Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disorder with a wide range of clinical symptoms. Enormous progress has been made in the immunological and genetic understanding of SLE. However, the biology of disease heterogeneity in SLE has remained largely unexplored. Human immune profiling studies, helped by recent technological advances especially in single-cell and “omics” analyses, are now shedding light on the cellular and molecular basis of clinical symptoms and disease flares in individual patients. Peripheral blood immunophenotyping analysis with flow cytometry or mass cytometry are identifying responsible cell subsets and markers characteristic of disease heterogeneity. Transcriptome analysis is discovering molecular networks responsible for disease activity, disease subtype and future relapse. In this review, we summarize recent advances in the immune profiling analysis of SLE patients and discuss how they will be used for future precision medicine.
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by a broad spectrum of clinical symptoms and arrays of autoantibodies, such as antinuclear antibodies (ANA) and anti-double strand DNA (dsDNA) antibodies. Both genetic and environmental factors contribute to the onset of SLE. Concordance rates of 20 to 30% in monozygotic twins imply the importance of environmental factors in genetically predisposed individuals [1]. SLE primarily affects women of childbearing age. The female-to-male ratio of about 9:1 suggest a role of female sex hormones [2]. Exposure to ultraviolet light exacerbates skin disease in SLE and can trigger disease onset by induction of apoptosis in the skin [3]. Other environmental factors including cigarette smoking, chemicals, drugs, viral infections and gut microbiota [4] can also act as environmental triggers for SLE.
In the pathophysiology of SLE, there is an imbalance between cellular apoptosis and disposal of apoptotic material [5]. During the course of apoptosis, nuclear antigens and nucleic acids become accessible to the immune system. Persistent apoptotic debris can stimulate an inflammatory response through the activation of nucleic acid recognition receptors, such as members of the Toll-like receptor (TLR) family. Nucleic acid recognition receptors are strongly associated with type I interferon (IFN) production. Type I IFNs and other cytokines promote B-cell differentiation and loss of tolerance. B cells respond to nucleic acids or nuclear antigens and produce autoantibodies against them. Tolerance checkpoints can be bypassed when B cells are activated by TLR9 [6] or BLyS/ BAFF, a cytokine promoting B-cell survival and development [7,8].
In the 1950s, corticosteroids were introduced for treatment of SLE, and the 5-year survival of SLE in developed countries gradually increased to >95%, plateauing in the 1990s [9]. Major causes of death in SLE patients shifted from lupus activity to infection or cardiovascular events that can be related to treatment [10]. In most patients, active disease is controlled by corticosteroid treatment combined with immunosuppressant drugs such as cyclophosphamide introduced in the 1970s, with mycophenolate mofetil becoming available in 2000s. Treatment guidelines or recommendations are available from academic societies around the world [11,12,13,14]. Treat-to-target has been proposed, with a target of clinical remission or low disease activity that predicts favourable long-term outcomes [15]. However, controlling disease activity life-long without or with use of a minimal amount of immunosuppressants is a clinical challenge. The goal of prolonged remission or low disease activity is achieved only in a small proportion of patients [16,17]. Unpredictable and frequent relapses of active SLE, flares, and difficulties in maintaining a state of remission have persuades patients and physicians to continue immunosuppressive treatment (with accompanying side effects) to control disease activity.
In addition to disease activity, SLE patients are stratified and treated according to organ involvement, such as the kidney. The pattern of autoantibodies is also correlated to some SLE phenotypes [18]. For example, anti-dsDNA antibodies are associated with lupus nephritis [19,20]. However, these clinical stratifications do not necessarily reflect the biological heterogeneity of SLE.
Belimumab, a monoclonal antibody against BLyS/ BAFF was approved as the first targeted biological therapy for SLE in 2011 [21,22,23]. At least 74 targeted treatments utilizing IFN, cytokines, chemokines, and B-cells are under clinical trial for SLE [24]. With increasing treatment options, identification of biomarkers that stratify SLE patients to adequate treatment is an urgent need.
A biomarker is a characteristic that can be objectively measured as an indicator of a normal or pathological biological processes, or as an indicator of response to therapy [25]. Immune profiling, or immune cell profiling, realized by technological advances in multi-dimensional analysis of immune cells, will enable deeper understanding of disease pathogenesis and heterogeneity and the discovery of novel biomarkers [26]. Prognostic biomarkers to forecast the natural course of the patient or predictive biomarkers to forecast the response to therapy are necessary for precisely tailored treatment, precision medicine, of SLE (Figure 1) [27].
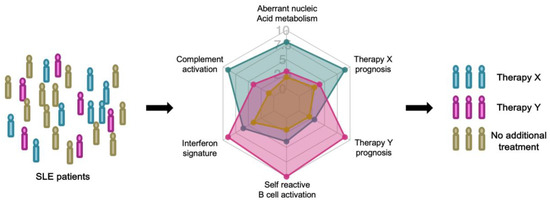
Figure 1.
Stratified treatment of systemic lupus erythematosus (SLE) patients based on prognostic and predictive biomarkers.
In the coming era of precision medicine, SLE patients will be stratified by immune profiling. Each patient will be longitudinally evaluated with prognostic biomarkers that predict the natural course of the disease. Prognostic biomarkers reflect immunological characteristics, such as a high interferon signature. When disease relapse is predicted with high confidence, the patient’s treatment can be guided by predictive biomarkers that project the response to each therapy.
2. Genetics of SLE and Immunological Insights
2.1. Monogenic SLE
Identification of causal mutations in rare monogenic lupus families or lupus-like autoimmune syndrome gave crucial insights into the pathogenesis of SLE. Lupus-prone families with primary defects of classical complement pathways (C1q, C1r/s, C2, C4A and C4B) revealed the importance of this pathway for lupus pathogenesis [28]. Complement is essential for opsonization and clearance of immune complexes and apoptotic cells. In addition, gene mutations involved in DNA processing during apoptosis cause lupus-like systemic autoimmunity [29]. Pediatric brain disease with a lupus-like symptom, Aicardi-Goutieres syndrome (AGS) is a prototypic example. AGS involves mutations in RNases (RNASEH2A, RNASEH2B, RNASEH2C), a DNase (TREX1), double-stranded RNA (dsRNA) editing (ADAR), dsRNA recognition and binding (IFIH1), and activation of the innate immune system (SAMHD1, IFIH1) [30]. Twelve of 20 AGS patients presented SLE-like characteristics, such as antinuclear antibodies (ANA), skin disease, thrombocytopenia, leukocytopenia, and arthritis [31]. AGS is now considered as a ‘type I interferonopathy’ because type I IFN production is constitutively upregulated in AGS patients and is directly relevant to pathogenesis [32]. Decreased DNase 1 activity by heterozygous nonsense DNASE1 mutation has been reported in 2 of 20 SLE patients [33]. Additionally, DNASE1L3 mutation causes familial SLE with positive ANA, anti-double strand DNA (dsDNA) antibodies, and anti-neutrophil cytoplasmic antibodies (ANCA) and low C3/C4 [34].
