Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
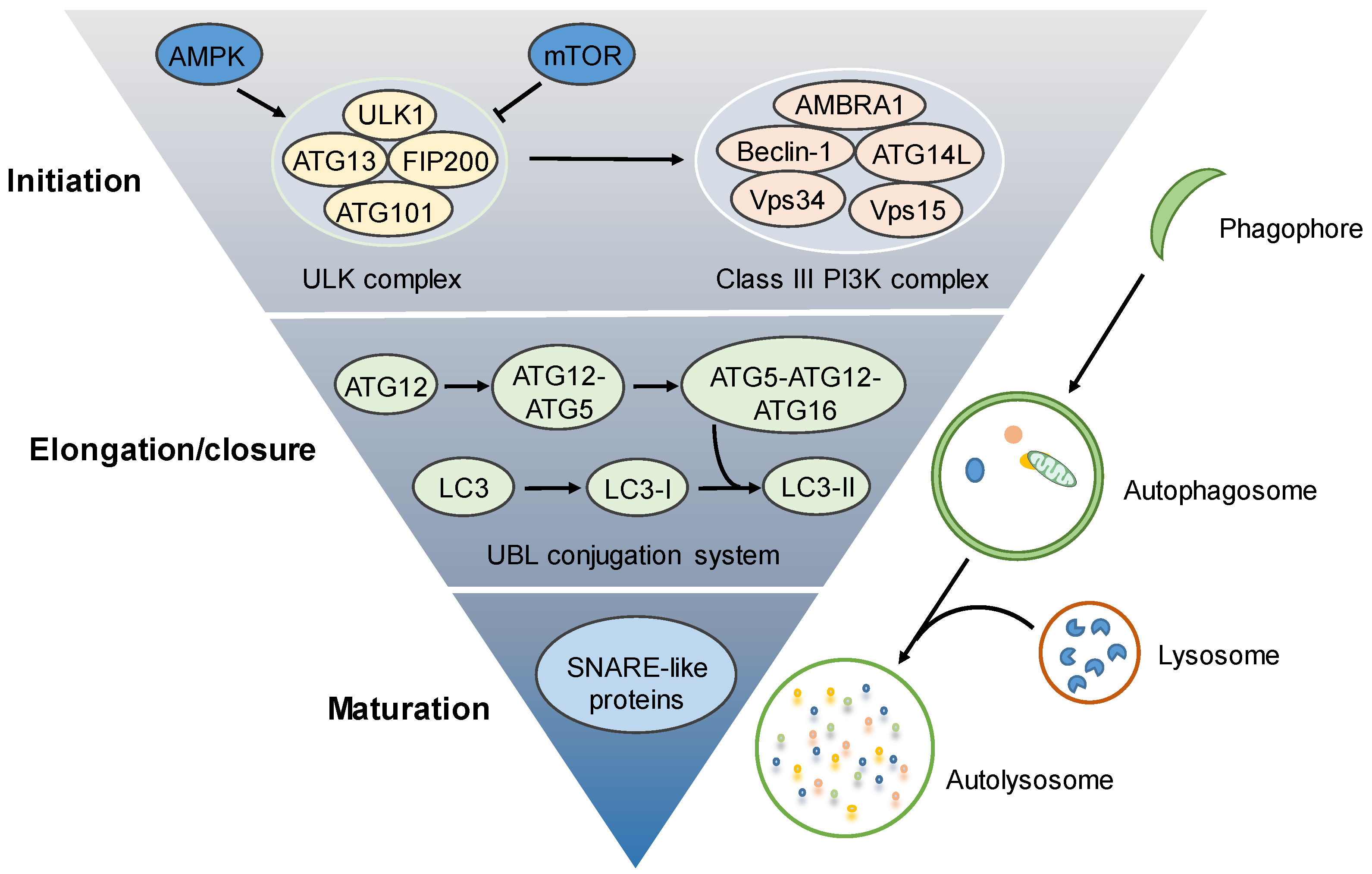
2. Molecular Regulation of the Autophagy Process
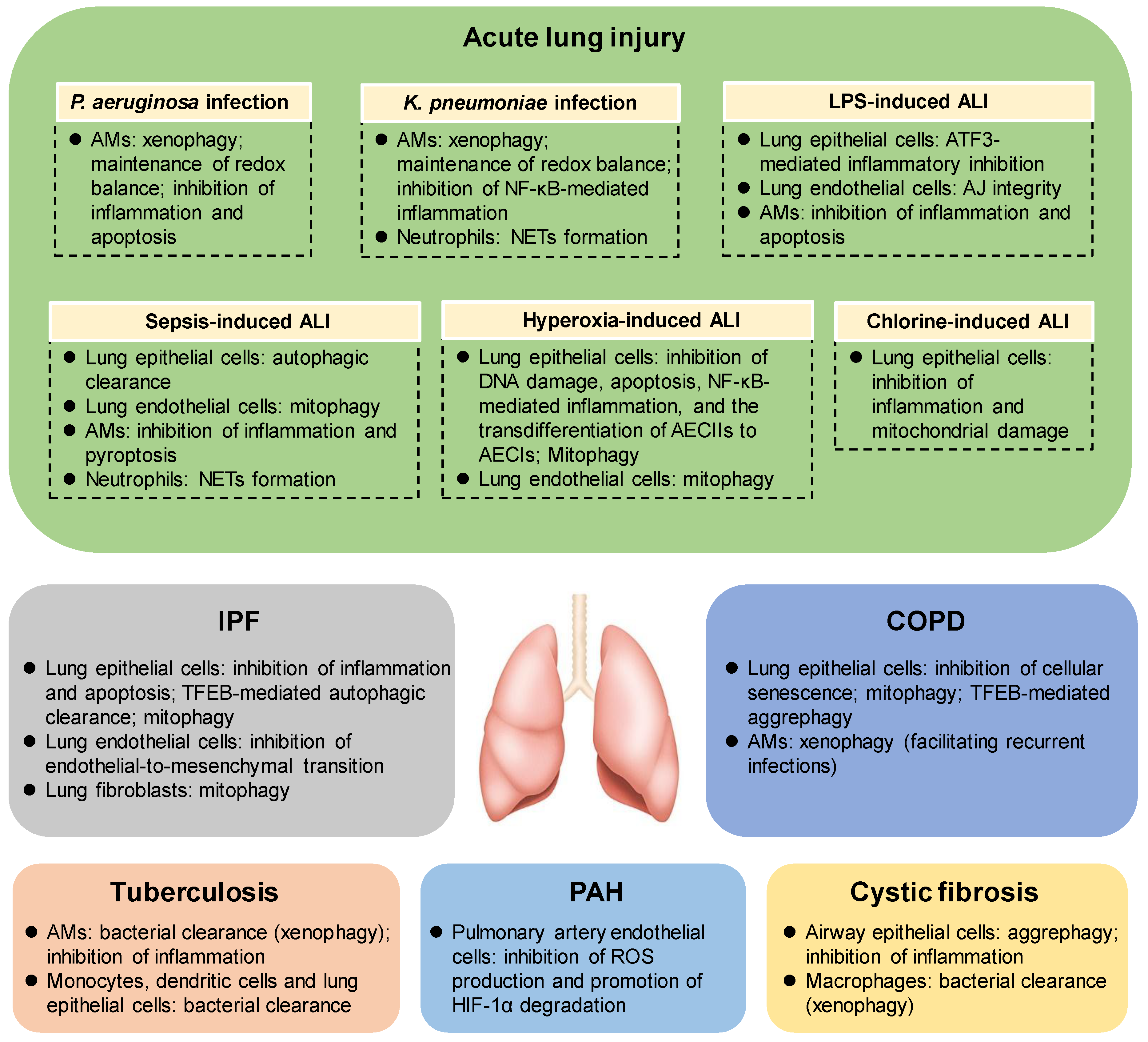
3. The Protective Roles of Autophagy in Acute Lung Injury (ALI)
3.1. The Protective Roles of Autophagy in Bacteria–Induced ALI
3.2. The Protective Roles of Autophagy in LPS–Induced ALI
3.3. The Protective Roles of Autophagy in Sepsis–Induced ALI
3.4. The Protective Roles of Autophagy in Hyperoxia–Induced ALI
3.5. The Protective Roles of Autophagy in Chlorine–Induced ALI
4. The Protective Roles of Autophagy in Idiopathic Pulmonary Fibrosis (IPF)
5. The Protective and Deleterious Roles of Autophagy in COPD
6. The Protective Roles of Autophagy in Tuberculosis
7. The Protective Roles of Autophagy in Cystic Fibrosis (CF)
8. The Protective Roles of Autophagy in Pulmonary Arterial Hypertension (PAH)
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rdedition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Mehrpour, M.; Botti, J.; Dupont, N.; Hamai, A.; Nascimbeni, A.C.; Codogno, P. Autophagy: A Druggable Process. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Nakahira, K.; Haspel, J.A.; Choi, A.M. Autophagy in pulmonary diseases. Annu. Rev. Physiol. 2012, 74, 377–401. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, J.; Matthay, M.A. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu. Rev. Physiol. 2013, 75, 593–615. [Google Scholar] [CrossRef]
- Nakahira, K.; Choi, A.M. Autophagy: A potential therapeutic target in lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L93–L107. [Google Scholar] [CrossRef]
- Nakahira, K.; Cloonan, S.M.; Mizumura, K.; Choi, A.M.; Ryter, S.W. Autophagy: A crucial moderator of redox balance, inflammation, and apoptosis in lung disease. Antioxid. Redox Signal. 2014, 20, 474–494. [Google Scholar] [CrossRef]
- Haspel, J.A.; Choi, A.M. Autophagy: A core cellular process with emerging links to pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1237–1246. [Google Scholar] [CrossRef]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Pabon Porras, M.A.; Choi, A.M. Autophagy in Pulmonary Diseases. Am. J. Respir. Crit. Care Med. 2016, 194, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, K.; Lei, Y.; Li, Q.; Nice, E.C.; Huang, C. Redox signaling: Potential arbitrator of autophagy and apoptosis in therapeutic response. Free Radic. Biol. Med. 2015, 89, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Renna, M.; Rubinsztein, D.C. Connections between SNAREs and autophagy. Trends Biochem. Sci. 2013, 38, 57–63. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Sui, X.; Liang, X.; Chen, L.; Guo, C.; Han, W.; Pan, H.; Li, X. Bacterial xenophagy and its possible role in cancer: A potential antimicrobial strategy for cancer prevention and treatment. Autophagy 2017, 13, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.; Choi, M.E.; Hashimoto, S.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Autophagy: Friend or Foe in Lung Disease? Ann. Am. Thorac. Soc. 2016, 13, S40–S47. [Google Scholar] [PubMed]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014, 10, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Lam, H.C.; Ryter, S.W.; Choi, A.M. “Ciliophagy”: The consumption of cilia components by autophagy. Autophagy 2014, 10, 532–534. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef]
