Revisiting Telomere Shortening in Cancer


Abstract
:
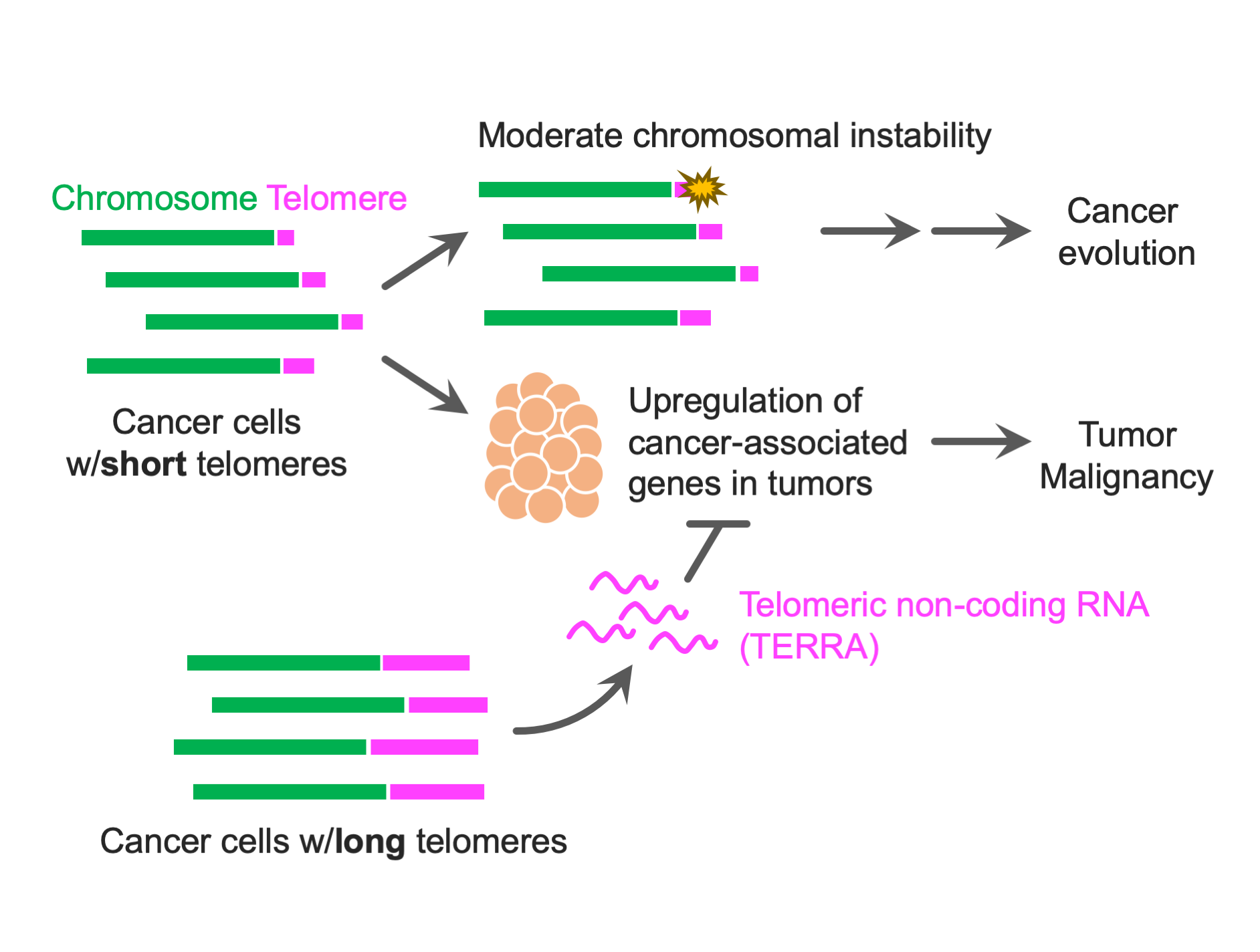{kind=link}
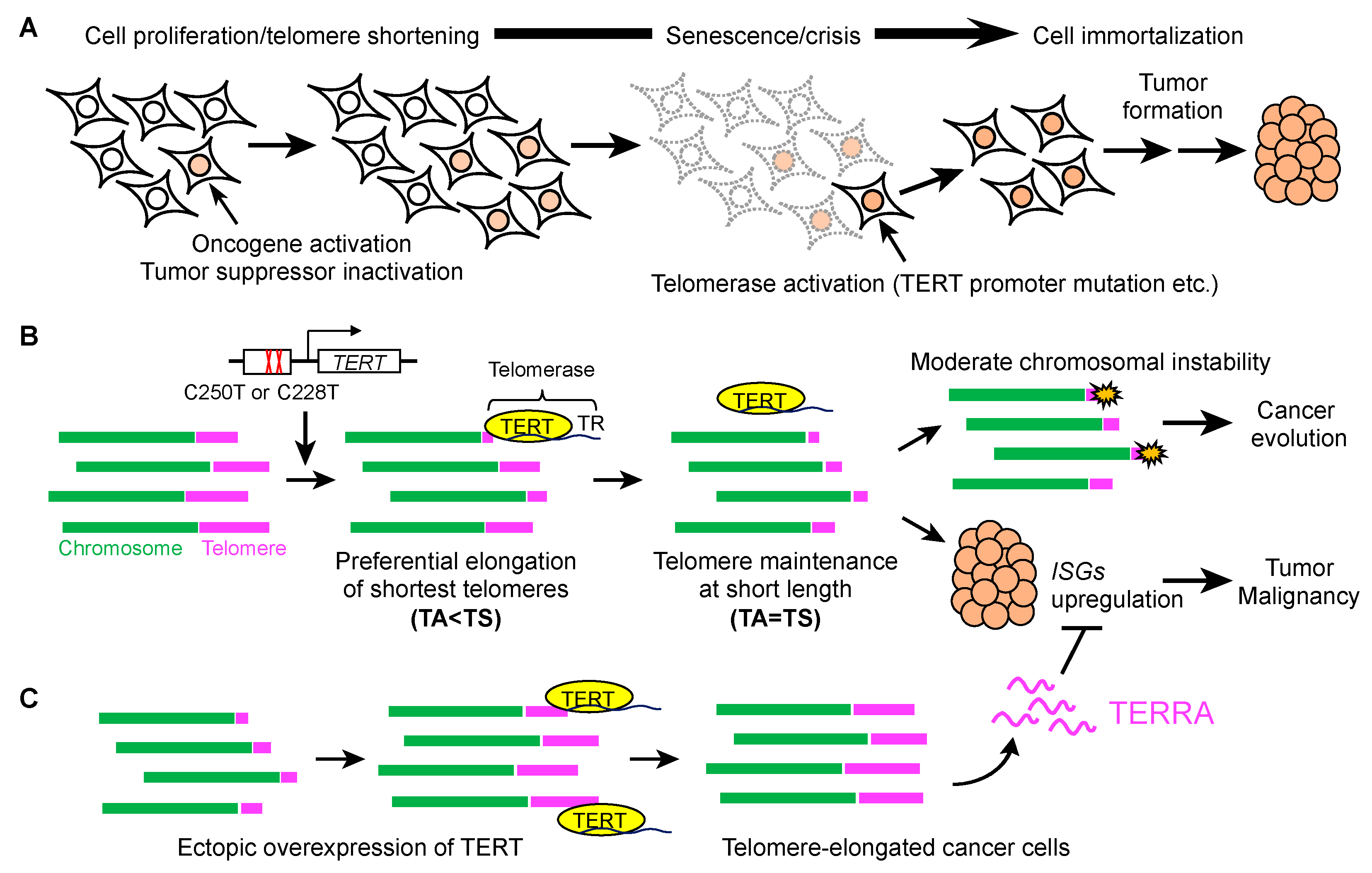{kind=link}
1. Introduction
2. TERT Promoter Mutations in Cancer
3. Epigenetic Regulation of TERT Expression
4. Telomere Shortening in Cancer
5. Potential Advantage of Shortened Telomeres in Cancer
6. Regulation of Gene Expression by Telomeres
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, H.D.; West, S.C.; Beattie, T.L. InTERTpreting telomerase structure and function. Nucleic Acids Res. 2010, 38, 5609–5622. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, D.; Wan, M.; Safari, A.; Kim, H.; Sun, W.; O’Connor, M.S.; Songyang, Z. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 2007, 445, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Counter, C.M.; Eaton, E.N.; Ellisen, L.W.; Steiner, P.; Caddle, S.D.; Ziaugra, L.; Beijersbergen, R.L.; Davidoff, M.J.; Liu, Q.; et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997, 90, 785–795. [Google Scholar] [CrossRef]
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999, 59, 551–557. [Google Scholar]
- Horikawa, I.; Cable, P.L.; Afshari, C.; Barrett, J.C. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999, 59, 826–830. [Google Scholar]
- Cong, Y.S.; Wen, J.; Bacchetti, S. The human telomerase catalytic subunit hTERT: Organization of the gene and characterization of the promoter. Hum. Mol. Genet. 1999, 8, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Fujiwara, T.; Inoue, M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008, 99, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Renkawitz, R.; Bartkuhn, M. Insulators and domains of gene expression. Curr. Opin. Genet. Dev. 2016, 37, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yu, N.K.; Kaang, B.K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Abdullaev, Z.; Guilleret, I.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007, 35, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Eldholm, V.; Haugen, A.; Zienolddiny, S. CTCF mediates the TERT enhancer-promoter interactions in lung cancer cells: Identification of a novel enhancer region involved in the regulation of TERT gene. Int. J. Cancer 2014, 134, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 1999, 274, 13085–13090. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, J.; Ohmichi, M.; Takahashi, T.; Ohshima, C.; Mabuchi, S.; Takahashi, K.; Igarashi, H.; Mori-Abe, A.; Saitoh, M.; Du, B.; et al. Raloxifene inhibits estrogen-induced up-regulation of telomerase activity in a human breast cancer cell line. J. Biol. Chem. 2003, 278, 43363–43372. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ohmichi, M.; Kawagoe, J.; Kyo, S.; Mabuchi, S.; Takahashi, T.; Ohshima, C.; Arimoto-Ishida, E.; Nishio, Y.; Inoue, M.; et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene 2004, 23, 4505–4515. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, J.R., 3rd; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Multani, A.S.; Cosme-Blanco, W.; Tahara, H.; Ma, J.; Pathak, S.; Deng, Y.; Chang, S. POT1b protects telomeres from end-to-end chromosomal fusions and aberrant homologous recombination. Embo J. 2006, 25, 5180–5190. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Daniels, J.P.; Takai, H.; de Lange, T. Recent expansion of the telomeric complex in rodents: Two distinct POT1 proteins protect mouse telomeres. Cell 2006, 126, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Multani, A.S.; He, H.; Cosme-Blanco, W.; Deng, Y.; Deng, J.M.; Bachilo, O.; Pathak, S.; Tahara, H.; Bailey, S.M.; et al. