Strategies to Circumvent Host Innate Immune Response by Hepatitis C Virus



Abstract
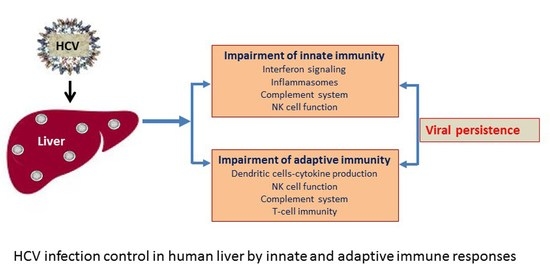
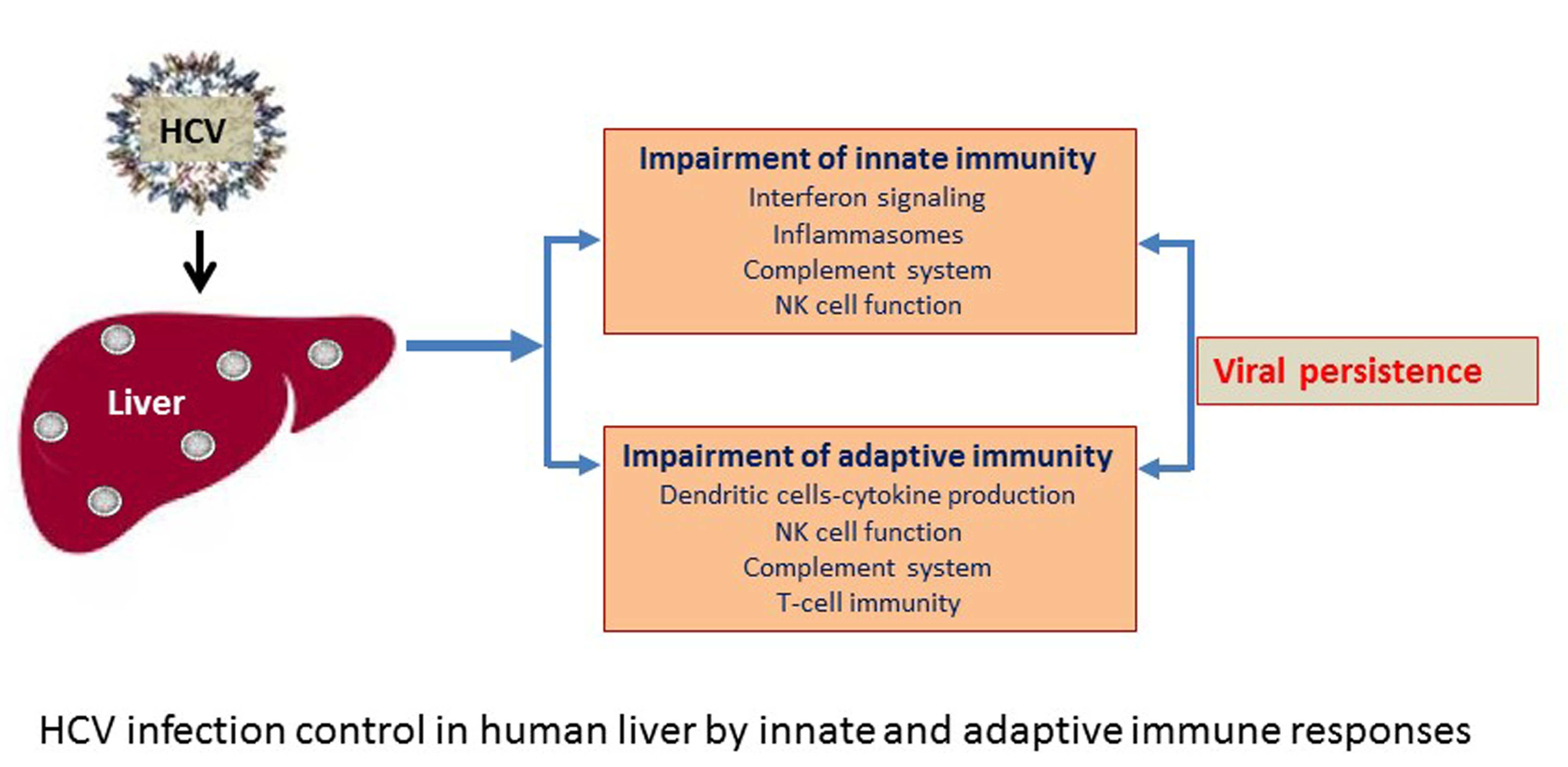{kind=link}
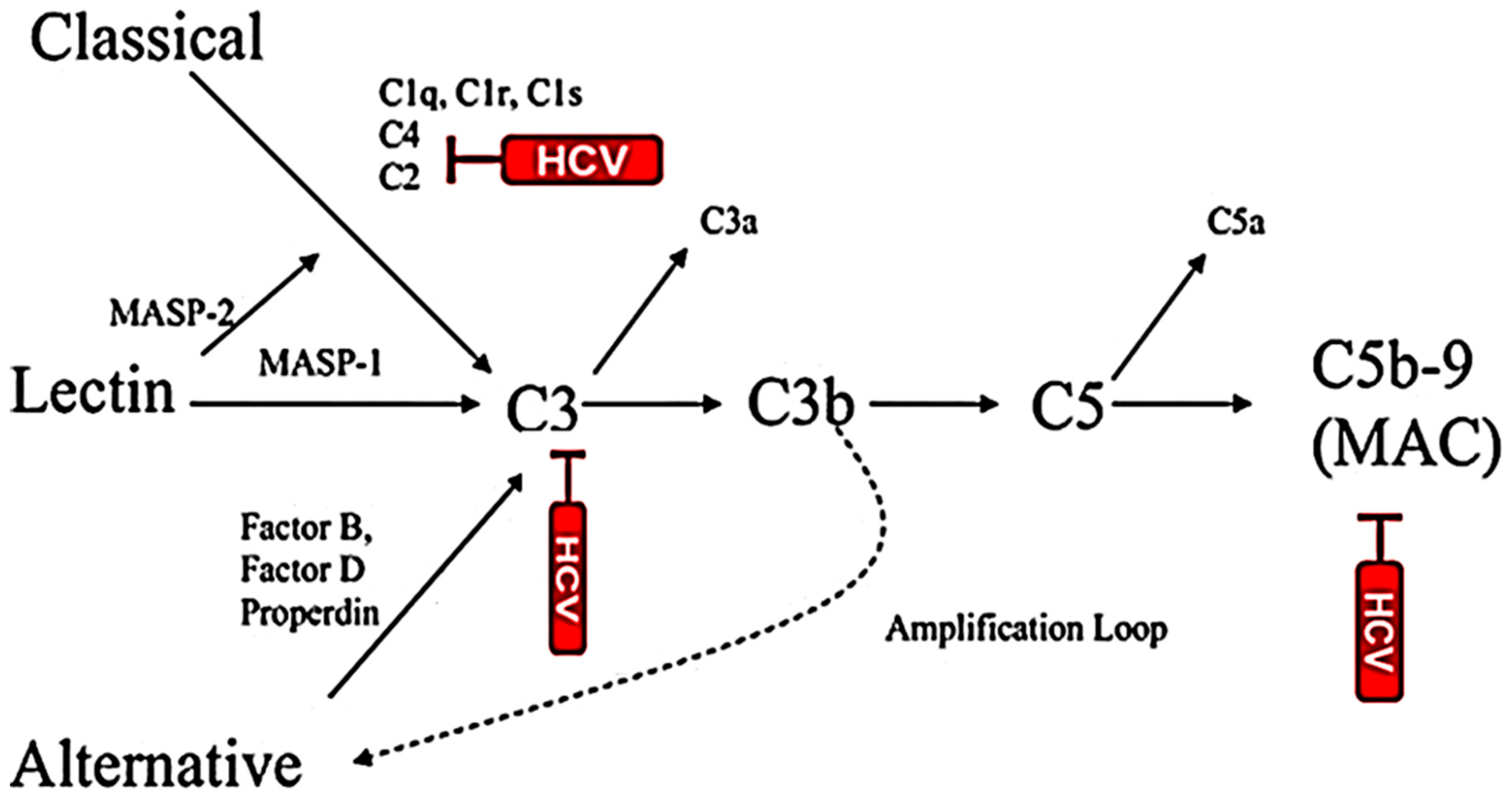{kind=link}
{kind=link}
1. Introduction
2. Endogenous Interferon Production
3. Induction of Proinflammatory Responses
4. Modulation of Natural Killer Cell Response
5. Activation of Complement System
6. Innate Immunity Helping Adaptive Immune Response
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cell cytotoxicity |
| APC | antigen-presenting cell |
| CDC | complement-dependent lysis |
| DAA | direct-acting antiviral |
| DAMP | danger-associated molecular patterns |
| DC | dendritic cells |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| IFN | interferon |
| ISG | interferon-stimulated genes |
| ISGF3 | interferon-stimulated gene factor 3 |
| ISRE | interferon-stimulated response element |
| MAC | membrane attack complex |
| MAIT | mucosal-associated invariant T |
| MASP | mannose-associated serine protease |
| mDCs | monocyte-derived dendritic cells |
| NK | natural killer |
| PAMP | pathogen-associated molecular patterns |
| PD-1 | programmed cell death protein-1 |
| PKR | protein kinase R |
| PRRs | pattern-recognition receptors |
| RIG-I | retinoic acid inducible gene-I |
| SVR | sustained virological response |
| TLR | toll-like receptor |
| UTR | untranslated region |
References
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Pfaender, S.; Brown, R.J.; Pietschmann, T.; Steinmann, E. Natural reservoirs for homologs of hepatitis C virus. Emerg. Microbes Infect. 2014, 3, e21. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P. Classification and Genomic Diversity of Enterically Transmitted Hepatitis Viruses. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef]
- Kanwal, F.; Hoang, T.; Kramer, J.R.; Asch, S.M.; Goetz, M.B.; Zeringue, A.; Richardson, P.; El-Serag, H.B. Increasing prevalence of HCC and cirrhosis in patients with chronic hepatitis C virus infection. Gastroenterology 2011, 140, 1182–1188.e1. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M. Viral hepatitis and liver cancer: The case of hepatitis C. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef]
- Pinter, M.; Sieghart, W. Long-term remission in advanced stage hepatocellular carcinoma? A chance for cure? MEMO 2018, 11, 185–192. [Google Scholar] [CrossRef]
- Sherman, M. Hepatocellular carcinoma: Epidemiology, surveillance, and diagnosis. Semin. Liver Dis. 2010, 30, 3–16. [Google Scholar] [CrossRef]
- Szabo, G.; Saha, B.; Bukong, T.N. Alcohol and HCV: Implications for liver cancer. Adv. Exp. Med. Biol. 2015, 815, 197–216. [Google Scholar]
