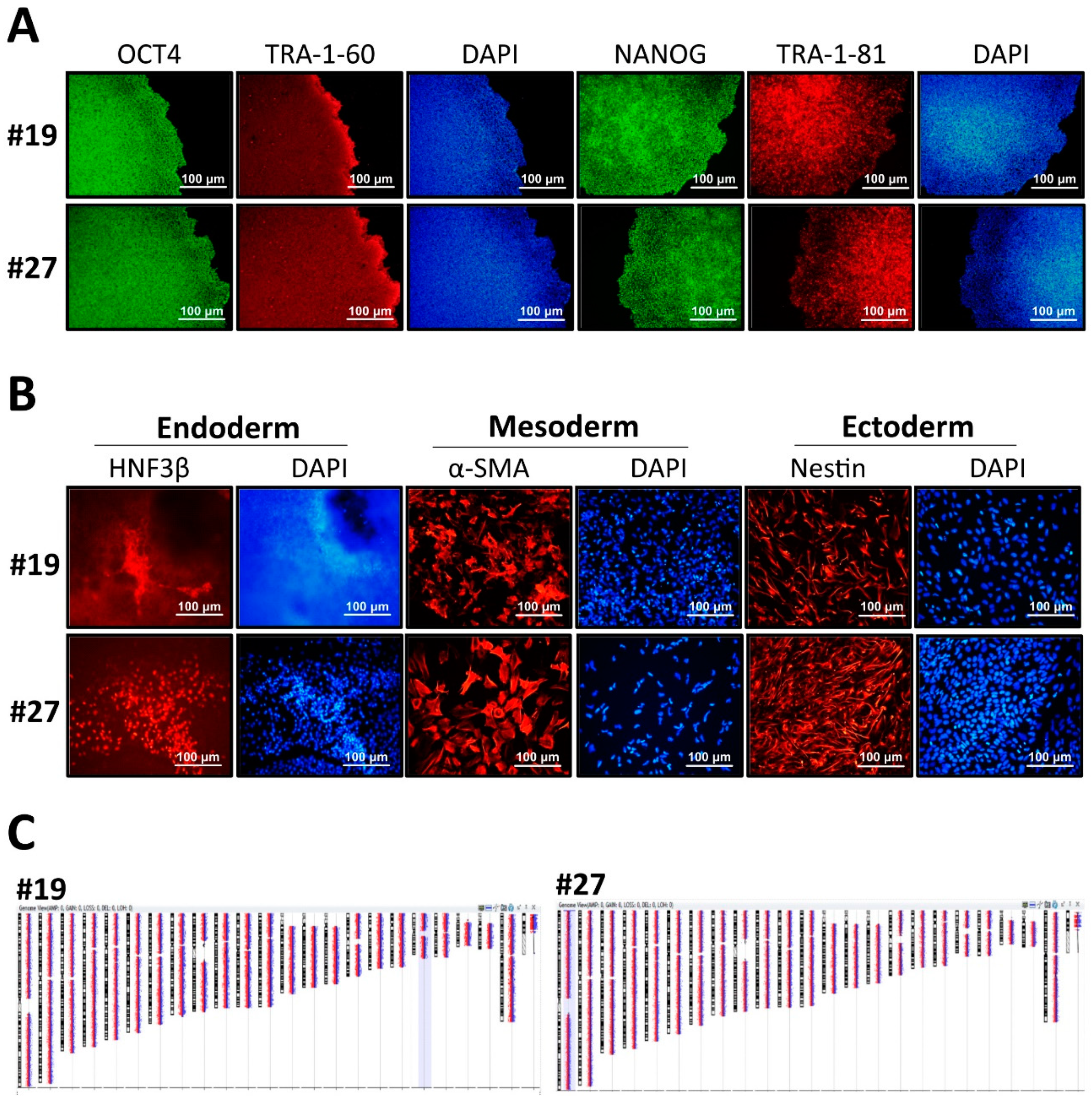
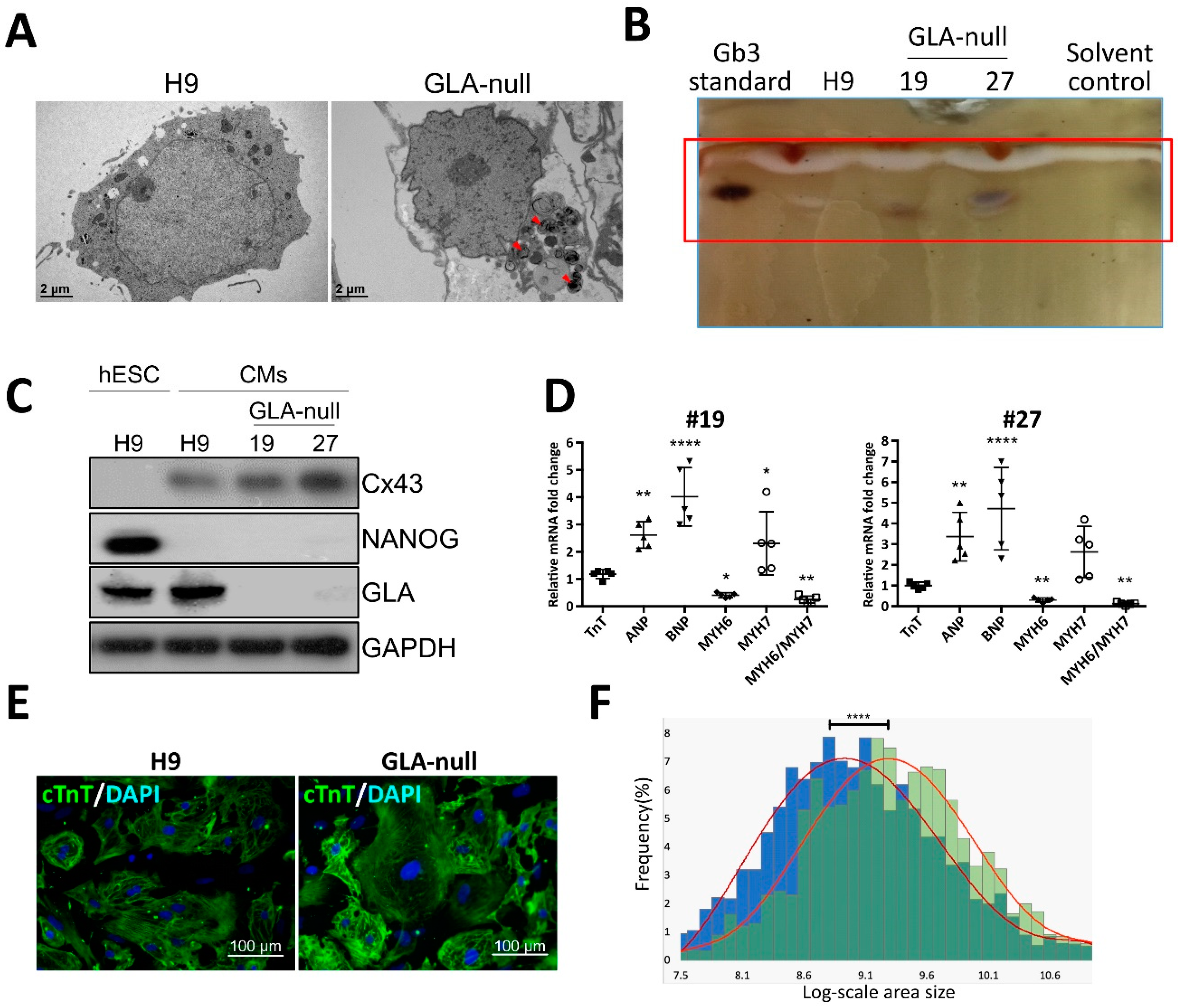
3.2. Characterization of GLA-Null hESC Clones
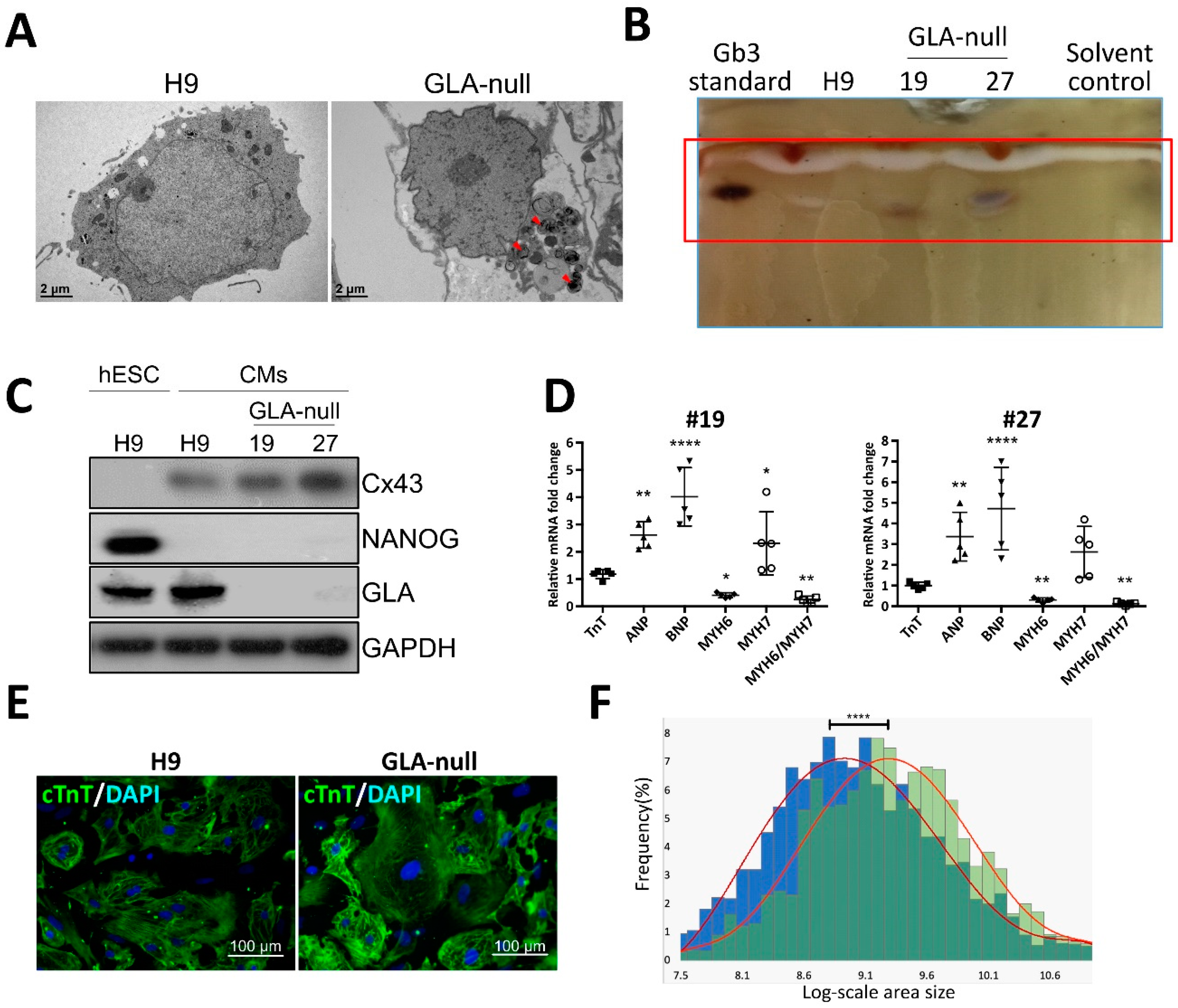
3.3. Recapitulation of FD-Specific Cardiac Abnormalities in GLA-Null Cardiomyocytes
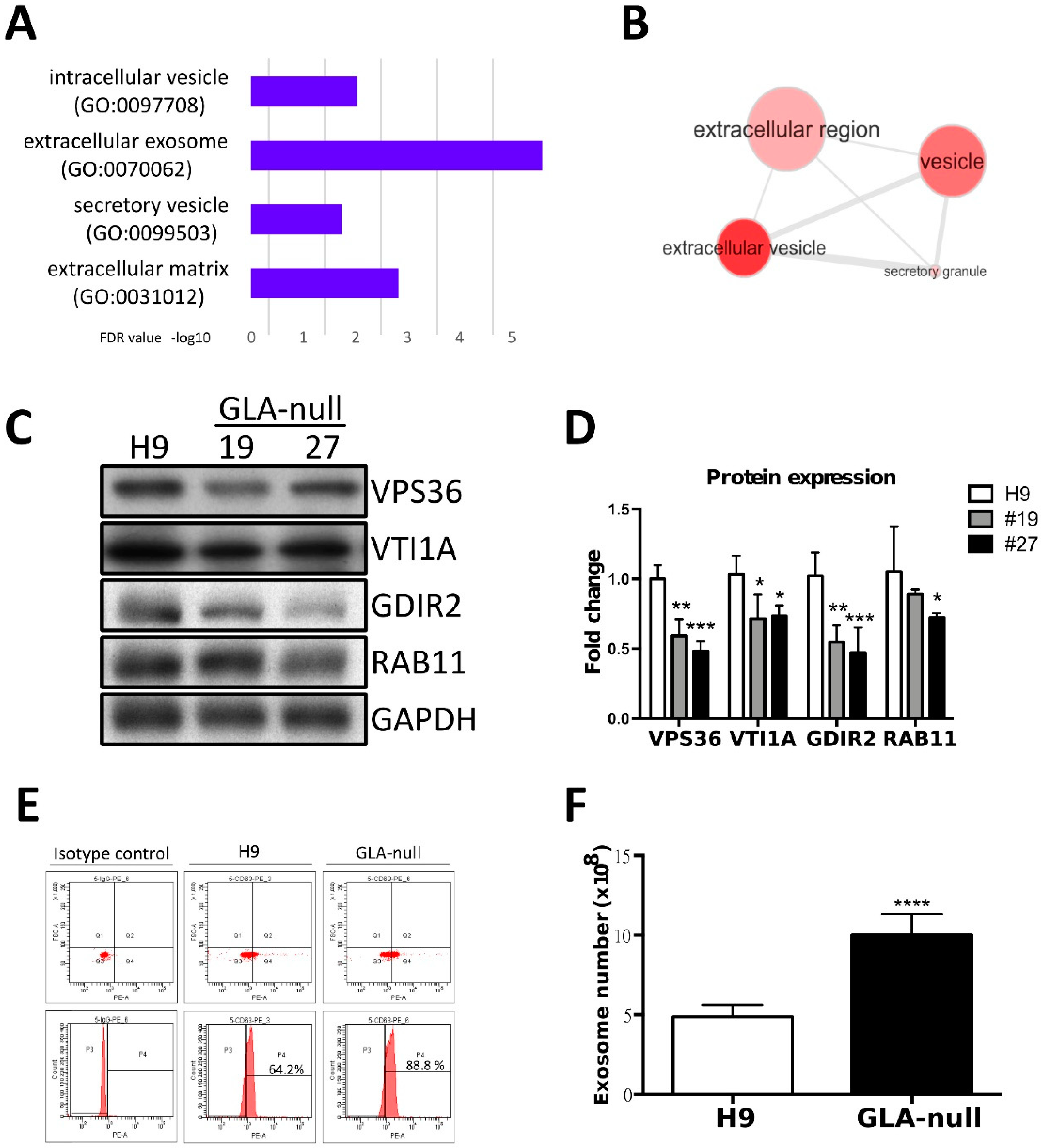
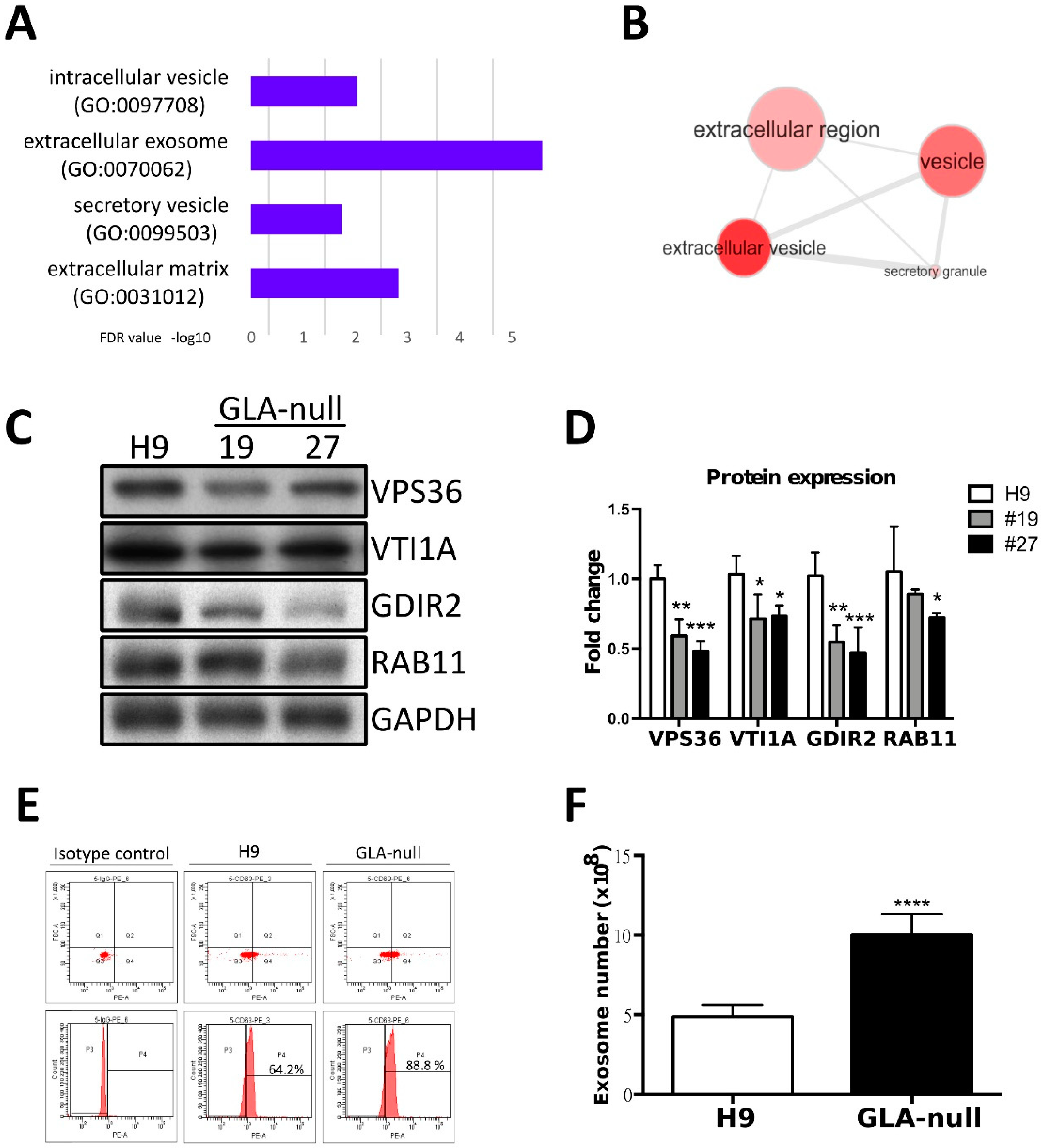
3.4. Proteomic Analysis of GLA-Null CMs
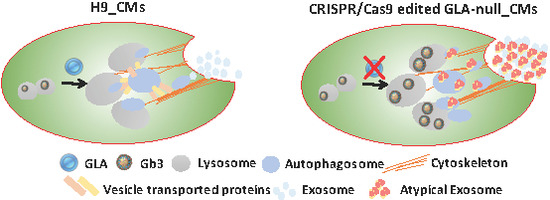
3.5. GLA-Null CMs Secrete More Exosomes Than H9 CMs
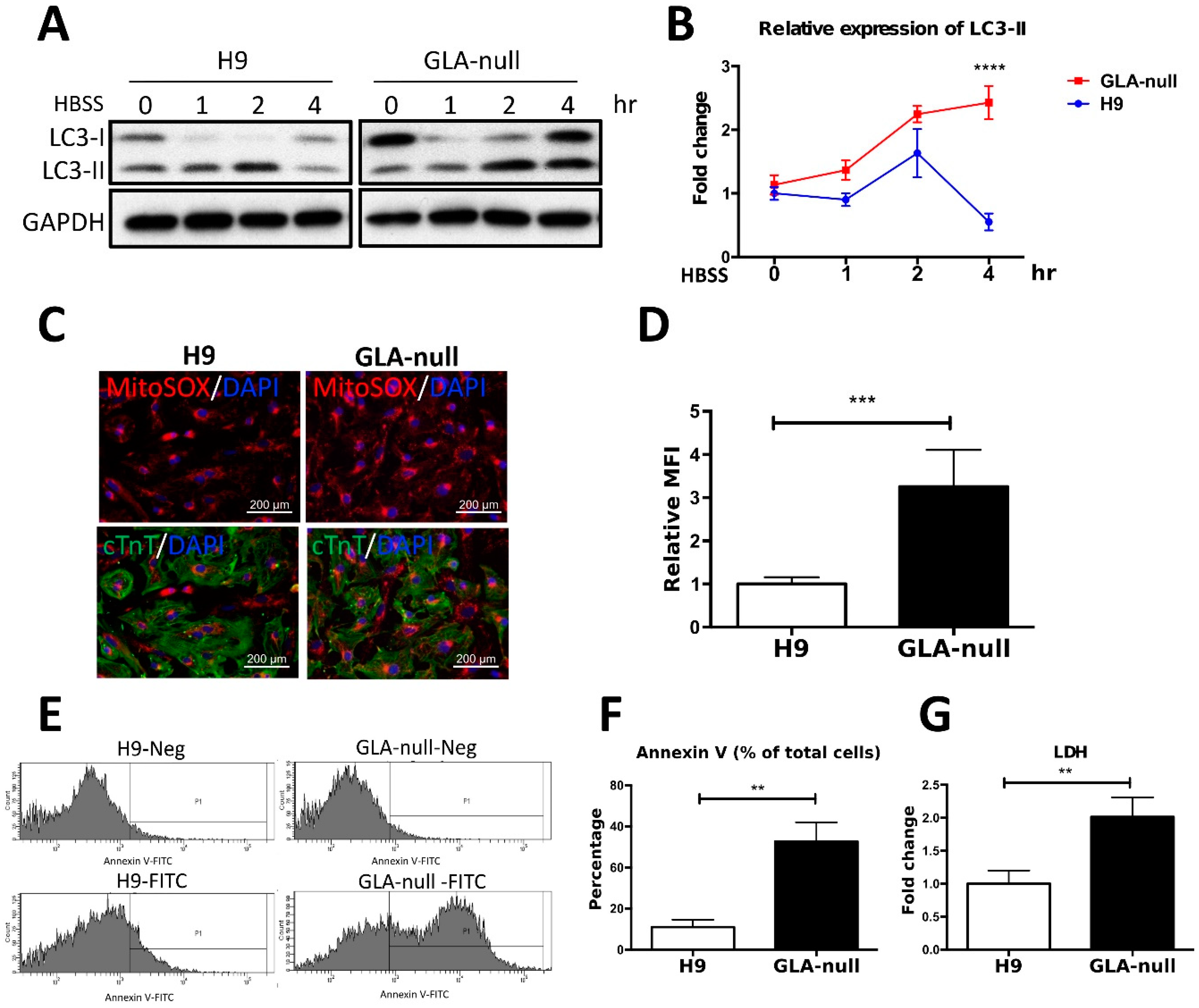
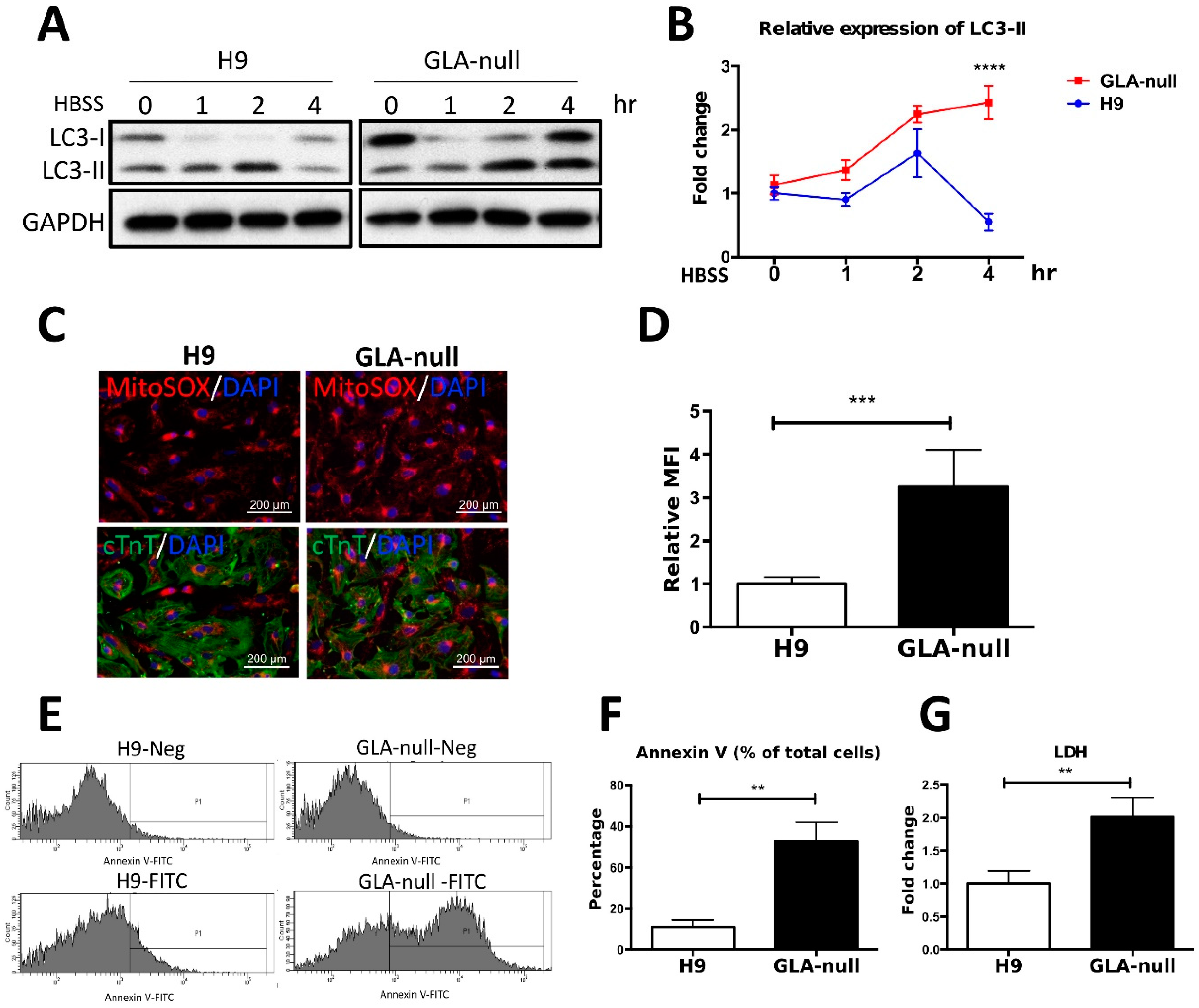
3.6. Vesicle Turnover Impairment Induces Cardiotoxicity in GLA-Null CMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| LSD | lysosomal storage disease |
| FD | Fabry disease |
| Gb3 | globotriaosylceramide |
| α-Gal A | α-galactosidase A |
| ERT | enzyme replacement therapy |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| sgRNA | single-guide RNA |
| Cas9 | CRISPR-associated protein |
| KO | knockout |
| hESCs | human embryonic stem cells |
| CMs | cardiomyocytes |
| MVEs | multivesicular endosomes |
References
- Roos, J.C.; Cox, T.M. Glycogen storage diseases and cardiomyopathy. N. Engl. J. Med. 2005, 352, 2553. [Google Scholar] [PubMed]
- von Scheidt, W.; Eng, C.M.; Fitzmaurice, T.F.; Erdmann, E.; Hubner, G.; Olsen, E.G.; Christomanou, H.; Kandolf, R.; Bishop, D.F.; Desnick, R.J. An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N. Engl. J. Med. 1991, 324, 395–399. [Google Scholar] [CrossRef]