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 2006, 126, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Van Ly, D.; Low, R.R.J.; Frolich, S.; Bartolec, T.K.; Kafer, G.R.; Pickett, H.A.; Gaus, K.; Cesare, A.J. Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell 2018, 71, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Stansel, R.M.; de Lange, T.; Griffith, J.D. T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. Embo J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef]
- Bianchi, A.; Smith, S.; Chong, L.; Elias, P.; de Lange, T. TRF1 is a dimer and bends telomeric DNA. Embo J. 1997, 16, 1785–1794. [Google Scholar] [CrossRef]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.Z.; Hockemeyer, D.; Krutchinsky, A.N.; Loayza, D.; Hooper, S.M.; Chait, B.T.; de Lange, T. POT1-interacting protein PIP1: A telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004, 18, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Safari, A.; O’Connor, M.S.; Chan, D.W.; Laegeler, A.; Qin, J.; Songyang, Z. PTOP interacts with POT1 and regulates its localization to telomeres. Nat. Cell Biol. 2004, 6, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Houghtaling, B.R.; Cuttonaro, L.; Chang, W.; Smith, S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 2004, 14, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Iwano, T.; Tachibana, M.; Shinkai, Y. Distinct roles of TRF1 in the regulation of telomere structure and lengthening. J. Biol. Chem. 2008, 283, 23981–23988. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, C.; Kurth, I.; Lingner, J. Human protection of telomeres 1 (POT1) is a negative regulator of telomerase activity in vitro. Mol. Cell. Biol. 2005, 25, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, J.; Bell, C.F.; Weidenfeld, I.; Zaug, A.J.; Leinwand, L.A.; Cech, T.R. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 2012, 492, 285–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L.Y.; Han, X.; Xie, W.; Kim, H.; Yang, D.; Liu, D.; Songyang, Z. Phosphorylation of TPP1 regulates cell cycle-dependent telomerase recruitment. Proc. Natl. Acad. Sci. USA 2013, 110, 5457–5462. [Google Scholar] [CrossRef]
- Hirai, Y.; Tamura, M.; Otani, J.; Ishikawa, F. NEK6-mediated phosphorylation of human TPP1 regulates telomere length through telomerase recruitment. Genes Cells 2016, 21, 874–889. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed]
- Koelsche, C.; Renner, M.; Hartmann, W.; Brandt, R.; Lehner, B.; Waldburger, N.; Alldinger, I.; Schmitt, T.; Egerer, G.; Penzel, R.; et al. TERT promoter hotspot mutations are recurrent in myxoid liposarcomas but rare in other soft tissue sarcoma entities. J. Exp. Clin. Cancer Res. 2014, 33, 33. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Reddel, R.R. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 2087. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.C.; Ayhan, A.; Maeda, D.; Kim, K.R.; Clarke, B.A.; Shaw, P.; Chui, M.H.; Rosen, B.; Shih Ie, M.; Wang, T.L. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J. Pathol. 2014, 232, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A.; Miller, J.J.; Tummala, S.S.; Penson, T.; Iafrate, A.J.; Juratli, T.A.; Cahill, D.P. TERT promoter wild-type glioblastomas show distinct clinical features and frequent PI3K pathway mutations. Acta Neuropathol. Commun. 2018, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Suryo Rahmanto, Y.; Jung, J.G.; Wu, R.C.; Kobayashi, Y.; Heaphy, C.M.; Meeker, A.K.; Wang, T.L.; Shih Ie, M. Inactivating ARID1A Tumor Suppressor Enhances TERT Transcription and Maintains Telomere Length in Cancer Cells. J. Biol. Chem. 2016, 291, 9690–9699. [Google Scholar] [CrossRef] [PubMed]