- Palumbo, E. Pegylated interferon and ribavirin treatment for hepatitis C virus infection. Ther. Adv. Chronic Dis. 2011, 2, 39–45. [Google Scholar] [CrossRef]
- AASLD/IDSA HCV Guidance Panel. Hepatitis C guidance: AASLD-IDSA recommendations for testing, managing, and treating adults infected with hepatitis C virus. Hepatology 2015, 62, 932–954. [Google Scholar] [CrossRef] [PubMed]
- Au, J.S.; Pockros, P.J. Novel therapeutic approaches for hepatitis C. Clin. Pharmacol. Ther. 2014, 95, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Ray, R. Hepatitis C virus manipulates humans as its favorite host for long term relationship. Hepatology 2019, 69, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.C.; Ray, R.B.; Ray, R. Hepatitis C virus infection: Establishment of chronicity and liver disease progression. EXCLI J. 2014, 13, 977–996. [Google Scholar] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Heim, M.H. Innate immunity and HCV. J. Hepatol. 2013, 58, 564–574. [Google Scholar] [CrossRef]
- Schnell, G.; Loo, Y.M.; Marcotrigiano, J.; Gale, M., Jr. Uridine composition of the poly-U/UC tract of HCV RNA defines non-self-recognition by RIG-I. PLoS Pathog. 2012, 8, e1002839. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.R. Emerging concepts in immunity to hepatitis C virus infection. J. Clin. Investig. 2013, 123, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.J., Jr.; Korth, M.J.; Katze, M.G. Repression of the PKR protein kinase by the hepatitis C virus NS5A protein: A potential mechanism of interferon resistance. Clin. Diagn. Virol. 1998, 10, 157–162. [Google Scholar] [CrossRef]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ludwig, S.; Ehrhardt, C.; Albrecht, U.; Erhardt, A.; Schaper, F.; Heinrich, P.C.; Haussinger, D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003, 17, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.H.; Lan, K.L.; Lee, W.P.; Sheu, M.L.; Chen, M.Y.; Lee, Y.L.; Yen, S.H.; Chang, F.Y.; Lee, S.D. HCV NS5A inhibits interferon-alpha signaling through suppression of STAT1 phosphorylation in hepatocyte-derived cell lines. J. Hepatol. 2007, 46, 759–767. [Google Scholar] [CrossRef]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef] [PubMed]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Dash, S.; Kanda, T.; Ray, R.; Ray, R.B. Hepatitis C virus infection impairs IRF-7 translocation and Alpha interferon synthesis in immortalized human hepatocytes. J. Virol. 2010, 84, 10991–10998. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R., Jr.; Ikeda, M.; Lemon, S.M.; Gale, M., Jr. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Gale, M., Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 2013, 19, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakami, T.; Yamamoto, C.; Ogura, Y.; Izumi, N.; Marumo, F.; Sato, C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 1996, 334, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Chang, S.; Kodys, K.; Mandrekar, P.; Bakis, G.; Cormier, M.; Szabo, G. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J. Immunol. 2006, 177, 6758–6768. [Google Scholar] [CrossRef]