- Brady, R.O.; Tallman, J.F.; Johnson, W.G.; Gal, A.E.; Leahy, W.R.; Quirk, J.M.; Dekaban, A.S. Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry’s disease. N. Engl. J. Med. 1973, 289, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Hendriksz, C.J.; Roberts, M.; Sharma, R. Observational clinical study of 22 adult-onset Pompe disease patients undergoing enzyme replacement therapy over 5years. Mol. Genet. Metab. 2016, 117, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.; Kasper, D.C.; Desnick, R.J. Correlation of Lyso-Gb3 levels in dried blood spots and sera from patients with classic and Later-Onset Fabry disease. Mol. Genet. Metab. 2017, 121, 320–324. [Google Scholar] [CrossRef]
- Roddy, T.P.; Nelson, B.C.; Sung, C.C.; Araghi, S.; Wilkens, D.; Zhang, X.K.; Thomas, J.J.; Richards, S.M. Liquid chromatography-tandem mass spectrometry quantification of globotriaosylceramide in plasma for long-term monitoring of Fabry patients treated with enzyme replacement therapy. Clin. Chem. 2005, 51, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, S.; Togawa, T.; Tsukimura, T.; Kodama, T.; Tanaka, T.; Doi, K.; Noiri, E.; Akai, Y.; Saito, Y.; Yoshino, M.; et al. Mutant alpha-galactosidase A with M296I does not cause elevation of the plasma globotriaosylsphingosine level. Mol. Genet. Metab. 