- Rusinek, D.; Pfeifer, A.; Krajewska, J.; Oczko-Wojciechowska, M.; Handkiewicz-Junak, D.; Pawlaczek, A.; Zebracka-Gala, J.; Kowalska, M.; Cyplinska, R.; Zembala-Nozynska, E.; et al. Coexistence of TERT Promoter Mutations and the BRAF V600E Alteration and Its Impact on Histopathological Features of Papillary Thyroid Carcinoma in a Selected Series of Polish Patients. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Hipskind, R.A.; Buscher, D.; Nordheim, A.; Baccarini, M. Ras/MAP kinase-dependent and -independent signaling pathways target distinct ternary complex factors. Genes Dev. 1994, 8, 1803–1816. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Shore, P.; Sharrocks, A.D.; Davis, R.J. Integration of MAP kinase signal transduction pathways at the serum response element. Science 1995, 269, 403–407. [Google Scholar] [CrossRef]
- Vallarelli, A.F.; Rachakonda, P.S.; Andre, J.; Heidenreich, B.; Riffaud, L.; Bensussan, A.; Kumar, R.; Dumaz, N. TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation. Oncotarget 2016, 7, 53127–53136. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef]
- Devereux, T.R.; Horikawa, I.; Anna, C.H.; Annab, L.A.; Afshari, C.A.; Barrett, J.C. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 1999, 59, 6087–6090. [Google Scholar]
- Shin, K.H.; Kang, M.K.; Dicterow, E.; Park, N.H. Hypermethylation of the hTERT promoter inhibits the expression of telomerase activity in normal oral fibroblasts and senescent normal oral keratinocytes. Br. J. Cancer 2003, 89, 1473–1478. [Google Scholar] [CrossRef]
- Dessain, S.K.; Yu, H.; Reddel, R.R.; Beijersbergen, R.L.; Weinberg, R.A. Methylation of the human telomerase gene CpG island. Cancer Res. 2000, 60, 537–541. [Google Scholar]
- Zinn, R.L.; Pruitt, K.; Eguchi, S.; Baylin, S.B.; Herman, J.G. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007, 67, 194–201. [Google Scholar] [CrossRef]
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nature Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Stern, J.L.; Theodorescu, D.; Vogelstein, B.; Papadopoulos, N.; Cech, T.R. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. 2015, 29, 2219–2224. [Google Scholar] [CrossRef]
- Huang, F.W.; Bielski, C.M.; Rinne, M.L.; Hahn, W.C.; Sellers, W.R.; Stegmeier, F.; Garraway, L.A.; Kryukov, G.V. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis 2015, 4, e176. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef]
- Garcia-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar] [CrossRef]
- Sommerfeld, H.J.; Meeker, A.K.; Piatyszek, M.A.; Bova, G.S.; Shay, J.W.; Coffey, D.S. Telomerase activity: A prevalent marker of malignant human prostate tissue. Cancer Res. 1996, 56, 218–222. [Google Scholar]
- Meeker, A.K.; Hicks, J.L.; Platz, E.A.; March, G.E.; Bennett, C.J.; Delannoy, M.J.; De Marzo, A.M. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res. 2002, 62, 6405–6409. [Google Scholar]
- Chiba, K.; Lorbeer, F.K.; Shain, A.H.; McSwiggen, D.T.; Schruf, E.; Oh, A.; Ryu, J.; Darzacq, X.; Bastian, B.C.; Hockemeyer, D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, 1416–1420. [Google Scholar] [CrossRef]