- Lang, J.D.; McArdle, P.J.; O’Reilly, P.J.; Matalon, S. Oxidant-antioxidant balance in acute lung injury. Chest 2002, 122, 314S–320S. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Wu, Y.F.; Xu, X.C.; Zhou, J.S.; Wang, Y.; Shen, H.H.; Chen, Z.H. Autophagy as a double-edged sword in pulmonary epithelial injury: A review and perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L207–L217. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Huang, C.; Fox, J.; Laturnus, D.; Carlson, E.; Zhang, B.; Yin, Q.; Gao, H.; Wu, M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J. Cell Sci. 2012, 125, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Tan, S.; Yu, M.; Jundt, M.C.; Zhang, S.; Wu, M. Annexin A2 Regulates Autophagy in Pseudomonas aeruginosa Infection through the Akt1-mTOR-ULK1/2 Signaling Pathway. J. Immunol. 2015, 195, 3901–3911. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Zhou, X.; Ye, Y.; Tan, S.; Zhang, S.; Li, R.; Yu, M.; Jundt, M.C.; Hidebrand, A.; et al. Lyn Delivers Bacteria to Lysosomes for Eradication through TLR2-Initiated Autophagy Related Phagocytosis. PLoS Pathog. 2016, 12, e1005363. [Google Scholar] [CrossRef] [PubMed]
- Jati, S.; Kundu, S.; Chakraborty, A.; Mahata, S.K.; Nizet, V.; Sen, M. Wnt5A Signaling Promotes Defense Against Bacterial Pathogens by Activating a Host Autophagy Circuit. Front. Immunol. 2018, 9, 679. [Google Scholar] [CrossRef]
- Li, X.; Ye, Y.; Zhou, X.; Huang, C.; Wu, M. Atg7 enhances host defense against infection via downregulation of superoxide but upregulation of nitric oxide. J. Immunol. 2015, 194, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Jabir, M.S.; Ritchie, N.D.; Li, D.; Bayes, H.K.; Tourlomousis, P.; Puleston, D.; Lupton, A.; Hopkins, L.; Simon, A.K.; Bryant, C.; et al. Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and beta-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe 2014, 15, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Jabir, M.S.; Hopkins, L.; Ritchie, N.D.; Ullah, I.; Bayes, H.K.; Li, D.; Tourlomousis, P.; Lupton, A.; Puleston, D.; Simon, A.K.; et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 2015, 11, 166–182. [Google Scholar] [CrossRef]
- Ye, Y.; Li, X.; Wang, W.; Ouedraogo, K.C.; Li, Y.; Gan, C.; Tan, S.; Zhou, X.; Wu, M. Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L355–L363. [Google Scholar] [CrossRef]