2012, 107, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Chiang, H.C.; Tseng, W.L.; Wu, P.; Chien, C.S.; Leu, H.B.; Yang, Y.P.; Wang, M.L.; Jong, Y.J.; Chen, C.H.; et al. Using CRISPR/Cas9-Mediated GLA Gene Knockout as an In Vitro Drug Screening Model for Fabry Disease. Int. J. Mol. Sci. 2016, 17, 2089. [Google Scholar] [CrossRef]
- Kuramoto, Y.; Naito, A.T.; Tojo, H.; Sakai, T.; Ito, M.; Shibamoto, M.; Nakagawa, A.; Higo, T.; Okada, K.; Yamaguchi, T.; et al. Generation of Fabry cardiomyopathy model for drug screening using induced pluripotent stem cell-derived cardiomyocytes from a female Fabry patient. J. Mol. Cell. Cardiol. 2018, 121, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, S.; Higuchi, T.; Otaka, M.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Okano, H.J.; Nakanishi, M.; Eto, Y. Morphological features of iPS cells generated from Fabry disease skin fibroblasts using Sendai virus vector (SeVdp). Mol. Genet. Metab. 2013, 109, 386–389. [Google Scholar] [CrossRef]
- Kajiwara, M.; Aoi, T.; Okita, K.; Takahashi, R.; Inoue, H.; Takayama, N.; Endo, H.; Eto, K.; Toguchida, J.; Uemoto, S.; et al. Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12538–12543. [Google Scholar] [CrossRef]
- Pan, X.; Ouyang, Y.; Wang, Z.; Ren, H.; Shen, P.; Wang, W.; Xu, Y.; Ni, L.; Yu, X.; Chen, X.; et al. Genotype: A Crucial but Not Unique Factor Affecting the Clinical Phenotypes in Fabry Disease. PLoS ONE 2016, 11, e0161330. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Sillence, D.J. Glucosylceramide modulates endolysosomal pH in Gaucher disease. Mol. Genet. Metab. 2013, 109, 194–200. [Google Scholar] [CrossRef]