- Wohnsland, A.; Hofmann, W.P.; Sarrazin, C. Viral determinants of resistance to treatment in patients with hepatitis C. Clin. Microbiol. Rev. 2007, 20, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Bochud, P.Y.; George, J. Host—hepatitis C viral interactions: The role of genetics. J. Hepatol. 2016, 65, S22–S32. [Google Scholar] [CrossRef]
- Matsuura, K.; Tanaka, Y. Host genetic variants influencing the clinical course of hepatitis C virus infection. J. Med. Virol. 2016, 88, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Hu, T.H.; Lu, S.N.; Wang, J.H.; Hung, C.H.; Chen, C.H.; Yen, Y.H. Peripheral blood toll-like receptor 4 correlates with rapid virological response to pegylated-interferon and ribavirin therapy in hepatitis C genotype 1 patients. BMC Gastroenterol. 2016, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Wang, N.; Liang, Y.; Devaraj, S.; Wang, J.; Lemon, S.M.; Li, K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J. Virol. 2009, 83, 9824–9834. [Google Scholar] [CrossRef]
- Takahasi, K.; Horiuchi, M.; Fujii, K.; Nakamura, S.; Noda, N.N.; Yoneyama, M.; Fujita, T.; Inagaki, F. Ser386 phosphorylation of transcription factor IRF-3 induces dimerization and association with CBP/p300 without overall conformational change. Genes Cells 2010, 15, 901–910. [Google Scholar] [CrossRef]
- Abe, T.; Kaname, Y.; Hamamoto, I.; Tsuda, Y.; Wen, X.; Taguwa, S.; Moriishi, K.; Takeuchi, O.; Kawai, T.; Kanto, T.; et al. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 2007, 81, 8953–8966. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Eng, H.L.; Lin, K.H.; Chang, C.H.; Hsieh, C.A.; Lin, Y.L.; Lin, T.M. TLR7 and TLR8 gene variations and susceptibility to hepatitis C virus infection. PLoS ONE 2011, 6, e26235. [Google Scholar] [CrossRef]
- Fernandez-Rodriguez, A.; Berenguer, J.; Jimenez-Sousa, M.A.; Garcia-Alvarez, M.; Aldamiz-Echevarria, T.; Pineda-Tenor, D.; Diez, C.; de la Barrera, J.; Bellon, J.M.; Briz, V.; et al. Toll-like receptor 8 (TLR8) polymorphisms are associated with non-progression of chronic hepatitis C in HIV/HCV coinfected patients. Infect. Genet. Evol. 2015, 36, 339–344. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Chattergoon, M.A.; Levine, J.S.; Latanich, R.; Osburn, W.O.; Thomas, D.L.; Cox, A.L. High plasma interleukin-18 levels mark the acute phase of hepatitis C virus infection. J. Infect. Dis. 2011, 204, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Mukherjee, A.; Ray, R.; Ray, R.B. Hepatitis C virus induces interleukin-1beta (IL-1beta)/IL-18 in circulatory and resident liver macrophages. J. Virol. 2013, 87, 12284–12290. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Chattergoon, M.A.; Latanich, R.; Quinn, J.; Winter, M.E.; Buckheit, R.W.; 3rd Blankson, J.N.; Pardoll, D.; Cox, A.L. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog. 2014, 10, e1004082. [Google Scholar] [CrossRef]
