MAP4K Family Kinases and DUSP Family Phosphatases in T-Cell Signaling and Systemic Lupus Erythematosus


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
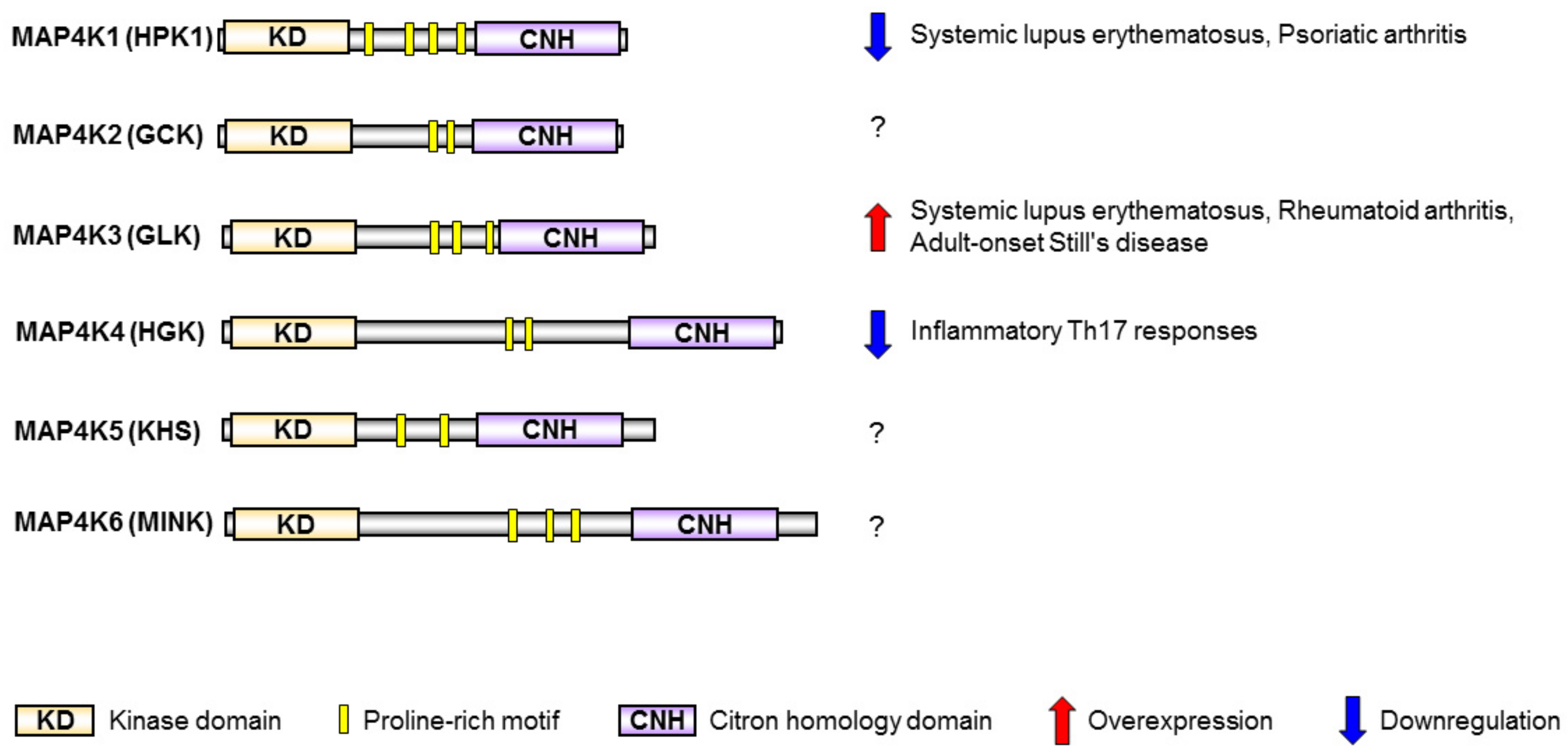
2. MAP4K Family Kinases Are Involved in T-Cell Activation and Human SLE
2.1. HPK1 Transcription Is Reduced in CD4+ T Cells of Human SLE Patients
2.2. GLK Is a Biomarker and Therapeutic Target for Human SLE
3. DUSP Family Phosphatases Are Involved in T-Cell Activation and Human SLE
3.1. DUSP22 Protein Level Is a Diagnostic and Prognostic Biomarker for SLE Nephritis
3.2. DUSP4 mRNA Level Is Increased in CD4+ T Cells of Human Juvenile-Onset SLE
3.3. DUSP23 mRNA Levels Are Increased in CD4+ T Cells of Human SLE
3.4. DUSP1, DUSP5, and DUSP14 Also Regulate T Cell-Mediated Autoimmune Responses in Mice
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MAP4K | MAP Kinase Kinase Kinase Kinase |
| DUSP | Dual-Specificity Phosphatase |
| HPK1 | Hematopoietic Progenitor Kinase 1 |
| GCK | Germinal Center Kinase |
| GLK | GCK-Like Kinase |
| HGK | HPK1/GCK-Like Kinase |
| KHS | Kinase Homologous to Sps1/Ste20 |
| MINK | Misshapen/Nck-Related Kinase |
| TCR | T-Cell Receptor |
| PKCθ | Protein Kinase C-theta |
| IKK | IκB Kinase |
| JMJD3 | Jumonji Domain-Containing Protein 3 |
| H3K27me3 | Histone H3 Lysine 27 Trimethylation |
| PRMT5 | Protein Arginine Methyltransferase 5 |
| CREMα | Camp Response Element Modulator α |
| CIA | Collagen-Induced Arthritis |
| EAE | Experimental Autoimmune Encephalomyelitis |
| SLE | Systemic Lupus Erythematosus |
| RA | Rheumatoid Arthritis |
| AOSD | Adult-Onset Still’s Disease |
| SLEDAI | SLE Disease Activity Index |
| SNP | Single-Nucleotide Polymorphism |
References
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Rich, S.S.; Raychaudhuri, S. Autoimmune diseases–connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet. 2016, 17, 160–174. [Google Scholar] [CrossRef]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef]