- Ye, Y.; Tan, S.; Zhou, X.; Li, X.; Jundt, M.C.; Lichter, N.; Hidebrand, A.; Dhasarathy, A.; Wu, M. Inhibition of p-IkappaBalpha Ubiquitylation by Autophagy-Related Gene 7 to Regulate Inflammatory Responses to Bacterial Infection. J. Infect. Dis. 2015, 212, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, J.K.; Sharma, A.; Sukumaran, P.; Sun, Y.; Mishra, B.B.; Singh, B.B.; Sharma, J. Oxidant sensor cation channel TRPM2 regulates neutrophil extracellular trap formation and protects against pneumoseptic bacterial infection. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Simonson, T.J.; Jondle, C.N.; Mishra, B.B.; Sharma, J. Mincle-Mediated Neutrophil Extracellular Trap Formation by Regulation of Autophagy. J. Infect. Dis. 2017, 215, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Lopez-Alonso, I.; Gonzalez-Lopez, A.; Amado-Rodriguez, L.; Batalla-Solis, E.; Astudillo, A.; Blazquez-Prieto, J.; Fernandez, A.F.; Galvan, J.A.; dos Santos, C.C.; et al. Defective autophagy impairs ATF3 activity and worsens lung injury during endotoxemia. J. Mol. Med. 2014, 92, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Sang, W.; Chen, S.; Chen, R.; Zhang, H.; Xue, F.; Li, Z.; Liu, Y.; Gong, Y.; Kong, X. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol. Lett. 2017, 271, 26–37. [Google Scholar] [CrossRef]
- Hu, Y.; Lou, J.; Mao, Y.Y.; Lai, T.W.; Liu, L.Y.; Zhu, C.; Zhang, C.; Liu, J.; Li, Y.Y.; Zhang, F.; et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy 2016, 12, 2286–2299. [Google Scholar] [CrossRef]
- Fan, K.; Lin, L.; Ai, Q.; Wan, J.; Dai, J.; Liu, G.; Tang, L.; Yang, Y.; Ge, P.; Jiang, R.; et al. Lipopolysaccharide-Induced Dephosphorylation of AMPK-Activated Protein Kinase Potentiates Inflammatory Injury via Repression of ULK1-Dependent Autophagy. Front. Immunol. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, J.; Ye, L.C.; Li, J.; Wu, Z.; Li, Y.; Li, C. Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J. Cell. Physiol. 2018, 233, 688–698. [Google Scholar] [CrossRef]
- Dong, W.; He, B.; Qian, H.; Liu, Q.; Wang, D.; Li, J.; Wei, Z.; Wang, Z.; Xu, Z.; Wu, G.; et al. RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury. Autophagy 2018, 14, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Slavin, S.A.; Leonard, A.; Grose, V.; Fazal, F.; Rahman, A. Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L388–L396. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Stripay, J.L.; Zhang, X.; Collage, R.D.; Hulver, M.; Carchman, E.H.; Howell, G.M.; Zuckerbraun, B.S.; Lee, J.S.; Rosengart, M.R. CaMKIalpha regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. J. Immunol. 2013, 190, 3620–3628. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, K.; Liao, L.; Zhang, T.; Yang, M.; Sun, C. Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respir. Res. 2018, 19, 243. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Kissoon, N.; Limmathurotsakul, D.; Bory, S.; Mutahunga, B.; Seymour, C.W.; Angus, D.C.; West, T.E. The global burden of sepsis: Barriers and potential solutions. Crit. Care 2018, 22, 232. [Google Scholar] [CrossRef]
- Yen, Y.T.; Yang, H.R.; Lo, H.C.; Hsieh, Y.C.; Tsai, S.C.; Hong, C.W.; Hsieh, C.H. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery 2013, 153, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; Yuan, S.S.; Hsu, C.; Cheng, Y.J.; Chang, Y.F.; Hsueh, H.W.; Lee, P.H.; Hsieh, Y.C. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann. Surg. 2013, 257, 352–363. [Google Scholar] [CrossRef]