- de la Mata, M.; Cotan, D.; Villanueva-Paz, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Suarez-Rivero, J.M.; Tiscornia, G.; Oropesa-Avila, M. Mitochondrial Dysfunction in Lysosomal Storage Disorders. Diseases 2016, 4, 31. [Google Scholar] [CrossRef]
- Miranda, A.M.; Lasiecka, Z.M.; Xu, Y.; Neufeld, J.; Shahriar, S.; Simoes, S.; Chan, R.B.; Oliveira, T.G.; Small, S.A.; Di Paolo, G. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun. 2018, 9, 291. [Google Scholar] [CrossRef]
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.; Xu, R.H.; Xu, C. Efficient differentiation of cardiomyocytes from human pluripotent stem cells with growth factors. Methods Mol. Biol. 2015, 1299, 115–131. [Google Scholar] [PubMed]
- Chien, K.R.; Zhu, H.; Knowlton, K.U.; Miller-Hance, W.; van-Bilsen, M.; O’Brien, T.X.; Evans, S.M. Transcriptional regulation during cardiac growth and development. Annu. Rev. Physiol. 1993, 55, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef]
- Al-Shahrour, F.; Diaz-Uriarte, R.; Dopazo, J. FatiGO: A web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef]
- Fader, C.M.; Colombo, M.I. Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy 2006, 2, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Kuno, A.; Hosoda, R.; Sebori, R.; Hayashi, T.; Sakuragi, H.; Tanabe, M.; Horio, Y. Resveratrol Ameliorates Mitophagy Disturbance and Improves Cardiac Pathophysiology of Dystrophin-deficient mdx Mice. Sci. Rep. 2018, 8, 15555. [Google Scholar] [CrossRef] [PubMed]