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef]
- Decker, K. Biologically active products of stimulated liver macrophages (Kupffer cells). Eur. J. Biochem. 1990, 192, 245–261. [Google Scholar] [CrossRef]
- Tacke, F.; Luedde, T.; Trautwein, C. Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol. 2009, 36, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Tian, Y.; Chan, S.T.; Kim, J.Y.; Cho, C.; Ou, J.H. TNF-alpha Induced by Hepatitis C Virus via TLR7 and TLR8 in Hepatocytes Supports Interferon Signaling via an Autocrine Mechanism. PLoS Pathog. 2015, 11, e1004937. [Google Scholar] [CrossRef]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Yin, M.; Fleckenstein, J.; Yang, S.Q.; Lin, H.Z.; Brenner, D.A.; Westwick, J.; Bagby, G.; Nelson, S. Tumor necrosis factor-alpha induces c-jun during the regenerative response to liver injury. Am. J. Physiol. 1994, 267, G552–G561. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, L.; Peng, Y.; Shi, X.; Chen, J.; Zhong, J.; Chen, X.; Cheng, G.; Deng, H. The hepatitis C virus protein NS3 suppresses TNF-alpha-stimulated activation of NF-kappaB by targeting, LUBAC. Sci. Signal. 2015, 8, ra118. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, M. Autocrine IL-6 signaling: A key event in tumorigenesis? Cancer Cell 2008, 13, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Di Fazio, I.; Romeo, M.A.; Restuccia, S.; Laurino, A.; Trovato, B.A. Elevation of interleukin 6 levels in patients with chronic hepatitis due to hepatitis C virus. J. Gastroenterol. 1997, 32, 211–215. [Google Scholar] [CrossRef]
- Koike, K.; Moriya, K. Metabolic aspects of hepatitis C viral infection: Steatohepatitis resembling but distinct from NASH. J. Gastroenterol. 2005, 40, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.C.; Meyer, K.; Peng, G.; Chatterjee, S.; Hoft, D.F.; Ray, R. Hepatitis C virus E2 envelope glycoprotein induces an immunoregulatory phenotype in macrophages. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Nellore, A.; Fishman, J.A. NK cells, innate immunity and hepatitis C infection after liver transplantation. Clin. Infect. Dis. 2011, 52, 369–377. [Google Scholar] [CrossRef]