- Mannam, P.; Shinn, A.S.; Srivastava, A.; Neamu, R.F.; Walker, W.E.; Bohanon, M.; Merkel, J.; Kang, M.J.; Dela Cruz, C.S.; Ahasic, A.M.; et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L604–L619. [Google Scholar] [CrossRef]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. 2017, 198, 3205–3213. [Google Scholar] [CrossRef]
- Park, S.Y.; Shrestha, S.; Youn, Y.J.; Kim, J.K.; Kim, S.Y.; Kim, H.J.; Park, S.H.; Ahn, W.G.; Kim, S.; Lee, M.G.; et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 577–589. [Google Scholar] [CrossRef]
- Liu, F.; Nie, C.; Zhao, N.; Wang, Y.; Liu, Y.; Li, Y.; Zeng, Z.; Ding, C.; Shao, Q.; Qing, C.; et al. MiR-155 Alleviates Septic Lung Injury by Inducing Autophagy Via Inhibition of Transforming Growth Factor-beta-Activated Binding Protein 2. Shock 2017, 48, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, N.; Chora, A.; Raquel, H.; Pejanovic, N.; Pereira, P.; Hartleben, B.; Neves-Costa, A.; Moita, C.; Pedroso, D.; Pinto, A.; et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity 2013, 39, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Ryter, S.W.; Xu, J.F.; Nakahira, K.; Kim, H.P.; Choi, A.M.; Kim, Y.S. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, S.J.; Coronata, A.A.; Fredenburgh, L.E.; Chung, S.W.; Perrella, M.A.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 2014, 20, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Dias-Freitas, F.; Metelo-Coimbra, C.; Roncon-Albuquerque, R., Jr. Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 2016, 119, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Zaher, T.E.; Miller, E.J.; Morrow, D.M.; Javdan, M.; Mantell, L.L. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic. Biol. Med. 2007, 42, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Jin, Y.; Lee, S.J.; Zhang, M.; Kim, H.P.; Stolz, D.B.; Ryter, S.W.; Choi, A.M. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am. J. Respir. Cell Mol. Biol. 2012, 46, 507–514. [Google Scholar] [CrossRef]
- Liang, X.; Wei, S.Q.; Lee, S.J.; Fung, J.K.; Zhang, M.; Tanaka, A.; Choi, A.M.; Jin, Y. p62 sequestosome 1/light chain 3b complex confers cytoprotection on lung epithelial cells after hyperoxia. Am. J. Respir. Cell Mol. Biol. 2013, 48, 489–496. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, S.; Yuan, L.J.; Wu, H.M.; Jiang, H.; Zhao, S.M.; Luo, G.; Xue, X.D. Autophagy regulates hyperoxia-induced intracellular accumulation of surfactant protein C in alveolar type II cells. Mol. Cell. Biochem. 2015, 408, 181–189. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, S.; Yuan, L.; Wu, H.; Jiang, H.; Luo, G. Hyperoxia-mediated LC3B activation contributes to the impaired transdifferentiation of type II alveolar epithelial cells (AECIIs) to type I cells (AECIs). Clin. Exp. Pharmacol. Physiol. 2016, 43, 834–843. [Google Scholar] [CrossRef]