- Hayflick, L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Wright, W.E.; Brasiskyte, D.; Piatyszek, M.A.; Shay, J.W. Experimental elongation of telomeres extends the lifespan of immortal x normal cell hybrids. Embo J. 1996, 15, 1734–1741. [Google Scholar] [CrossRef]
- Vaziri, H.; West, M.D.; Allsopp, R.C.; Davison, T.S.; Wu, Y.S.; Arrowsmith, C.H.; Poirier, G.G.; Benchimol, S. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. Embo J. 1997, 16, 6018–6033. [Google Scholar] [CrossRef]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar] [CrossRef]
- Preto, A.; Singhrao, S.K.; Haughton, M.F.; Kipling, D.; Wynford-Thomas, D.; Jones, C.J. Telomere erosion triggers growth arrest but not cell death in human cancer cells retaining wild-type p53: Implications for antitelomerase therapy. Oncogene 2004, 23, 4136–4145. [Google Scholar] [CrossRef]
- Chiba, K.; Johnson, J.Z.; Vogan, J.M.; Wagner, T.; Boyle, J.M.; Hockemeyer, D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife 2015, 4. [Google Scholar] [CrossRef]
- Yi, X.; Shay, J.W.; Wright, W.E. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 2001, 29, 4818–4825. [Google Scholar] [CrossRef]
- Xi, L.; Schmidt, J.C.; Zaug, A.J.; Ascarrunz, D.R.; Cech, T.R. A novel two-step genome editing strategy with CRISPR-Cas9 provides new insights into telomerase action and TERT gene expression. Genome Biol. 2015, 16, 231. [Google Scholar] [CrossRef]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Ouellette, M.M.; Liao, M.; Herbert, B.S.; Johnson, M.; Holt, S.E.; Liss, H.S.; Shay, J.W.; Wright, W.E. Subsenescent telomere lengths in fibroblasts immortalized by limiting amounts of telomerase. J. Biol. Chem. 2000, 275, 10072–10076. [Google Scholar] [CrossRef]
- Samper, E.; Flores, J.M.; Blasco, M.A. Restoration of telomerase activity rescues chromosomal instability and premature aging in Terc-/- mice with short telomeres. EMBO Rep. 2001, 2, 800–807. [Google Scholar] [CrossRef]
- Chang, M.; Arneric, M.; Lingner, J. Telomerase repeat addition processivity is increased at critically short telomeres in a Tel1-dependent manner in Saccharomyces cerevisiae. Genes Dev. 2007, 21, 2485–2494. [Google Scholar] [CrossRef]
- Liu, Q.; Turner, K.M.; Alfred Yung, W.K.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncol. 2014, 16, 1313–1323. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Nehyba, J.; Bose, H.R., Jr. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol. Cell. Biol. 2012, 32, 4283–4296. [Google Scholar] [CrossRef]
- Wong, M.S.; Wright, W.E.; Shay, J.W. Alternative splicing regulation of telomerase: A new paradigm? Trends Genet. 2014, 30, 430–438. [Google Scholar] [CrossRef]
- Colgin, L.M.; Wilkinson, C.; Englezou, A.; Kilian, A.; Robinson, M.O.; Reddel, R.R. The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia 2000, 2, 426–432. [Google Scholar] [CrossRef]
- Lincz, L.F.; Mudge, L.M.; Scorgie, F.E.; Sakoff, J.A.; Hamilton, C.S.; Seldon, M. Quantification of hTERT splice variants in melanoma by SYBR green real-time polymerase chain reaction indicates a negative regulatory role for the beta deletion variant. Neoplasia 2008, 10, 1131–1137. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative Splicing of hTERT Pre-mRNA: A Potential Strategy for the Regulation of Telomerase Activity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Saeboe-Larssen, S.; Fossberg, E.; Gaudernack, G. Characterization of novel alternative splicing sites in human telomerase reverse transcriptase (hTERT): Analysis of expression and mutual correlation in mRNA isoforms from normal and tumour tissues. BMC Mol. Biol. 2006, 7, 26. [Google Scholar] [CrossRef]