- Krueger, P.D.; Lassen, M.G.; Qiao, H.; Hahn, Y.S. Regulation of NK cell repertoire and function in the liver. Crit. Rev. Immunol. 2011, 31, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, G.; Martin, M.P.; Gao, X.; Carrington, M.; Rehermann, B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J. Clin. Investig. 2008, 118, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Cariani, E.; Pilli, M.; Zerbini, A.; Rota, C.; Olivani, A.; Zanelli, P.; Zanetti, A.; Trenti, T.; Ferrari, C.; Missale, G. HLA and killer immunoglobulin-like receptor genes as outcome predictors of hepatitis C virus-related hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5465–5473. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef]
- Velazquez, V.M.; Hon, H.; Ibegbu, C.; Knechtle, S.J.; Kirk, A.D.; Grakoui, A. Hepatic enrichment and activation of myeloid dendritic cells during chronic hepatitis C virus infection. Hepatology 2012, 56, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef]
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Crotta, S.; Stilla, A.; Wack, A.; D’Andrea, A.; Nuti, S.; D’Oro, U.; Mosca, M.; Filliponi, F.; Brunetto, R.M.; Bonino, F.; et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 2002, 195, 35–41. [Google Scholar] [CrossRef]
- Tseng, C.T.; Klimpel, G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 2002, 195, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Groh, V.; Spies, T.; Suzuki, T.; Miyagi, T.; Hayashi, N. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J. Immunol. 2003, 171, 5423–5429. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Rosen, H.R. Natural killer cells: Primary target for hepatitis C virus immune evasion strategies? Liver Transpl. 2006, 12, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Cheent, K.; Khakoo, S.I. Natural killer cells and hepatitis C: Action and reaction. Gut 2011, 60, 268–278. [Google Scholar] [CrossRef]
- Yoon, J.C.; Lim, J.B.; Park, J.H.; Lee, J.M. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J. Virol. 2011, 85, 12557–12569. [Google Scholar] [CrossRef]
- Kim, H.; Bose, S.K.; Meyer, K.; Ray, R. Hepatitis C virus impairs natural killer cell-mediated augmentation of complement synthesis. J. Virol. 2014, 88, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; He, X.; Ma, H.; Hou, N.; Wei, C.; Song, T.; Zhang, Y.; Sun, L.; Ma, Q.; Zhong, H. Hepatitis C virus infection downregulates the ligands of the activating receptor, NKG2D. Cell. Mol. Immunol. 2008, 5, 475–478. [Google Scholar] [CrossRef]
- Cosgrove, C.; Berger, C.T.; Kroy, D.C.; Cheney, P.C.; Ghebremichael, M.; Aneja, J.; Tomlinson, M.; Kim, A.Y.; Lauer, G.M.; Alter, G. Chronic HCV infection affects the NK cell phenotype in the blood more than in the liver. PLoS ONE 2014, 9, e105950. [Google Scholar] [CrossRef] [PubMed]