- Ye, Y.; Lin, P.; Zhang, W.; Tan, S.; Zhou, X.; Li, R.; Pu, Q.; Koff, J.L.; Dhasarathy, A.; Ma, F.; et al. DNA Repair Interacts with Autophagy To Regulate Inflammatory Responses to Pulmonary Hyperoxia. J. Immunol. 2017, 198, 2844–2853. [Google Scholar] [CrossRef]
- Narala, V.R.; Fukumoto, J.; Hernandez-Cuervo, H.; Patil, S.S.; Krishnamurthy, S.; Breitzig, M.; Galam, L.; Soundararajan, R.; Lockey, R.F.; Kolliputi, N. Akap1 genetic deletion increases the severity of hyperoxia-induced acute lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L860–L870. [Google Scholar] [CrossRef]
- Ma, C.; Beyer, A.M.; Durand, M.; Clough, A.V.; Zhu, D.; Norwood Toro, L.; Terashvili, M.; Ebben, J.D.; Hill, R.B.; Audi, S.H.; et al. Hyperoxia Causes Mitochondrial Fragmentation in Pulmonary Endothelial Cells by Increasing Expression of Pro-Fission Proteins. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 622–635. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, G.; Sauler, M.; Lee, P.J. Lung endothelial HO-1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J. 2013, 27, 4041–4058. [Google Scholar] [CrossRef]
- Zhang, Y.; Sauler, M.; Shinn, A.S.; Gong, H.; Haslip, M.; Shan, P.; Mannam, P.; Lee, P.J. Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J. Immunol. 2014, 192, 5296–5304. [Google Scholar] [CrossRef]
- Evans, R.B. Chlorine: State of the art. Lung 2005, 183, 151–167. [Google Scholar] [CrossRef]
- Carlisle, M.; Lam, A.; Svendsen, E.R.; Aggarwal, S.; Matalon, S. Chlorine-induced cardiopulmonary injury. Ann. N. Y. Acad. Sci. 2016, 1374, 159–167. [Google Scholar] [CrossRef]
- Jurkuvenaite, A.; Benavides, G.A.; Komarova, S.; Doran, S.F.; Johnson, M.; Aggarwal, S.; Zhang, J.; Darley-Usmar, V.M.; Matalon, S. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic. Biol. Med. 2015, 85, 83–94. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Gui, Y.S.; Wang, L.; Tian, X.; Li, X.; Ma, A.; Zhou, W.; Zeng, N.; Zhang, J.; Cai, B.; Zhang, H.; et al. mTOR Overactivation and Compromised Autophagy in the Pathogenesis of Pulmonary Fibrosis. PLoS ONE 2015, 10, e0138625. [Google Scholar] [CrossRef]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, T.; Lei, Y.; Li, X.; Jiang, J.; Lan, J.; Liu, Y.; Chen, H.; Gao, W.; Xie, N.; et al. Identification of ANXA2 (annexin A2) as a specific bleomycin target to induce pulmonary fibrosis by impeding TFEB-mediated autophagic flux. Autophagy 2018, 14, 269–282. [Google Scholar] [CrossRef]
- Mi, S.; Li, Z.; Yang, H.Z.; Liu, H.; Wang, J.P.; Ma, Y.G.; Wang, X.X.; Liu, H.Z.; Sun, W.; Hu, Z.W. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J. Immunol. 2011, 187, 3003–3014. [Google Scholar] [CrossRef]
- Liu, H.; Mi, S.; Li, Z.; Hua, F.; Hu, Z.W. Interleukin 17A inhibits autophagy through activation of PIK3CA to interrupt the GSK3B-mediated degradation of BCL2 in lung epithelial cells. Autophagy 2013, 9, 730–742. [Google Scholar] [CrossRef]
- Cabrera, S.; Maciel, M.; Herrera, I.; Nava, T.; Vergara, F.; Gaxiola, M.; Lopez-Otin, C.; Selman, M.; Pardo, A. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy 2015, 11, 670–684. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef]