- Rha, S.Y.; Jeung, H.C.; Park, K.H.; Kim, J.J.; Chung, H.C. Changes of telomerase activity by alternative splicing of full-length and beta variants of hTERT in breast cancer patients. Oncol. Res. 2009, 18, 213–220. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Tissue-specific alternate splicing of human telomerase reverse transcriptase (hTERT) influences telomere lengths during human development. Int. J. Cancer 2001, 91, 644–649. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998, 58, 4168–4172. [Google Scholar]
- Ozden, S.; Tiber, P.M.; Ozgen, Z.; Ozyurt, H.; Serakinci, N.; Orun, O. Expression of TRF2 and its prognostic relevance in advanced stage cervical cancer patients. Biol. Res. 2014, 47, 61. [Google Scholar] [CrossRef]
- Panero, J.; Stanganelli, C.; Arbelbide, J.; Fantl, D.B.; Kohan, D.; Garcia Rivello, H.; Rabinovich, G.A.; Slavutsky, I. Expression profile of shelterin components in plasma cell disorders. Clinical significance of POT1 overexpression. Blood Cells Mol. Dis. 2014, 52, 134–139. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Li, F.; Lin, W.; Liang, Y.; Ma, Z. Expression of Telomere Repeat Binding Factor 1 and TRF2 in Prostate Cancer and Correlation with Clinical Parameters. BioMed. Res. Int. 2017, 2017, 9764752. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Y.; Zou, M.; Yang, S.; Liang, X.Q. Expression of TRF1, TRF2, TIN2, TERT, KU70, and BRCA1 proteins is associated with telomere shortening and may contribute to multistage carcinogenesis of gastric cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1407–1414. [Google Scholar] [CrossRef]
- Oh, B.K.; Kim, Y.J.; Park, C.; Park, Y.N. Up-regulation of telomere-binding proteins, TRF1, TRF2, and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am. J. Pathol. 2005, 166, 73–80. [Google Scholar] [CrossRef]
- Gay-Bellile, M.; Romero, P.; Cayre, A.; Veronese, L.; Privat, M.; Singh, S.; Combes, P.; Kwiatkowski, F.; Abrial, C.; Bignon, Y.J.; et al. ERCC1 and telomere status in breast tumours treated with neoadjuvant chemotherapy and their association with patient prognosis. J. Pathol. Clin. Res. 2016, 2, 234–246. [Google Scholar] [CrossRef]
- Butler, K.S.; Hines, W.C.; Heaphy, C.M.; Griffith, J.K. Coordinate regulation between expression levels of telomere-binding proteins and telomere length in breast carcinomas. Cancer Med. 2012, 1, 165–175. [Google Scholar] [CrossRef]
- De Bie, J.; Demeyer, S.; Alberti-Servera, L.; Geerdens, E.; Segers, H.; Broux, M.; De Keersmaecker, K.; Michaux, L.; Vandenberghe, P.; Voet, T.; et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 1358–1369. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Y.; Di, J.; Su, Z.; Yang, H.; Jiang, B.; Wang, Z.; Zhuang, M.; Bai, F.; Su, X. Multi-region and single-cell sequencing reveal variable genomic heterogeneity in rectal cancer. BMC Cancer 2017, 17, 787. [Google Scholar] [CrossRef]
- Winterhoff, B.J.; Maile, M.; Mitra, A.K.; Sebe, A.; Bazzaro, M.; Geller, M.A.; Abrahante, J.E.; Klein, M.; Hellweg, R.; Mullany, S.A.; et al. Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells. Gynecologic Oncol. 2017, 144, 598–606. [Google Scholar] [CrossRef]
- Huang, E.E.; Tedone, E.; O’Hara, R.; Cornelius, C.; Lai, T.P.; Ludlow, A.; Wright, W.E.; Shay, J.W. The maintenance of telomere length in CD28+ T cells during T lymphocyte stimulation. Sci. Rep. 2017, 7, 6785. [Google Scholar] [CrossRef]