A homozygous missense mutation in PRKCD, encoding protein kinase δ (PKCδ) was reported in a family of juvenile-onset SLE [35]. PKCδ is a serine/threonine kinase that has been implicated in the negative selection of self-reactive B cells and control of cell proliferation in mice [36,37]. The missense mutation of PRKCD in affected families resulted in reduced activity of PKCδ, leading to resistance to apoptosis and increased B-cell proliferation.
These insights from Mendelian SLE or related syndrome families highlight the central role of nucleic acid metabolism, the complement pathway, and self-reactive B cells in human SLE pathogenesis.
2.2. Polygenic SLE
A genome-wide association study (GWAS) consists of hypothesis-free screening for linkage between loci and common multifactorial diseases, such as SLE. The association between GWAS-identified common single nucleotide polymorphisms (SNPs) and targeted traits is statistically robust. However, the effect size of most individual loci is small, as shown by typical odds ratios of identified loci ranging from 1.1 to 1.5 in large scale GWASs. Most of the identified SNPs lie in noncoding regions and affect gene expression through transcriptional or epigenetic modifications.
More than 100 loci have been shown to be robustly associated to SLE, especially in European and/or Asians GWASs [38,39]. Some of the reported genes are related to aberrant recognition of self-nucleic acid (NCF1, NCF2, FCGR2A, ITGAM and others), type I IFN overproduction/TLR signaling (IFIH1, IRF5, TNFAIP3 and others) and defective immune cell signaling (BLK, TNFSF13B and others). In other cases, the immunological function is unknown.
The human leukocyte antigen (HLA) region encodes more than 120 functional genes, such as HLA molecules involved in antigen presentation, complement components C2 and C4 and cytokine TNF-α [38,39]. Most of the genes in this region are immune-related and have a strong linkage equilibrium. HLA-DR and -DQ loci are consistently associated with SLE in different ethnic populations. The involvement of non-HLA class III region genes has also been strongly supported by GWAS results [40,41,42].
Expression quantitative trait loci (eQTL) analysis links each locus to variations of gene expression in each cell or tissue type. eQTL analysis of GWAS-identified loci with cell type-specific regulation of disease loci, such as BLK in B cells and JAZF1 in T cells [40]. SLE GWAS SNPs are enriched for B cell- and T-cell-specific gene expression and epigenetic enhancer marks [41,43]. Genetic risk score calculated by adding cumulative SLE-associated risk alleles weighted by SLE risk odds ratios revealed a higher genetic risk in non-European than European individuals, which may help explain the increased prevalence of SLE in non-Europeans [44].
SLE is a clinically heterogeneous disease and some phenotype-related loci have been reported, such as PDGRFA in lupus nephritis and ITGAM in arthritis [45,46]. However, the genetic architecture of subphenotypes of SLE is not fully elucidated. Reanalysis of existing GWAS with clinical subphenotypes may identify novel loci in association.
3. Immune Profiling of SLE
SLE is an autoimmune disease mediated by both innate and adaptive immune systems. Therefore, profiling of immune cells is a promising approach for biomarker discovery. Immunological memory enables immune systems to specifically and efficiently recognize antigens that they encountered, sometimes for a lifetime. Memory T cells and B cells that are long-lived and specific for particular antigens are the classical cells responsible for immune memory [47,48]. Defects in immune tolerance cause this efficient immune system to provoke autoimmunity that typically lasts a lifetime. Profiling of immune cells can reflect its history (for example, by analyzing autoantibody repertoire). Additionally, immune profiling can reflect the current status of an immunological system related to disease activity and future responses to treatment (for example, by analyzing IFN signature or cell subset frequencies).
Several methods have been developed to identify human immune cells. Flow cytometry or mass cytometry, targeting pre-specified marker proteins, allows quantification of immune cell composition at single-cell resolution. Transcriptome analysis, by microarray or RNA-sequencing (RNA-seq), allows genome-wide messenger RNA expression level quantification, methods that are fruitful for identifying pathways or modules of genes that are related to disease activity or prognosis. Proteome analysis can profile every protein or targeted proteins, such as autoantigens, thus reflecting immune system activation in SLE.
3.1. Flow Cytometry
Flow cytometry is a popular and powerful tool to characterize immune cell populations and functions. It utilizes fluorescent markers to label cells, and each cell is subjected to laser illumination to characterize expressed antigens/fluorochromes. Today, multicolor flow cytometry can simultaneously analyze more than 10 protein markers and a maximum of 50 markers on a single cell. Cell membrane permeabilization enables detection of not only cell surface markers, but also of cytokines and transcription factors in the cytoplasm and nucleus.
Kubo et al. performed flow cytometric phenotyping of circulating T cells, B cells and dendritic cell from 143 SLE patients and classified the patients into three clusters [49]. Patients with a high percentage of follicular helper T cells (Tfh) were more resistant to treatment with cyclophosphamide, mycophenolate mofetil, or calcineurin inhibitors, in addition to high-dose glucocorticoids. Tfh cells are essential for B-cell maturation and autoantibody production [50]. The proportions of CXCR5+ CCR7low PD-1high Tfh cells or CXCR5high ICOShigh PD-1high Tfh cells were also reported to be associated with disease activity in SLE [51,52]. OX40 ligand on myeloid antigen-presenting cells, induced by immune complexes containing RNA, is reportedly involved in the mechanism promoting Tfh responses in SLE [53].
Rituximab, an anti-CD20 monoclonal antibody targeting B cells, is a treatment option for severe lupus. However, randomized control trials did not confirm the efficacy [54,55]. Yusof et al. reported that B-cell depletion at 6 weeks predicts clinical response to rituximab treatment in SLE with an odds ratio of 3.22 in multivariate analysis [56]. Monitoring of B-cell depletion and individually tailored rituximab treatment was also tried in ANCA-associated vasculitis and resulted in fewer rituximab infusions with similar clinical efficacy [57]. Measuring disease-relevant cell population frequency is a candidate approach to the monitoring of immune system status or to predict treatment response.