- Yang, H.Z.; Wang, J.P.; Mi, S.; Liu, H.Z.; Cui, B.; Yan, H.M.; Yan, J.; Li, Z.; Liu, H.; Hua, F.; et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am. J. Pathol. 2012, 180, 275–292. [Google Scholar] [CrossRef]
- Ricci, A.; Cherubini, E.; Scozzi, D.; Pietrangeli, V.; Tabbi, L.; Raffa, S.; Leone, L.; Visco, V.; Torrisi, M.R.; Bruno, P.; et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J. Cell. Physiol. 2013, 228, 1516–1524. [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Alvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Im, J.; Hergert, P.; Nho, R.S. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L552–L561. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef]
- Kurita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Ichikawa, A.; Saito, N.; Kadota, T.; Tsubouchi, K.; Sato, N.; Yoshida, M.; et al. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir. Res. 2017, 18, 114. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef]
- Ryter, S.W.; Chen, Z.H.; Kim, H.P.; Choi, A.M. Autophagy in chronic obstructive pulmonary disease: Homeostatic or pathogenic mechanism? Autophagy 2009, 5, 235–237. [Google Scholar] [CrossRef]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef]
- Kim, H.P.; Wang, X.; Chen, Z.H.; Lee, S.J.; Huang, M.H.; Wang, Y.; Ryter, S.W.; Choi, A.M. Autophagic proteins regulate cigarette smoke-induced apoptosis: Protective role of heme oxygenase-1. Autophagy 2008, 4, 887–895. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhou, J.S.; Huang, H.Q.; Li, Z.Y.; Xu, X.C.; Lai, T.W.; Hu, Y.; Zhou, H.B.; Chen, H.P.; et al. MTOR Suppresses Cigarette Smoke-Induced Epithelial Cell Death and Airway Inflammation in Chronic Obstructive Pulmonary Disease. J. Immunol. 2018, 200, 2571–2580. [Google Scholar] [CrossRef]
- Hou, H.H.; Cheng, S.L.; Chung, K.P.; Kuo, M.Y.; Yeh, C.C.; Chang, B.E.; Lu, H.H.; Wang, H.C.; Yu, C.J. Elastase induces lung epithelial cell autophagy through placental growth factor: A new insight of emphysema pathogenesis. Autophagy 2014, 10, 1509–1521. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, H.; Strulovici-Barel, Y.; Al-Hijji, M.; Ou, X.; Salit, J.; Walters, M.S.; Staudt, M.R.; Kaner, R.J.; Crystal, R.G. Role of OSGIN1 in mediating smoking-induced autophagy in the human airway epithelium. Autophagy 2017, 13, 1205–1220. [Google Scholar] [CrossRef]
- Chen, Z.H.; Lam, H.C.; Jin, Y.; Kim, H.P.; Cao, J.; Lee, S.J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.; Sun, H.; Lu, R.; Zhang, C.; Gao, N.; Meng, Q.; Wu, S.; Wang, S.; Aschner, M.; et al. Taurine ameliorates particulate matter-induced emphysema by switching on mitochondrial NADH dehydrogenase genes. Proc. Natl. Acad. Sci. USA 2017, 114, E9655–E9664. [Google Scholar] [CrossRef]
- Lv, X.X.; Liu, S.S.; Li, K.; Cui, B.; Liu, C.; Hu, Z.W. Cigarette smoke promotes COPD by activating platelet-activating factor receptor and inducing neutrophil autophagic death in mice. Oncotarget 2017, 8, 74720–74735. [Google Scholar] [CrossRef]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012, 1, 630–641. [Google Scholar] [CrossRef]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of Cigarette Smoke-Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 159–173. [Google Scholar] [CrossRef]