- Hawkridge, A.M.; Muddiman, D.C. Mass spectrometry-based biomarker discovery: Toward a global proteome index of individuality. Annu. Rev. Anal. Chem. 2009, 2, 265–277. [Google Scholar] [CrossRef]
- Provencal, P.; Boutin, M.; Dworski, S.; Au, B.; Medin, J.A.; Auray-Blais, C. Relative distribution of Gb3 isoforms/analogs in NOD/SCID/Fabry mice tissues determined by tandem mass spectrometry. Bioanalysis 2016, 8, 1793–1807. [Google Scholar] [CrossRef]
- Kuchar, L.; Faltyskova, H.; Krasny, L.; Dobrovolny, R.; Hulkova, H.; Ledvinova, J.; Volny, M.; Strohalm, M.; Lemr, K.; Kryspinova, L.; et al. Fabry disease: Renal sphingolipid distribution in the alpha-Gal A knockout mouse model by mass spectrometric and immunohistochemical imaging. Anal. Bioanal. Chem. 2015, 407, 2283–2291. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J.A.; Na, H.Y.; Cho, S.E.; Park, S.; Jung, S.C.; Suh, S.H. Globotriaosylceramide induces lysosomal degradation of endothelial KCa3.1 in fabry disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 81–89. [Google Scholar] [CrossRef]