Additional immune cell subpopulations are gaining attention in the pathophysiology of SLE. Low-density granulocytes constitute a distinct pro-inflammatory neutrophil subset found in SLE, and they exhibit enhanced spontaneous NETosis [58]. SLE low-density granulocytes release pro-inflammatory oxidized mitochondrial DNA in a mitochondrial reactive oxygen species- (ROS) dependent manner [59]. NETs opsonized by autoantibodies can stimulate plasmacytoid dendritic cells (pDCs) to synthesize IFN-α. In healthy individuals, pDCs drive the differentiation of CD19+ CD24hi CD38hi immature B cells into IL-10-producing CD24+CD38hi regulatory B (Breg) cells and plasmablasts by the release of IFN-α and CD40 engagement [60]. In SLE, pDCs fail to induce Breg cells because of the high IFN-α concentration. CXCR5− CXCR3+ PD1hi CD4+ helper T cells (Th10 cells), distinct from Tfh cells, help B cells through interleukin-10 and succinate and are expanded in SLE blood and kidneys [61]. A subset of CD19+ IgD+ CD27− naïve B cells persist in the circulation for months and differentiate into autoantibody secreting cells [62]. Additionally, autoreactive CD27− IgD− CXCR5− CD11c+ (DN2) B cells were recently shown to be disease-relevant in SLE patients [63]. DN2 cells, characterized by high transcription factor T-bet expression, are derived from naïve B cells and generate autoreactive plasma cells likely through extrafollicular differentiation. They show similarities to previously reported age- or autoimmune-associated B cells (ABCs) [64,65].
Diamond et al. developed a flow cytometric assay to identify ANA-reacting B-cells using biotinylated nuclear extracts [66,67]. Malkiel et al. showed an impairment of anergy induction in ANA+ naïve B cells in SLE patients, assessed by the percentage of IgMlow ANA+ naïve B cells, and restoration with belimumab treatment [66]. Suurmond et al. observed increased numbers of ANA+ IgG+ plasma cells in SLE patients, as well as in lupus-prone MRL/lpr and NZB/W mice. This increase was suggested to be the result of aberrant IgG+ plasma cell expansion, not by impaired antigen-specific tolerance checkpoints [67]. While the clinical utility of ANA+ B-cell analysis has not been assessed, profiling of autoantibody-specific B-cells is an attractive approach to monitoring B-cell immunity in SLE.
3.2. Mass Cytometry
As an alternative to flow cytometry, mass cytometry, also known as Cytometry by time-of-flight (CyTOF), uses heavy metal isotopes to label antibodies, and the labeled cells are analyzed by high-throughput spectrometry on a single-cell level. Typically, it analyzes >40 markers simultaneously and significantly augments the ability to evaluate complex cellular systems in high dimension. In flow cytometry, fluorophore emission spectra overlap, which makes it difficult to resolve the colors in multicolor analysis. This feature limits the number of measurable parameters and “compensation” is needed to account for spillover of light among them. In contrast, mass cytometry is able to discriminate isotopes of different atomic weights with high accuracy and the need for compensation is reduced [68]. However, it cannot recover living cells after analysis because cells are atomized and ionized for analysis.
Application of this new technology to leukemia and carcinoma has successfully identified potential biomarkers to forecast prognosis [69,70,71]. Mass cytometry is also being used in studies of rheumatology. Mass cytometric analysis of peripheral blood or synovial cells of rheumatoid arthritis patients identified candidate disease-associated T-cell populations, PD-1hi CXCR5− peripheral helper T cells and CD27− HLA-DR+ effector memory T cells [72,73].
Using mass cytometry, O’Gorman et al. performed systematic ex vivo Toll-like receptor activation analysis of SLE [74]. Among various immune cell subsets analyzed, CD14hi monocytes exhibited the most polyfunctional cytokine expression patterns, with more than 80 distinct cytokine combinations. Eight newly diagnosed untreated SLE patients shared a distinct monocytic chemokine signature compared to healthy volunteers. They also analyzed 10 newly diagnosed and untreated pediatric SLE patients with mass cytometry and identified a distinct monocyte signature characterized by MCP1, Mip1β and IL-1RA [75]. The cytokine signature was induced in monocytes from healthy volunteers by adding plasma from clinically active SLE patients and was abrogated by selective Janus kinase (JAK) inhibition. This signature was partially abrogated by interferon-α/β receptor (IFNAR) blockade, suggesting a role of IFN pathway for this monocyte signature. These promising results show the utility of mass cytometry in unveiling the pathophysiology of SLE. Larger sample size analysis is needed to dissect disease heterogeneity and discover prognostic and predictive biomarkers in SLE.
3.3. Microarray and RNA-seq
Initially, microarrays were used to study the transcriptome, the full range of messenger RNAs (mRNAs), or all RNAs in various cell populations [76]. It utilizes hybridization between fluorescently labelled complementary DNA (cDNA) with custom-made microarrays or commercial high-density oligo microarrays. They are relatively inexpensive, and they rely on known genomic sequences. RNA-seq directly determines cDNA sequences with next generation sequencers. RNA-seq offers several advantages over microarrays. RNA-seq is not dependent on prior knowledge of genomic sequences, and the dynamic range to quantify gene expression level is superior to microarrays. It can also be applied to analyze different isoforms and allele-specific expression [77]. RNA-seq is expected to substitute for microarrays in the near future. Most of the existing SLE transcriptome data are based upon microarray analysis.
Early microarray analysis led to the development of gene expression signatures, such as the IFN signature, i.e., the group of transcripts modified by IFN exposure. In 2003, two groups almost simultaneously identified IFN-induced genes by microarray comparisons of peripheral blood mononuclear cells (PBMC) in active SLE [78,79]. The expression of IFN signature genes was correlated to disease activity, as confirmed by later cross-sectional analyses [80,81,82]. An IFN signature is inducible by type I IFNs (IFNα or IFNβ). Anifrolumab, a type I IFN receptor antagonist, showed significant, although not strong, decreases of disease activity in moderate to severe SLE in a phase II clinical trial [83]. Anifrolumab showed a greater effect in patients with a high IFN signature. This clinical efficacy confirmed the role of IFN signaling as a disease-relevant pathway of SLE. The importance of this pathway is also noted in other autoimmune diseases, such as rheumatoid arthritis, Sjogren syndrome and systemic sclerosis [84,85,86].
Chaussabel et al. applied modular analysis that identified sets of coordinately expressed transcripts to PBMC microarray data from 239 individuals and found two SLE disease activity-related transcriptional modules, IFN-inducible and neutrophil genes [87]. Their analysis also revealed complex IFN signatures in SLE that are composed of three modules and involve both IFNα signature and also IFNβ and IFNγ [88]. Recently, a two-score system for IFN status has also been proposed, based on factor analysis of 31 IFN-stimulated genes [89,90]. Large scale IFN signaling network analysis (by the Immunological Genome Project Consortium) showed five regulatory modules of IFN signaling [91]. Most SLE-associated genes were from one cluster and appeared to require TYK2. These genes were sensitive to TYK2 deletion in mice. TYK2 is a JAK kinase activated by IFN binding to the IFNAR receptor and one of the GWAS-identified locus for SLE [38].