- Bodas, M.; Pehote, G.; Silverberg, D.; Gulbins, E.; Vij, N. Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and COPD-emphysema pathogenesis. Free Radic. Biol. Med. 2018, 131, 81–97. [Google Scholar] [CrossRef]
- Bodas, M.; Patel, N.; Silverberg, D.; Walworth, K.; Vij, N. Master Autophagy Regulator Transcription Factor EB Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Antioxid. Redox Signal. 2017, 27, 150–167. [Google Scholar] [CrossRef]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2018. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef]
- Chandra, P.; Kumar, D. Selective autophagy gets more selective: Uncoupling of autophagy flux and xenophagy flux in Mycobacterium tuberculosis-infected macrophages. Autophagy 2016, 12, 608–609. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef]
- Castillo, E.F.; Dekonenko, A.; Arko-Mensah, J.; Mandell, M.A.; Dupont, N.; Jiang, S.; Delgado-Vargas, M.; Timmins, G.S.; Bhattacharya, D.; Yang, H.; et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, E3168–E3176. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, H.M.; Park, K.S.; Shin, D.M.; Kim, T.S.; Kim, Y.S.; Suh, H.W.; Kim, S.Y.; Kim, I.S.; Kim, J.M.; et al. MIR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy 2017, 13, 423–441. [Google Scholar] [CrossRef]
- Tateosian, N.L.; Pellegrini, J.M.; Amiano, N.O.; Rolandelli, A.; Casco, N.; Palmero, D.J.; Colombo, M.I.; Garcia, V.E. IL17A augments autophagy in Mycobacterium tuberculosis-infected monocytes from patients with active tuberculosis in association with the severity of the disease. Autophagy 2017, 13, 1191–1204. [Google Scholar] [CrossRef]
- Etna, M.P.; Sinigaglia, A.; Grassi, A.; Giacomini, E.; Romagnoli, A.; Pardini, M.; Severa, M.; Cruciani, M.; Rizzo, F.; Anastasiadou, E.; et al. Mycobacterium tuberculosis-induced miR-155 subverts autophagy by targeting ATG3 in human dendritic cells. PLoS Pathog. 2018, 14, e1006790. [Google Scholar] [CrossRef]
- Fine, K.L.; Metcalfe, M.G.; White, E.; Virji, M.; Karls, R.K.; Quinn, F.D. Involvement of the autophagy pathway in trafficking of Mycobacterium tuberculosis bacilli through cultured human type II epithelial cells. Cell. Microbiol. 2012, 14, 1402–1414. [Google Scholar] [CrossRef]
- Romagnoli, A.; Etna, M.P.; Giacomini, E.; Pardini, M.; Remoli, M.E.; Corazzari, M.; Falasca, L.; Goletti, D.; Gafa, V.; Simeone, R.; et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 2012, 8, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stallings, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef]