- Escudero, C.A.; Lazo, O.M.; Galleguillos, C.; Parraguez, J.I.; Lopez-Verrilli, M.A.; Cabeza, C.; Leon, L.; Saeed, U.; Retamal, C.; Gonzalez, A.; et al. The p75 neurotrophin receptor evades the endolysosomal route in neuronal cells, favouring multivesicular bodies specialised for exosomal release. J. Cell Sci. 2014, 127, 1966–1979. [Google Scholar] [CrossRef]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in PBMC isolated from patients with Fabry disease: Preliminary findings. Mol. Biosyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef]
- Wei, L.; Imanaka-Yoshida, K.; Wang, L.; Zhan, S.; Schneider, M.D.; DeMayo, F.J.; Schwartz, R.J. Inhibition of Rho family GTPases by Rho GDP dissociation inhibitor disrupts cardiac morphogenesis and inhibits cardiomyocyte proliferation. Development 2002, 129, 1705–1714. [Google Scholar]
- Wei, L.; Taffet, G.E.; Khoury, D.S.; Bo, J.; Li, Y.; Yatani, A.; Delaughter, M.C.; Klevitsky, R.; Hewett, T.E.; Robbins, J.; et al. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. FASEB J. 2004, 18, 857–859. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Kaibuchi, K.; Hori, Y.; Fujioka, H.; Araki, S.; Ueda, T.; Kikuchi, A.; Takai, Y. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 1990, 5, 1321–1328. [Google Scholar]
- Aikawa, R.; Komuro, I.; Nagai, R.; Yazaki, Y. Rho plays an important role in angiotensin II-induced hypertrophic responses in cardiac myocytes. Mol. Cell. Biochem. 2000, 212, 177–182. [Google Scholar] [CrossRef]
- Aikawa, R.; Komuro, I.; Yamazaki, T.; Zou, Y.; Kudoh, S.; Zhu, W.; Kadowaki, T.; Yazaki, Y. Rho family small G proteins play critical roles in mechanical stress-induced hypertrophic responses in cardiac myocytes. Circ. Res. 1999, 84, 458–466. [Google Scholar] [CrossRef]