Banchereau et al. transcriptionally profiled 156 pediatric SLE patients longitudinally [92]. With 924 whole blood samples analyzed with microarrays, the plasmablast signature was the most robust disease activity biomarker in their analysis. While IFN response and plasmablast signature were involved in all patients, the neutrophil module was only activated in patients with active nephritis. They proposed a model of gradual disease progression, with early increases in IFN response and differentiation of B cells into plasmablasts, and late kidney disease and full-blown systemic inflammation fueled by myeloid cells, including neutrophils. They stratified SLE patients into seven clusters based on personalized immune-monitoring correlates of disease activity. Interestingly, the disease activity-related correlates differed from cluster to cluster, showing high molecular heterogeneity in SLE patients.
One limitation of whole blood transcriptome analysis is the fact that the differences in transcriptomes are largely influenced by the composition of immune cells in each sample. For example, a plasmablast signature can reflect increased frequency of plasmablasts and it is difficult to assess the qualitative difference in B-cell differentiation. To address qualitative changes in each cell subset, analysis of purified populations is needed. Lyons et al. isolated CD4 and CD8 T cells, B cells, monocytes and neutrophils from SLE and vasculitis patients and performed microarrays together with whole PBMC analysis. A substantial number of differentially expressed genes was only identified with purified cells and discrimination between patient groups was improved with purified monocytes [93]. McKinney et al. analyzed microarrays of purified CD8 and CD4 T cells with network and module analysis. They found that CD8 T-cell exhaustion negatively correlated with CD4 T-cell co-stimulation and that it indicated a better prognosis in SLE and in ANCA-associated vasculitis patients [94,95]. A candidate prognosis marker in the transcriptome of unseparated PBMCs, KAT2B, was also identified in their analysis. Their study emphasizes the utility of transcriptomic analysis of purified cell subsets.
To guide the initial therapy of active lupus nephritis, kidney biopsy and histological evaluation is the gold-standard [96]. Parikh et al. analyzed the expression of 511 immune-response genes with NanoString in kidney biopsies from 19 patients [97]. Genes responsible for clinical responses included IFN pathway genes, while complement genes were mainly found in non-responders. Transcriptome profiling of biopsy specimens may provide additional biomarkers for clinical histology, although access to affected organs, such as kidney, is more limited than blood sampling.
Recently, several groups in this field have shown the utility of RNA-seq. Shi et al. performed RNA-seq of both coding and noncoding RNAs of monocytes from 9 SLE patients [98]. They found significantly altered splicing patterns and novel transcripts. Rai et al. compared peripheral blood RNA-seq data from 28 SLE patients [99]. Multiple cytokine signaling pathways were specifically dysregulated in anti-dsDNA+ patients, whereas IFN signaling was predominantly dysregulated in anti-extractable nuclear antigen (ENA)+ patients. Zhang et al. performed RNA-seq in combination with H3K4me3 Chip-seq in T cells, B cells and monocytes from 6 SLE patients [100]. Although their analysis was limited by the small number of participants, it showed both shared and cell-specific changes in gene expression and epigenetics.
3.4. Single-Cell Expression Profiles and RNA-seq
Application of RNA-seq to single cells (single-cell RNA-seq) allows comprehensive analysis of immune cells at single-cell resolution [101,102]. In combination with plate-based or droplet-based single-cell isolation, hundreds, thousands, and more cells can be simultaneously analyzed for their transcriptome. It can be used to identify and characterize disease related immune cell subsets. The transcriptional signatures of these immune cells enable the identification of novel biomarkers. Additionally, immune repertoire analysis of single cells can identify T-cell or B-cell receptor pairs.
Jin et al. analyzed single-cell gene expression profiles using a monocyte-related transcription panel to assess SLE patient monocytes [103]. Unsupervised hierarchical clustering of SLE monocytes demonstrated that independent clusters of cells were related to disease activity, type I IFN and medication use. Accelerating Medicines Partnership (AMP) in Rheumatoid Arthritis and Lupus Network is a public-private partnership created to develop new ways of identifying and validating promising biological targets for diagnostics and drug development. In an early report from the AMP project, Der et al. applied single-cell RNA-seq to kidney and skin biopsy samples from 16 lupus nephritis patients [81]. Keratinocytes from the skin of those patients also revealed upregulation of IFN-inducible genes. With greater sample sizes, their analysis may help to identify disease-relevant or disease subtype-related populations that only exist in affected organs.
3.5. Autoantibody Repertoire and Proteomics
More than 180 autoantibodies have been found in the blood of SLE patients and different patients may exhibit different autoantibody profiles [104]. Autoantigen arrays are a high-throughput autoantibody screening platform based on antigen-antibody reactions. The arrays are produced by immobilizing hundreds or more diverse autoantigens on the coated surface of glass slides. The arrays are reacted with diluted samples, such as serum, and the autoantibodies bound to their corresponding antigens are detected with the fluorophore-conjugated second antibodies. Li et al. constructed protein microarray bearing about 30 antigens known to be expressed in the kidney and identified five distinct clusters of IgG autoreactivity in the sera of lupus patients, and two of the clusters showed association with disease activity [105]. Huang et al. used protein arrays containing over 5000 recombinant human proteins to profile the autoantibodies in the sera of SLE patients. Four novel antigens including CLIC2, were identified as potential targets of autoantibodies in SLE [106]. ELISA experiments confirmed the presence of autoantibody to CLIC2 in 28% of SLE patients. Kinloch et al. screened cloned activated B cells isolated from renal biopsy specimens of lupus nephritis patients. They found that vimentin was a dominant autoantigen targeted in lupus tubulointerstitial nephritis [107]. These results show that protein arrays can contribute to the discovery of novel autoantibodies. Detection of certain patterns of autoantibodies may help to forecast disease activity or response to therapy.
Autoantibodies, such as anti-dsDNA antibodies and anti-Ro antibodies, are present many years before the diagnosis of SLE and progressively accumulate until clinical onset [108]. Recent analysis with multiplex bead-based antigen detection for 398 antigens, showed that the number of epitopes recognized by autoantibodies generally do not change over six years in established SLE patients [109]. dsDNA and histone H3 autoantibodies were notable exceptions and constituted disease activity markers. An increase in antibodies to the U1-RNP epitopes at the time of new organ involvement was noted, suggesting intramolecular epitope spreading may be more sensitive to disease activity.