- Hahn, W.C.; Stewart, S.A.; Brooks, M.W.; York, S.G.; Eaton, E.; Kurachi, A.; Beijersbergen, R.L.; Knoll, J.H.; Meyerson, M.; Weinberg, R.A. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 1999, 5, 1164–1170. [Google Scholar] [CrossRef]
- Seimiya, H.; Muramatsu, Y.; Ohishi, T.; Tsuruo, T. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 2005, 7, 25–37. [Google Scholar] [CrossRef]
- Seimiya, H.; Oh-hara, T.; Suzuki, T.; Naasani, I.; Shimazaki, T.; Tsuchiya, K.; Tsuruo, T. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Mol. Cancer Ther. 2002, 1, 657–665. [Google Scholar]
- Frink, R.E.; Peyton, M.; Schiller, J.H.; Gazdar, A.F.; Shay, J.W.; Minna, J.D. Telomerase inhibitor imetelstat has preclinical activity across the spectrum of non-small cell lung cancer oncogenotypes in a telomere length dependent manner. Oncotarget 2016, 7, 31639–31651. [Google Scholar] [CrossRef]
- Fujiwara, C.; Muramatsu, Y.; Nishii, M.; Tokunaka, K.; Tahara, H.; Ueno, M.; Yamori, T.; Sugimoto, Y.; Seimiya, H. Cell-based chemical fingerprinting identifies telomeres and lamin A as modifiers of DNA damage response in cancer cells. Sci. Rep. 2018, 8, 14827. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef]
- Shore, D.; Bianchi, A. Telomere length regulation: Coupling DNA end processing to feedback regulation of telomerase. Embo J. 2009, 28, 2309–2322. [Google Scholar] [CrossRef]
- Marcand, S.; Gilson, E.; Shore, D. A protein-counting mechanism for telomere length regulation in yeast. Science 1997, 275, 986–990. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef]
- Hirashima, K.; Migita, T.; Sato, S.; Muramatsu, Y.; Ishikawa, Y.; Seimiya, H. Telomere length influences cancer cell differentiation in vivo. Mol. Cell. Biol. 2013, 33, 2988–2995. [Google Scholar] [CrossRef]
- Derycke, L.D.; Bracke, M.E. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 2004, 48, 463–476. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Chin, Y.E.; Kitagawa, M.; Su, W.C.; You, Z.H.; Iwamoto, Y.; Fu, X.Y. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Horvath, C.M.; Wen, Z.; Schreiber, R.D.; Darnell, J.E., Jr. Transcriptionally active STAT1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 7673–7678. [Google Scholar] [CrossRef]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar]
- Greenwood, C.; Metodieva, G.; Al-Janabi, K.; Lausen, B.; Alldridge, L.; Leng, L.; Bucala, R.; Fernandez, N.; Metodiev, M.V. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J. Proteom. 2012, 75, 3031–3040. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Z.; Xu, Q.; Zhang, Y.; Zhai, Y.; Bai, J.; Liu, M.; Hui, Z.; Xu, N. Inhibition of STAT1 sensitizes renal cell carcinoma cells to radiotherapy and chemotherapy. Cancer Biol. Therapy 2012, 13, 401–407. [Google Scholar] [CrossRef]
- Tsai, M.H.; Cook, J.A.; Chandramouli, G.V.; DeGraff, W.; Yan, H.; Zhao, S.; Coleman, C.N.; Mitchell, J.B.; Chuang, E.Y. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007, 67, 3845–3852. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Beckett, M.; Labay, E.; Darga, T.; Roizman, B.; Weichselbaum, R.R. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1714–1719. [Google Scholar] [CrossRef]
- Roberts, D.; Schick, J.; Conway, S.; Biade, S.; Laub, P.B.; Stevenson, J.P.; Hamilton, T.C.; O’Dwyer, P.J.; Johnson, S.W. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br. J. Cancer 2005, 92, 1149–1158. [Google Scholar] [CrossRef]