- Sene, D.; Levasseur, F.; Abel, M.; Lambert, M.; Camous, X.; Hernandez, C.; Pene, V.; Rosenberg, A.R.; Jouvin-Marche, E.; Marche, P.N.; et al. Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. 2010, 6, e1001184. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Koo, G.C.; Salmon, J.E. Complement regulates conventional DC-mediated NK-cell activation by inducing TGF-beta1 in Gr-1+ myeloid cells. Eur. J. Immunol. 2012, 42, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Gao, B. The complement system in liver diseases. Cell. Mol. Immunol. 2006, 3, 333–340. [Google Scholar]
- Ghebrehiwet, B.; Lim, B.L.; Kumar, R.; Feng, X.; Peerschke, E.I. gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol. Rev. 2001, 180, 65–77. [Google Scholar] [CrossRef]
- Sansonno, D.; Tucci, F.A.; Ghebrehiwet, B.; Lauletta, G.; Peerschke, E.I.; Conteduca, V.; Russi, S.; Gatti, P.; Sansonno, L.; Dammacco, F. Role of the receptor for the globular domain of C1q protein in the pathogenesis of hepatitis C virus-related cryoglobulin vascular damage. J. Immunol. 2009, 183, 6013–6020. [Google Scholar] [CrossRef]
- Cummings, K.L.; Rosen, H.R.; Hahn, Y.S. Frequency of gC1qR+CD4+ T cells increases during acute hepatitis C virus infection and remains elevated in patients with chronic infection. Clin. Immunol. 2009, 132, 401–411. [Google Scholar] [CrossRef]
- Ali, O.S.; Abo-Shadi, M.A.; Hammad, L.N. The biological significance of serum complements C3 and C4 in HCV-related chronic liver diseases and hepatocellular carcinoma. Egypt. J. Immunol. 2005, 12, 91–99. [Google Scholar]
- Dumestre-Perard, C.; Ponard, D.; Drouet, C.; Leroy, V.; Zarski, J.P.; Dutertre, N.; Colomb, M.G. Complement C4 monitoring in the follow-up of chronic hepatitis C treatment. Clin. Exp. Immunol. 2002, 127, 131–136. [Google Scholar] [CrossRef]
- Mazumdar, B.; Kim, H.; Meyer, K.; Bose, S.K.; Di Bisceglie, A.M.; Ray, R.B.; Ray, R. Hepatitis C virus proteins inhibit C3 complement production. J. Virol. 2012, 86, 2221–2228. [Google Scholar] [CrossRef]
- Mazumdar, B.; Kim, H.; Meyer, K.; Bose, S.K.; Di Bisceglie, A.M.; Ray, R.B.; Diamond, M.S.; Atkinson, J.P.; Ray, R. Hepatitis C virus infection upregulates CD55 expression on the hepatocyte surface and promotes association with virus particles. J. Virol. 2013, 87, 7902–7910. [Google Scholar] [CrossRef]
- Kim, H.; Meyer, K.; Di Bisceglie, A.M.; Ray, R. Inhibition of c3 convertase activity by hepatitis C virus as an additional lesion in the regulation of complement components. PLoS ONE 2014, 9, e101422. [Google Scholar] [CrossRef]
- Brockman, M.A.; Knipe, D.M. Herpes simplex virus as a tool to define the role of complement in the immune response to peripheral infection. Vaccine 2008, 26 (Suppl. S8), I94–I99. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Li, K.; Anderson, K.; Farrar, C.A.; Lu, B.; Smith, R.A.; Sacks, S.H.; Zhou, W. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood 2008, 111, 2452–2461. [Google Scholar] [CrossRef]