Of note, autoantigen microarrays and bead-based multiplex analysis are both “biased” proteomic analyses to the extent that they target only known peptides [110]. Typical “unbiased” proteomic analysis with mass spectrometry first separates proteins using gel-based techniques and identifies proteins with mass spectrometry [111]. More than 241 SLE candidate proteomic biomarkers have been discovered with mass spectrometry analysis and 28 candidate biomarkers were validated in independent cohorts or studies. Ferreira et al. identified candidate lupus diagnosis and activity markers with two-dimensional differential gel electrophoresis and mass spectrometry [112]. Limited numbers of lupus participants, typically 5 to 20, and variable experimental conditions in each study warrant validation of these candidate protein biomarkers.
4. Toward Precision Medicine in SLE
At present, no validated biological biomarker exists to predict disease course and treatment response with high reliability and reproducibility [113]. Conventional disease assessment methods, including the use of acute phase markers, such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), and anti-dsDNA antibodies are of limited sensitivity and specificity.
Future enhancements of prognostic and predictive biomarkers will improve the assessment and clinical management of SLE (Figure 1). It will help the diagnosis, evaluation of disease activity and therapeutic decisions. Precise biological evaluation of disease activity by use of prognostic biomarkers will quantitatively evaluate the possibility of future relapse or organ damage. In that way, clinical decisions regarding intensification or tapering of treatment can be improved. Predictive biomarkers will forecast the clinical response and possible adverse reactions and will aid clinical decision making based upon the available treatment options.
Transcriptome analysis of whole blood or targeted immune cells is a promising way to identify gene expression biomarkers. There have been recent successes in discovering gene modules closely related to disease activity, disease subtype, or future relapse [92,94,95]. The bulk transcriptome reflects the average expression of individual cells. A small cell fraction may be responsible for the transcriptome difference. For example, a CD8 T-cell exhaustion signature can be the result of an expanded small fraction of exhausted CD8 T cells. To fully characterize a disease-relevant small cell subset that is responsible for transcriptome change, deeper immune phenotyping, probably by mass cytometry or single-cell RNA-seq in combination with bulk transcriptome analysis is needed. Additionally, quantifying the expression of a small number of mRNAs, as is done in IFN signature assays [89,90], may serve as an alternative approach to monitor transcriptome signatures.
Reproducibility is one of the most important issues for clinical application of biomarkers to precision medicine. Thus, candidate biomarkers must be assessed with a standardized protocol. For example, standardization of immune cell phenotyping has been proposed [114]. However, researchers continue to identify new candidate immune cell populations that drive SLE including Th10 cells and DN2/ABCs described above. These examples of the diversity of candidate biomarker populations provoke a dilemma between standardized and exploratory research protocols. Exploratory immune profiling “omics” analysis needs to be followed by standardized multi-center validation studies, targeting a small number of candidate biomarkers. Identification of reliable and reproducible biomarkers is the key to realize precision or personalized medicine in SLE.
5. Conclusions
The heterogeneous nature of SLE is being elucidated by human immune profiling studies. Disease-relevant immune subsets identified by immunophenotyping analysis, or gene network signatures identified by transcriptome analysis, if validated by independent study, may serve as novel biomarkers to realize future precision medicine in SLE. Prognostic biomarkers will reflect immunological abnormalities in each patient and will forecast their natural course. Importantly, predictive biomarkers will forecast the response to treatment options. These biomarkers will improve and guide future clinical management of SLE.
Author Contributions
Y.N., H.S., and K.F. contributed to writing the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
K.F. received financial support or fees from Astellas, BMS, Daiichi-Sankyo, Mitsubishi Tanabe, Pfizer, Ayumi, Takeda, Chugai, Eisai, Taisho Toyama, UCB, Janssen, Eli Lilly and NIPPON KAYAKU. All other authors declare no conflicts of interest.
References
- Wahren-Herlenius, M.; Dorner, T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013, 382, 819–831. [Google Scholar] [CrossRef]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Zandman-Goddard, G.; Solomon, M.; Rosman, Z.; Peeva, E.; Shoenfeld, Y. Environment and lupus-related diseases. Lupus 2012, 21, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Manfredo Vieira, S.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, T.; Youinou, P.; Pers, J.O.; Jamin, C. TLR9 drives the development of transitional B cells towards the marginal zone pathway and promotes autoimmunity. J. Autoimmun. 2012, 39, 173–179. [Google Scholar] [CrossRef]
- Lesley, R.; Xu, Y.; Kalled, S.L.; Hess, D.M.; Schwab, S.R.; Shu, H.B.; Cyster, J.G. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 2004, 20, 441–453. [Google Scholar] [CrossRef]
- Thien, M.; Phan, T.G.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 2004, 20, 785–798. [Google Scholar] [CrossRef]
- Tektonidou, M.G.; Lewandowski, L.B.; Hu, J.; Dasgupta, A.; Ward, M.M. Survival in adults and children with systemic lupus erythematosus: A systematic review and Bayesian meta-analysis of studies from 1950 to 2016. Ann. Rheum. Dis. 2017, 76, 2009–2016. [Google Scholar] [CrossRef]
- Ocampo-Piraquive, V.; Nieto-Aristizabal, I.; Canas, C.A.; Tobon, G.J. Mortality in systemic lupus erythematosus: Causes, predictors and interventions. Expert Rev. Clin. Immunol. 2018, 14, 1043–1053. [Google Scholar] [CrossRef]