- Fryknas, M.; Dhar, S.; Oberg, F.; Rickardson, L.; Rydaker, M.; Goransson, H.; Gustafsson, M.; Pettersson, U.; Nygren, P.; Larsson, R.; et al. STAT1 signaling is associated with acquired crossresistance to doxorubicin and radiation in myeloma cell lines. Int. J. Cancer 2007, 120, 189–195. [Google Scholar] [CrossRef]
- Rickardson, L.; Fryknas, M.; Dhar, S.; Lovborg, H.; Gullbo, J.; Rydaker, M.; Nygren, P.; Gustafsson, M.G.; Larsson, R.; Isaksson, A. Identification of molecular mechanisms for cellular drug resistance by combining drug activity and gene expression profiles. Br. J. Cancer 2005, 93, 483–492. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Roach, P.; Pitroda, S.P.; Golden, D.W.; Bhayani, M.; Shao, M.Y.; Darga, T.E.; Beveridge, M.G.; Sood, R.F.; Sutton, H.G.; et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS ONE 2009, 4, e5821. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Qadir, A.S.; Ceppi, P.; Brockway, S.; Law, C.; Mu, L.; Khodarev, N.N.; Kim, J.; Zhao, J.C.; Putzbach, W.; Murmann, A.E.; et al. CD95/Fas increases stemness in cancer cells by inducing a STAT1-dependent type I interferon response. Cell Rep. 2017, 18, 2373–2386. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Martin, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Zhang, H.; Zhu, M.; Chen, F.; Hu, Y.; Liu, H.; Zhu, H. Interferon-stimulated gene 15 (ISG15) is a trigger for tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget 2014, 5, 8429–8441. [Google Scholar] [CrossRef]
- Forys, J.T.; Kuzmicki, C.E.; Saporita, A.J.; Winkeler, C.L.; Maggi, L.B., Jr.; Weber, J.D. ARF and p53 coordinate tumor suppression of an oncogenic IFN-beta-STAT1-ISG15 signaling axis. Cell Rep. 2014, 7, 514–526. [Google Scholar] [CrossRef]
- Duarte, C.W.; Willey, C.D.; Zhi, D.; Cui, X.; Harris, J.J.; Vaughan, L.K.; Mehta, T.; McCubrey, R.O.; Khodarev, N.N.; Weichselbaum, R.R.; et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS ONE 2012, 7, e29653. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.E.; Kim, Y.H.; Kleihues, P.; Ohgaki, H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.L.; McDonnell, J.; Muntoni, A.; Henson, J.D.; Hegi, M.E.; von Deimling, A.; Wheeler, H.R.; Cook, R.J.; Biggs, M.T.; Little, N.S.; et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J. Neuropathol. Exp. Neurol. 2010, 69, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, D.E.; Aparicio, O.M.; Billington, B.L.; Zakian, V.A. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 1990, 63, 751–762. [Google Scholar] [CrossRef]
- Lou, Z.; Wei, J.; Riethman, H.; Baur, J.A.; Voglauer, R.; Shay, J.W.; Wright, W.E. Telomere length regulates ISG15 expression in human cells. Aging 2009, 1, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.K.; Sharma, S.; Sengupta, S.; Saha, D.; Kumar, P.; Hussain, T.; Srivastava, V.; Roy, S.D.; Shay, J.W.; Chowdhury, S. Telomere length-dependent transcription and epigenetic modifications in promoters remote from telomere ends. PLos Genet. 2018, 14, e1007782. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.J.; López-Silanes, I.; Megías, D.; Fraga, M.F.; Castells-García, Á.; Blasco, M.A. TERRA recruitment of polycomb to telomeres is essential for histone trymethylation marks at telomeric heterochromatin. Nat. Commun. 2018, 9, 1548. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Feuerhahn, S.; Lingner, J. TERRA-reinforced association of LSD1 with MRE11 promotes processing of uncapped telomeres. Cell Rep. 2014, 6, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Stong, N.; Plasschaert, R.; Moczan, A.; Chen, H.S.; Hu, S.; Wikramasinghe, P.; Davuluri, R.V.; Bartolomei, M.S.; et al. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. Embo J. 2012, 31, 4165–4178. [Google Scholar] [CrossRef]