- Li, K.; Okemefuna, A.I.; Gor, J.; Hannan, J.P.; Asokan, R.; Holers, V.M.; Perkins, S.J. Solution structure of the complex formed between human complement C3d and full-length complement receptor type 2. J. Mol. Biol. 2008, 384, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C. Complement and humoral immunity. Vaccine 2008, 26 (Suppl. S8), I28–I33. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.C.; Kim, H.; Meyer, K.; Di Bisceglie, A.M.; Ray, R. Distinct CD55 Isoform Synthesis and Inhibition of Complement-Dependent Cytolysis by Hepatitis C Virus. J. Immunol. 2016, 197, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Mikesch, J.H.; Buerger, H.; Simon, R.; Brandt, B. Decay-accelerating factor (CD55): A versatile acting molecule in human malignancies. Biochim. Biophys. Acta 2006, 1766, 42–52. [Google Scholar] [CrossRef]
- Ochoa, M.C.; Minute, L.; Rodriguez, I.; Garasa, S.; Perez-Ruiz, E.; Inoges, S.; Melero, I.; Berraondo, P. Antibody-dependent cell cytotoxicity: Immunotherapy strategies enhancing effector NK cells. Immunol. Cell. Biol. 2017, 95, 347–355. [Google Scholar] [CrossRef]
- Sarobe, P.; Lasarte, J.J.; Zabaleta, A.; Arribillaga, L.; Arina, A.; Melero, I.; Borras-Cuesta, F.; Prieto, J. Hepatitis C virus structural proteins impair dendritic cell maturation and inhibit in vivo induction of cellular immune responses. J. Virol. 2003, 77, 10862–10871. [Google Scholar] [CrossRef] [PubMed]
- Kanto, T.; Hayashi, N.; Takehara, T.; Tatsumi, T.; Kuzushita, N.; Ito, A.; Sasaki, Y.; Kasahara, A.; Hori, M. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J. Immunol. 1999, 162, 5584–5591. [Google Scholar]
- Zimmermann, M.; Flechsig, C.; La Monica, N.; Tripodi, M.; Adler, G.; Dikopoulos, N. Hepatitis C virus core protein impairs in vitro priming of specific T cell responses by dendritic cells and hepatocytes. J. Hepatol. 2008, 48, 51–60. [Google Scholar] [CrossRef]
- Salou, M.; Franciszkiewicz, K.; Lantz, O. MAIT cells in infectious diseases. Curr. Opin. Immunol. 2017, 48, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Barathan, M.; Mohamed, R.; Vadivelu, J.; Chang, L.Y.; Saeidi, A.; Yong, Y.K.; Ravishankar Ram, M.; Gopal, K.; Velu, V.; Larsson, M.; et al. Peripheral loss of CD8(+) CD161(++) TCRValpha7.2(+) mucosal-associated invariant T cells in chronic hepatitis C virus-infected patients. Eur. J. Clin. Investig. 2016, 46, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Cruise, M.W.; Lassen, M.G.; Hahn, Y.S. Blockade of PD-1/B7-H1 interaction restores effector CD8+ T cell responses in a hepatitis C virus core murine model. J. Immunol. 2008, 180, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Palmer, B.; Klarquist, J.; Mengshol, J.A.; Castelblanco, N.; Rosen, H.R. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J. Virol. 2007, 81, 9249–9258. [Google Scholar] [CrossRef] [PubMed]