- Del Re, D.P.; Miyamoto, S.; Brown, J.H. RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J. Biol. Chem. 2007, 282, 8069–8078. [Google Scholar] [CrossRef]
- Sah, V.P.; Minamisawa, S.; Tam, S.P.; Wu, T.H.; Dorn, G.W., 2nd; Ross, J., Jr.; Chien, K.R.; Brown, J.H. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J. Clin. Investig. 1999, 103, 1627–1634. [Google Scholar] [CrossRef]
- Liebau, M.C.; Braun, F.; Hopker, K.; Weitbrecht, C.; Bartels, V.; Muller, R.U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef]
- Chevrier, M.; Brakch, N.; Celine, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerriere, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef]
- Beer, L.; Zimmermann, M.; Mitterbauer, A.; Ellinger, A.; Gruber, F.; Narzt, M.S.; Zellner, M.; Gyongyosi, M.; Madlener, S.; Simader, E.; et al. Analysis of the Secretome of Apoptotic Peripheral Blood Mononuclear Cells: Impact of Released Proteins and Exosomes for Tissue Regeneration. Sci. Rep. 2015, 5, 16662. [Google Scholar] [CrossRef]
- Kanemoto, S.; Nitani, R.; Murakami, T.; Kaneko, M.; Asada, R.; Matsuhisa, K.; Saito, A.; Imaizumi, K. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2016, 480, 166–172. [Google Scholar] [CrossRef]