- Bertsias, G.; Ioannidis, J.P.; Boletis, J.; Bombardieri, S.; Cervera, R.; Dostal, C.; Font, J.; Gilboe, I.M.; Houssiau, F.; Huizinga, T.; et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann. Rheum. Dis. 2008, 67, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.H.; McMahon, M.A.; Wilkinson, A.; Wallace, W.D.; Daikh, D.I.; Fitzgerald, J.D.; Karpouzas, G.A.; Merrill, J.T.; Wallace, D.J.; Yazdany, J.; et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res. 2012, 64, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Bertsias, G.K.; Tektonidou, M.; Amoura, Z.; Aringer, M.; Bajema, I.; Berden, J.H.; Boletis, J.; Cervera, R.; Dorner, T.; Doria, A.; et al. Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann. Rheum. Dis. 2012, 71, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Yap, D.Y.; Navarra, S.V.; Liu, Z.H.; Zhao, M.H.; Lu, L.; Takeuchi, T.; Avihingsanon, Y.; Yu, X.Q.; Lapid, E.A.; et al. Overview of lupus nephritis management guidelines and perspective from Asia. Int. J. Rheum. Dis. 2013, 16, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, R.F.; Mosca, M.; Bertsias, G.; Isenberg, D.; Kuhn, A.; Lerstrom, K.; Aringer, M.; Bootsma, H.; Boumpas, D.; Bruce, I.N.; et al. Treat-to-target in systemic lupus erythematosus: Recommendations from an international task force. Ann. Rheum. Dis. 2014, 73, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Zen, M.; Iaccarino, L.; Gatto, M.; Bettio, S.; Nalotto, L.; Ghirardello, A.; Punzi, L.; Doria, A. Prolonged remission in Caucasian patients with SLE: Prevalence and outcomes. Ann. Rheum. Dis. 2015, 74, 2117–2122. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Feletar, M.; Bruce, I.N.; Ibanez, D.; Gladman, D.D. Prolonged remission in systemic lupus erythematosus. J. Rheumatol. 2005, 32, 1467–1472. [Google Scholar] [PubMed]
- To, C.H.; Petri, M. Is antibody clustering predictive of clinical subsets and damage in systemic lupus erythematosus? Arthritis Rheum. 2005, 52, 4003–4010. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Kanayama, Y.; Amastu, K.; Negoro, N.; Kohda, S.; Takeda, T.; Inoue, T. Significance of enzyme linked immunosorbent assay (ELISA) for antibodies to double stranded and single stranded DNA in patients with lupus nephritis: Correlation with severity of renal histology. Ann. Rheum. Dis. 1993, 52, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.A.; Garton, M.; Reichlin, M.W.; Reichlin, M. Long-term follow-up of autoantibody profiles in black female lupus patients and clinical comparison with Caucasian and Asian patients. Br. J. Rheumatol. 1997, 36, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- Tesar, V.; Hruskova, Z. Belimumab in the management of systemic lupus erythematosus—An update. Expert Opin. Biol. Ther. 2017, 17, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Felten, R.; Dervovic, E.; Chasset, F.; Gottenberg, J.E.; Sibilia, J.; Scher, F.; Arnaud, L. The 2018 pipeline of targeted therapies under clinical development for Systemic Lupus Erythematosus: A systematic review of trials. Autoimmun. Rev. 2018, 17, 781–790. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ermann, J.; Rao, D.A.; Teslovich, N.C.; Brenner, M.B.; Raychaudhuri, S. Immune cell profiling to guide therapeutic decisions in rheumatic diseases. Nat. Rev. Rheumatol. 2015, 11, 541–551. [Google Scholar] [CrossRef]
- Mirnezami, R.; Nicholson, J.; Darzi, A. Preparing for precision medicine. N. Engl. J. Med. 2012, 366, 489–491. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Niewold, T.B. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J. Autoimmun. 2015, 64, 125–136. [Google Scholar] [CrossRef]
- Saeed, M. Lupus pathobiology based on genomics. Immunogenetics 2017, 69, 1–12. [Google Scholar] [CrossRef]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167a, 296–312. [Google Scholar] [CrossRef]
- Ramantani, G.; Kohlhase, J.; Hertzberg, C.; Innes, A.M.; Engel, K.; Hunger, S.; Borozdin, W.; Mah, J.K.; Ungerath, K.; Walkenhorst, H.; et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum. 2010, 62, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Belot, A.; Kasher, P.R.; Trotter, E.W.; Foray, A.P.; Debaud, A.L.; Rice, G.I.; Szynkiewicz, M.; Zabot, M.T.; Rouvet, I.; Bhaskar, S.S.; et al. Protein kinase cdelta deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis Rheum. 2013, 65, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Mecklenbrauker, I.; Saijo, K.; Zheng, N.Y.; Leitges, M.; Tarakhovsky, A. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature 2002, 416, 860–865. [Google Scholar] [CrossRef]
- Miyamoto, A.; Nakayama, K.; Imaki, H.; Hirose, S.; Jiang, Y.; Abe, M.; Tsukiyama, T.; Nagahama, H.; Ohno, S.; Hatakeyama, S.; et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature 2002, 416, 865–869. [Google Scholar] [CrossRef]
- Deng, Y.; Tsao, B.P. Updates in Lupus Genetics. Curr. Rheumatol. Rep. 2017, 19, 68. [Google Scholar] [CrossRef]
- Cui, Y.; Sheng, Y.; Zhang, X. Genetic susceptibility to SLE: Recent progress from GWAS. J. Autoimmun. 2013, 41, 25–33. [Google Scholar] [CrossRef]
- Bentham, J.; Morris, D.L.; Graham, D.S.C.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martin, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef]
- Sun, C.; Molineros, J.E.; Looger, L.L.; Zhou, X.J.; Kim, K.; Okada, Y.; Ma, J.; Qi, Y.Y.; Kim-Howard, X.; Motghare, P.; et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat. Genet. 2016, 48, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.L.; Zidovetzki, R.; Alarcón-Riquelme, M.E.; Tsao, B.P.; Criswell, L.A.; Kimberly, R.P.; Harley, J.B.; Sivils, K.L.; Vyse, T.J.; Gaffney, P.M.; et al. GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immun. 2014, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef]
- Morris, D.L.; Sheng, Y.; Zhang, Y.; Wang, Y.F.; Zhu, Z.; Tombleson, P.; Chen, L.; Cunninghame Graham, D.S.; Bentham, J.; Roberts, A.L.; et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016, 48, 940–946. [Google Scholar] [CrossRef]
- Chung, S.A.; Brown, E.E.; Williams, A.H.; Ramos, P.S.; Berthier, C.C.; Bhangale, T.; Alarcon-Riquelme, M.E.; Behrens, T.W.; Criswell, L.A.; Graham, D.C.; et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J. Am. Soc. Nephrol. 2014, 25, 2859–2870. [Google Scholar] [CrossRef]
- Taylor, K.E.; Chung, S.A.; Graham, R.R.; Ortmann, W.A.; Lee, A.T.; Langefeld, C.D.; Jacob, C.O.; Kamboh, M.I.; Alarcon-Riquelme, M.E.; Tsao, B.P.; et al. Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011, 7, e1001311. [Google Scholar] [CrossRef]