- Redon, S.; Zemp, I.; Lingner, J. A three-state model for the regulation of telomerase by TERRA and hnRNPA1. Nucleic Acids Res. 2013, 41, 9117–9128. [Google Scholar] [CrossRef]
- Hirashima, K.; Seimiya, H. Telomeric repeat-containing RNA/G-quadruplex-forming sequences cause genome-wide alteration of gene expression in human cancer cells in vivo. Nucleic Acids Res. 2015, 43, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, Z.; Dahmane, N.; Tsai, K.; Wang, P.; Williams, D.R.; Kossenkov, A.V.; Showe, L.C.; Zhang, R.; Huang, Q.; et al. Telomeric repeat-containing RNA (TERRA) constitutes a nucleoprotein component of extracellular inflammatory exosomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6293–E6300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lieberman, P.M. The crosstalk of telomere dysfunction and inflammation through cell-free TERRA containing exosomes. RNA Biol. 2016, 13, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Arons, E.; Zhou, H.; Edelman, D.C.; Gomez, A.; Steinberg, S.M.; Petersen, D.; Wang, Y.; Meltzer, P.S.; Kreitman, R.J. Impact of telomere length on survival in classic and variant hairy cell leukemia. Leuk. Res. 2015, 39, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Weischer, M.; Nordestgaard, B.G.; Cawthon, R.M.; Freiberg, J.J.; Tybjaerg-Hansen, A.; Bojesen, S.E. Short telomere length, cancer survival, and cancer risk in 47,102 individuals. J. Natl. Cancer Inst. 2013, 105, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Seow, W.J.; Cawthon, R.M.; Purdue, M.P.; Hu, W.; Gao, Y.T.; Huang, W.Y.; Weinstein, S.J.; Ji, B.T.; Virtamo, J.; Hosgood, H.D., 3rd; et al. Telomere length in white blood cell DNA and lung cancer: A pooled analysis of three prospective cohorts. Cancer Res. 2014, 74, 4090–4098. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Hann, H.W.; Ye, Z.; Hann, R.S.; Lai, Y.; Wang, C.; Li, L.; Myers, R.E.; Li, B.; Xing, J.; et al. Prospective and longitudinal evaluations of telomere length of circulating DNA as a risk predictor of hepatocellular carcinoma in HBV patients. Carcinogenesis 2017, 38, 439–446. [Google Scholar] [CrossRef]
- Nezu, T.; Hosomi, N.; Takahashi, T.; Anno, K.; Aoki, S.; Shimamoto, A.; Maruyama, H.; Hayashi, T.; Matsumoto, M.; Tahara, H. Telomere G-tail length is a promising biomarker related to white matter lesions and endothelial dysfunction in patients with cardiovascular risk: A cross-sectional study. eBioMedicine 2015, 2, 960–967. [Google Scholar] [CrossRef]
- Seimiya, H. Predicting risk at the end of the end: Telomere G-tail as a biomarker. eBioMedicine 2015, 2, 804–805. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamoto, K.; Seimiya, H. Revisiting Telomere Shortening in Cancer. Cells 2019, 8, 107. https://doi.org/10.3390/cells8020107
Okamoto K, Seimiya H. Revisiting Telomere Shortening in Cancer. Cells. 2019; 8(2):107. https://doi.org/10.3390/cells8020107
Chicago/Turabian StyleOkamoto, Keiji, and Hiroyuki Seimiya. 2019. "Revisiting Telomere Shortening in Cancer" Cells 8, no. 2: 107. https://doi.org/10.3390/cells8020107
APA StyleOkamoto, K., & Seimiya, H. (2019). Revisiting Telomere Shortening in Cancer. Cells, 8(2), 107. https://doi.org/10.3390/cells8020107