- Villella, V.R.; Esposito, S.; Bruscia, E.M.; Maiuri, M.C.; Raia, V.; Kroemer, G.; Maiuri, L. Targeting the Intracellular Environment in Cystic Fibrosis: Restoring Autophagy as a Novel Strategy to Circumvent the CFTR Defect. Front. Pharmacol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Junkins, R.D.; McCormick, C.; Lin, T.J. The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy 2014, 10, 538–547. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Villella, V.R.; Esposito, S.; Gavina, M.; Russo, I.; Silano, M.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; Scholte, B.; et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DeltaF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012, 8, 1657–1672. [Google Scholar] [CrossRef]
- Mayer, M.L.; Blohmke, C.J.; Falsafi, R.; Fjell, C.D.; Madera, L.; Turvey, S.E.; Hancock, R.E. Rescue of dysfunctional autophagy attenuates hyperinflammatory responses from cystic fibrosis cells. J. Immunol. 2013, 190, 1227–1238. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Brown, K.; Shang, S.; Tamayo, M.H.; Hegyi, K.; Grimsey, N.J.; Cusens, D.; Coulter, S.; Cooper, J.; et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Investig. 2011, 121, 3554–3563. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Khweek, A.A.; Akhter, A.; Caution, K.; Kotrange, S.; Abdelaziz, D.H.; Newland, C.; Rosales-Reyes, R.; Kopp, B.; McCoy, K.; et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011, 7, 1359–1370. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Khweek, A.A.; Akhter, A.; Caution, K.; Tazi, M.; Hassan, H.; Zhang, Y.; Rowland, P.D.; Malhotra, S.; Aeffner, F.; et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr DeltaF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J. Biol. Chem. 2013, 288, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Potoka, K.C.; Champion, H.C.; Mora, A.L.; Gladwin, M.T. Pulmonary arterial hypertension: The clinical syndrome. Circ. Res. 2014, 115, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Smith, A.; Guo, L.; Alastalo, T.P.; Li, M.; Sawada, H.; Liu, X.; Chen, Z.H.; Ifedigbo, E.; Jin, Y.; et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2011, 183, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Wang, X.; Tian, X.; Zhang, S.; Yang, F.; Guo, H.; Fan, R.; Feng, N.; Jia, M.; et al. The Protective Effects of Kappa-Opioid Receptor Stimulation in Hypoxic Pulmonary Hypertension Involve Inhibition of Autophagy Through the AMPK-MTOR Pathway. Cell. Physiol. Biochem. 2017, 44, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Chen, Y.; Zhang, P.; Lin, P.; Xie, N.; Wu, M. Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases. Cells 2019, 8, 123. https://doi.org/10.3390/cells8020123
Wang K, Chen Y, Zhang P, Lin P, Xie N, Wu M. Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases. Cells. 2019; 8(2):123. https://doi.org/10.3390/cells8020123
Chicago/Turabian StyleWang, Kui, Yi Chen, Pengju Zhang, Ping Lin, Na Xie, and Min Wu. 2019. "Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases" Cells 8, no. 2: 123. https://doi.org/10.3390/cells8020123
APA StyleWang, K., Chen, Y., Zhang, P., Lin, P., Xie, N., & Wu, M. (2019). Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases. Cells, 8(2), 123. https://doi.org/10.3390/cells8020123