- Ahmed, R.; Gray, D. Immunological memory and protective immunity: Understanding their relation. Science 1996, 272, 54–60. [Google Scholar] [CrossRef]
- Farber, D.L.; Netea, M.G.; Radbruch, A.; Rajewsky, K.; Zinkernagel, R.M. Immunological memory: Lessons from the past and a look to the future. Nat Rev Immunol. 2016, 16, 124–128. [Google Scholar] [CrossRef]
- Kubo, S.; Nakayamada, S.; Yoshikawa, M.; Miyazaki, Y.; Sakata, K.; Nakano, K.; Hanami, K.; Iwata, S.; Miyagawa, I.; Saito, K.; et al. Peripheral Immunophenotyping Identifies Three Subgroups Based on T Cell Heterogeneity in Lupus Patients. Arthritis Rheumatol. 2017, 69, 2029–2037. [Google Scholar] [CrossRef]
- Craft, J.E. Follicular helper T cells in immunity and systemic autoimmunity. Nat. Rev. Rheumatol. 2012, 8, 337–347. [Google Scholar] [CrossRef]
- Choi, J.Y.; Ho, J.H.; Pasoto, S.G.; Bunin, V.; Kim, S.T.; Carrasco, S.; Borba, E.F.; Goncalves, C.R.; Costa, P.R.; Kallas, E.G.; et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: Association with disease activity. Arthritis Rheumatol. 2015, 67, 988–999. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tsai, L.M.; Leong, Y.A.; Hu, X.; Ma, C.S.; Chevalier, N.; Sun, X.; Vandenberg, K.; Rockman, S.; Ding, Y.; et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, C.; Schmitt, N.; Contin-Bordes, C.; Liu, Y.; Narayanan, P.; Seneschal, J.; Maurouard, T.; Dougall, D.; Davizon, E.S.; Dumortier, H.; et al. OX40 Ligand Contributes to Human Lupus Pathogenesis by Promoting T Follicular Helper Response. Immunity 2015, 42, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Md Yusof, M.Y.; Shaw, D.; El-Sherbiny, Y.M.; Dunn, E.; Rawstron, A.C.; Emery, P.; Vital, E.M. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.; Terrier, B.; Perrodeau, E.; Cohen, P.; Faguer, S.; Huart, A.; Hamidou, M.; Agard, C.; Bonnotte, B.; Samson, M.; et al. Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: Results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann. Rheum. Dis. 2018, 77, 1143–1149. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef]
- Caielli, S.; Veiga, D.T.; Balasubramanian, P.; Athale, S.; Domic, B.; Murat, E.; Banchereau, R.; Xu, Z.; Chandra, M.; Chung, C.H.; et al. A CD4(+) T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat. Med. 2018. [Google Scholar] [CrossRef]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 2015, 16, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Tipton, C.M.; Hom, J.R.; Fucile, C.F.; Rosenberg, A.F.; Sanz, I. Understanding B-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: A B-cell immunomics approach. Immunol. Rev. 2018, 284, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Phalke, S.; Marrack, P. Age (autoimmunity) associated B cells (ABCs) and their relatives. Curr. Opin. Immunol. 2018, 55, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Malkiel, S.; Jeganathan, V.; Wolfson, S.; Manjarrez Orduno, N.; Marasco, E.; Aranow, C.; Mackay, M.; Gregersen, P.K.; Diamond, B. Checkpoints for Autoreactive B Cells in the Peripheral Blood of Lupus Patients Assessed by Flow Cytometry. Arthritis Rheumatol. 2016, 68, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Suurmond, J.; Atisha-Fregoso, Y.; Marasco, E.; Barlev, A.N.; Ahmed, N.; Calderon, S.A.; Wong, M.Y.; Mackay, M.C.; Aranow, C.; Diamond, B. Loss of an IgG plasma cell checkpoint in lupus. J. Allergy Clin. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir el, A.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017, 542, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, C.Y.; Rao, D.A.; Teslovich, N.C.; Korsunsky, I.; Hannes, S.K.; Slowikowski, K.; Gurish, M.F.; Donlin, L.T.; Lederer, J.A.; Weinblatt, M.E.; et al. Mixed-effects association of single cells identifies an expanded effector CD4(+) T cell subset in rheumatoid arthritis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, W.E.; Hsieh, E.W.; Savig, E.S.; Gherardini, P.F.; Hernandez, J.D.; Hansmann, L.; Balboni, I.M.; Utz, P.J.; Bendall, S.C.; Fantl, W.J.; et al. Single-cell systems-level analysis of human Toll-like receptor activation defines a chemokine signature in patients with systemic lupus erythematosus. J. Allergy Clin. Immunol. 2015, 136, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, W.E.; Kong, D.S.; Balboni, I.M.; Rudra, P.; Bolen, C.R.; Ghosh, D.; Davis, M.M.; Nolan, G.P.; Hsieh, E.W. Mass cytometry identifies a distinct monocyte cytokine signature shared by clinically heterogeneous pediatric SLE patients. J. Autoimmun. 2017. [Google Scholar] [CrossRef]
- Hyatt, G.; Melamed, R.; Park, R.; Seguritan, R.; Laplace, C.; Poirot, L.; Zucchelli, S.; Obst, R.; Matos, M.; Venanzi, E.; et al. Gene expression microarrays: Glimpses of the immunological genome. Nat. Immunol. 2006, 7, 686–691. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nature Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Nikpour, M.; Dempsey, A.A.; Urowitz, M.B.; Gladman, D.D.; Barnes, D.A. Association of a gene expression profile from whole blood with disease activity in systemic lupus erythaematosus. Ann. Rheum. Dis. 2008, 67, 1069–1075. [Google Scholar] [CrossRef]
- Feng, X.; Wu, H.; Grossman, J.M.; Hanvivadhanakul, P.; FitzGerald, J.D.; Park, G.S.; Dong, X.; Chen, W.; Kim, M.H.; Weng, H.H.; et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2951–2962. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K. Type I interferon in the pathogenesis of lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Van der Pouw Kraan, T.C.; Wijbrandts, C.A.; van Baarsen, L.G.; Voskuyl, A.E.; Rustenburg, F.; Baggen, J.M.; Ibrahim, S.M.; Fero, M.; Dijkmans, B.A.; Tak, P.P.; et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: Assignment of a type I interferon signature in a subpopulation of patients. Ann. Rheum. Dis. 2007, 66, 1008–1014. [Google Scholar] [CrossRef]
- Brkic, Z.; Maria, N.I.; van Helden-Meeuwsen, C.G.; van de Merwe, J.P.; van Daele, P.L.; Dalm, V.A.; Wildenberg, M.E.; Beumer, W.; Drexhage, H.A.; Versnel, M.A. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann. Rheum. Dis. 2013, 72, 728–735. [Google Scholar] [CrossRef]
- Brkic, Z.; van Bon, L.; Cossu, M.; van Helden-Meeuwsen, C.G.; Vonk, M.C.; Knaapen, H.; van den Berg, W.; Dalm, V.A.; Van Daele, P.L.; Severino, A.; et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann. Rheum. Dis. 2016, 75, 1567–1573. [Google Scholar] [CrossRef]