- Radziewicz, H.; Ibegbu, C.C.; Fernandez, M.L.; Workowski, K.A.; Obideen, K.; Wehbi, M.; Hanson, H.L.; Steinberg, J.P.; Masopust, D.; Wherry, E.J.; et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 2007, 81, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Raziorrouh, B.; Ulsenheimer, A.; Schraut, W.; Heeg, M.; Kurktschiev, P.; Zachoval, R.; Jung, M.C.; Thimme, R.; Neumann-Haefelin, C.; Horster, S.; et al. Inhibitory molecules that regulate expansion and restoration of HCV-specific CD4+ T cells in patients with chronic infection. Gastroenterology 2011, 141, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Perry, C.J.; Tsui, Y.C.; Staron, M.M.; Parish, I.A.; Dominguez, C.X.; Rosenberg, D.W.; Kaech, S.M. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 2015, 21, 327–334. [Google Scholar] [CrossRef]
- Kared, H.; Fabre, T.; Bedard, N.; Bruneau, J.; Shoukry, N.H. Galectin-9 and IL-21 mediate cross-regulation between Th17 and Treg cells during acute hepatitis C. PLoS Pathog. 2013, 9, e1003422. [Google Scholar] [CrossRef]
- Ren, J.P.; Zhao, J.; Dai, J.; Griffin, J.W.; Wang, L.; Wu, X.Y.; Morrison, Z.D.; Li, G.Y.; El Gazzar, M.; Ning, S.B.; et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology 2016, 148, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Budhu, A.; Chen, S.; Zhou, X.; Popescu, N.C.; Valerie, K.; Wang, X.W. Effect of hepatitis C virus core protein on the molecular profiling of human B lymphocytes. Mol. Med. 2006, 12, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.Q.; Prayther, D.; Trabue, C.; Dong, Z.P.; Moorman, J. Differential regulation of SOCS-1 signalling in B and T lymphocytes by hepatitis C virus core protein. Immunology 2008, 125, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Ceccherini-Silberstein, F.; Cento, V.; Di Maio, V.C.; Perno, C.F.; Craxi, A. Viral resistance in HCV infection. Curr. Opin. Virol. 2018, 32, 115–127. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patra, T.; Ray, R.B.; Ray, R. Strategies to Circumvent Host Innate Immune Response by Hepatitis C Virus. Cells 2019, 8, 274. https://doi.org/10.3390/cells8030274
Patra T, Ray RB, Ray R. Strategies to Circumvent Host Innate Immune Response by Hepatitis C Virus. Cells. 2019; 8(3):274. https://doi.org/10.3390/cells8030274
Chicago/Turabian StylePatra, Tapas, Ratna B. Ray, and Ranjit Ray. 2019. "Strategies to Circumvent Host Innate Immune Response by Hepatitis C Virus" Cells 8, no. 3: 274. https://doi.org/10.3390/cells8030274
APA StylePatra, T., Ray, R. B., & Ray, R. (2019). Strategies to Circumvent Host Innate Immune Response by Hepatitis C Virus. Cells, 8(3), 274. https://doi.org/10.3390/cells8030274