- Stoorvogel, W. Functional transfer of microRNA by exosomes. Blood 2012, 119, 646–648. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic Stem Cell-Derived Exosomes Promote Endogenous Repair Mechanisms and Enhance Cardiac Function Following Myocardial Infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Li, Y.; Chen, L.; Wang, X.; Guo, W.; Zhang, X.; Qin, G.; He, S.H.; Zimmerman, A.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69. [Google Scholar] [CrossRef]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.-Y.; Chien, C.-S.; Yarmishyn, A.A.; Chou, S.-J.; Yang, Y.-P.; Wang, M.-L.; Wang, C.-Y.; Leu, H.-B.; Yu, W.-C.; Chang, Y.-L.; et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. https://doi.org/10.3390/cells8040327
Song H-Y, Chien C-S, Yarmishyn AA, Chou S-J, Yang Y-P, Wang M-L, Wang C-Y, Leu H-B, Yu W-C, Chang Y-L, et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells. 2019; 8(4):327. https://doi.org/10.3390/cells8040327
Chicago/Turabian StyleSong, Hui-Yung, Chian-Shiu Chien, Aliaksandr A. Yarmishyn, Shih-Jie Chou, Yi-Ping Yang, Mong-Lien Wang, Chien-Ying Wang, Hsin-Bang Leu, Wen-Chung Yu, Yuh-Lih Chang, and et al. 2019. "Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy" Cells 8, no. 4: 327. https://doi.org/10.3390/cells8040327
APA StyleSong, H.-Y., Chien, C.-S., Yarmishyn, A. A., Chou, S.-J., Yang, Y.-P., Wang, M.-L., Wang, C.-Y., Leu, H.-B., Yu, W.-C., Chang, Y.-L., & Chiou, S.-H. (2019). Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells, 8(4), 327. https://doi.org/10.3390/cells8040327