- Chaussabel, D.; Quinn, C.; Shen, J.; Patel, P.; Glaser, C.; Baldwin, N.; Stichweh, D.; Blankenship, D.; Li, L.; Munagala, I.; et al. A modular analysis framework for blood genomics studies: Application to systemic lupus erythematosus. Immunity 2008, 29, 150–164. [Google Scholar] [CrossRef]
- Chiche, L.; Jourde-Chiche, N.; Whalen, E.; Presnell, S.; Gersuk, V.; Dang, K.; Anguiano, E.; Quinn, C.; Burtey, S.; Berland, Y.; et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014, 66, 1583–1595. [Google Scholar] [CrossRef]
- El-Sherbiny, Y.M.; Psarras, A.; Md Yusof, M.Y.; Hensor, E.M.A.; Tooze, R.; Doody, G.; Mohamed, A.A.A.; McGonagle, D.; Wittmann, M.; Emery, P.; et al. A novel two-score system for interferon status segregates autoimmune diseases and correlates with clinical features. Sci. Rep. 2018, 8, 5793. [Google Scholar] [CrossRef]
- Md Yusof, M.Y.; Psarras, A.; El-Sherbiny, Y.M.; Hensor, E.M.A.; Dutton, K.; Ul-Hassan, S.; Zayat, A.S.; Shalbaf, M.; Alase, A.; Wittmann, M.; et al. Prediction of autoimmune connective tissue disease in an at-risk cohort: Prognostic value of a novel two-score system for interferon status. Ann. Rheum. Dis. 2018, 77, 1432–1439. [Google Scholar] [CrossRef]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoite, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.Y.; Feng, T.; et al. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Lyons, P.A.; McKinney, E.F.; Rayner, T.F.; Hatton, A.; Woffendin, H.B.; Koukoulaki, M.; Freeman, T.C.; Jayne, D.R.; Chaudhry, A.N.; Smith, K.G. Novel expression signatures identified by transcriptional analysis of separated leucocyte subsets in systemic lupus erythematosus and vasculitis. Ann. Rheum. Dis. 2010, 69, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Lyons, P.A.; Carr, E.J.; Hollis, J.L.; Jayne, D.R.; Willcocks, L.C.; Koukoulaki, M.; Brazma, A.; Jovanovic, V.; Kemeny, D.M.; et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat. Med. 2010, 16, 586–591. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.; Lyons, P.A.; Smith, K.G. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Weening, J.J.; D’Agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Balow, J.E.; Bruijn, J.A.; Cook, T.; Ferrario, F.; et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J. Am. Soc. Nephrol. 2004, 15, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.V.; Malvar, A.; Song, H.; Alberton, V.; Lococo, B.; Vance, J.; Zhang, J.; Yu, L.; Rovin, B.H. Characterising the immune profile of the kidney biopsy at lupus nephritis flare differentiates early treatment responders from non-responders. Lupus Sci. Med. 2015, 2, e000112. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, Z.; Yu, A.M.; Wang, W.; Wei, Z.; Akhter, E.; Maurer, K.; Costa Reis, P.; Song, L.; Petri, M.; et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PLoS ONE 2014, 9, e93846. [Google Scholar] [CrossRef]
- Rai, R.; Chauhan, S.K.; Singh, V.V.; Rai, M.; Rai, G. RNA-seq Analysis Reveals Unique Transcriptome Signatures in Systemic Lupus Erythematosus Patients with Distinct Autoantibody Specificities. PLoS ONE 2016, 11, e0166312. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, L.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Overall Downregulation of mRNAs and Enrichment of H3K4me3 Change Near Genome-Wide Association Study Signals in Systemic Lupus Erythematosus: Cell-Specific Effects. Front. Immunol. 2018, 9, 497. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Fan, W.; Jensen, M.A.; Dorschner, J.M.; Bonadurer, G.F.; Vsetecka, D.M.; Amin, S.; Makol, A.; Ernste, F.; Osborn, T.; et al. Single-cell gene expression patterns in lupus monocytes independently indicate disease activity, interferon and therapy. Lupus Sci. Med. 2017, 4, e000202. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Yan, M.; Zuo, X.; Li, Q.Z. Autoantigen Microarray for High-throughput Autoantibody Profiling in Systemic Lupus Erythematosus. Genom. Proteom. Bioinform. 2015, 13, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Z.; Xie, C.; Wu, T.; Mackay, M.; Aranow, C.; Putterman, C.; Mohan, C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J. Clin. Investig. 2005, 115, 3428–3439. [Google Scholar] [CrossRef]
- Huang, W.; Hu, C.; Zeng, H.; Li, P.; Guo, L.; Zeng, X.; Liu, G.; Zhang, F.; Li, Y.; Wu, L. Novel systemic lupus erythematosus autoantigens identified by human protein microarray technology. Biochem. Biophys. Res. Commun. 2012, 418, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Kinloch, A.J.; Chang, A.; Ko, K.; Henry Dunand, C.J.; Henderson, S.; Maienschein-Cline, M.; Kaverina, N.; Rovin, B.H.; Salgado Ferrer, M.; Wolfgeher, D.; et al. Vimentin is a dominant target of in situ humoral immunity in human lupus tubulointerstitial nephritis. Arthritis Rheumatol. 2014, 66, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef]
- Vordenbaumen, S.; Brinks, R.; Hoyer, A.; Fischer-Betz, R.; Pongratz, G.; Lowin, T.; Zucht, H.D.; Budde, P.; Bleck, E.; Schulz-Knappe, P.; et al. Comprehensive longitudinal surveillance of the IgG autoantibody repertoire in established systemic lupus erythematosus. Arthritis Rheumatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fathman, C.G.; Soares, L.; Chan, S.M.; Utz, P.J. An array of possibilities for the study of autoimmunity. Nature 2005, 435, 605–611. [Google Scholar] [CrossRef]
- Nicolaou, O.; Kousios, A.; Hadjisavvas, A.; Lauwerys, B.; Sokratous, K.; Kyriacou, K. Biomarkers of systemic lupus erythematosus identified using mass spectrometry-based proteomics: A systematic review. J. Cell Mol. Med. 2017, 21, 993–1012. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.A.R.; de Andrade, H.M.; de Pádua, P.M.; Carvalho, M.D.G.; Pires, S.D.F.; Oliveira, I.H.R.; Lima, B.S.S.; Fialho Júnior, L.C.; Cicarini, W.B.; Chapeourouge, D.A.; et al. Identification of potential biomarkers for systemic lupus erythematosus diagnosis using two-dimensional differential gel electrophoresis (2D-DIGE) and mass spectrometry. Autoimmunity 2017, 50, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Illei, G.G.; Tackey, E.; Lapteva, L.; Lipsky, P.E. Biomarkers in systemic lupus erythematosus. I. General overview of biomarkers and their applicability. Arthritis Rheum. 2004, 50, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).