- Martin, J.C.; Baeten, D.L.; Josien, R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin. Immunol. 2014, 154, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chu, Y.; Yang, X.; Gao, D.; Zhu, L.; Yang, X.; Wan, L.; Li, M. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Ines, L.; Couto, M.; Pedreiro, S.; Santos, C.; Magalhaes, M.; Santos, P.; Velada, I.; Almeida, A.; Carvalheiro, T.; et al. Frequency and functional activity of Th17, Tc17 and other T-cell subsets in systemic lupus erythematosus. Cell. Immunol. 2010, 264, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Lopez, P.; Rodriguez-Carrio, J.; Caminal-Montero, L.; Mozo, L.; Suarez, A. A pathogenic IFNα, BLyS and IL-17 axis in systemic lupus erythematosus patients. Sci. Rep. 2016, 6, 20651. [Google Scholar] [CrossRef]
- Shah, K.; Lee, W.W.; Lee, S.H.; Kim, S.H.; Kang, S.W.; Craft, J.; Kang, I. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R53. [Google Scholar] [CrossRef]
- Chuang, H.C.; Chen, Y.M.; Chen, M.H.; Hung, W.T.; Yang, H.Y.; Tseng, Y.H.; Tan, T.H. AhR-RORγt complex is a therapeutic target for MAP4K3/GLKhighIL-17Ahigh subpopulation of systemic lupus erythematosus. FASEB J. 2019, 33, 11469–11480. [Google Scholar] [CrossRef]
- Chuang, H.C.; Tsai, C.Y.; Hsueh, C.H.; Tan, T.H. GLK-IKKβ signaling induces dimerization and translocation of AhR-RORγt complex in IL-17A induction and autoimmune disease. Sci. Adv. 2018, 4, eaat5401. [Google Scholar] [CrossRef]
- Chuang, H.C.; Tan, T.H. MAP4K3/GLK in autoimmune disease, cancer and aging. J. Biomed. Sci. 2019, 26, 82. [Google Scholar] [CrossRef] [PubMed]
- Corbett, M.; Soares, M.; Jhuti, G.; Rice, S.; Spackman, E.; Sideris, E.; Moe-Byrne, T.; Fox, D.; Marzo-Ortega, H.; Kay, L.; et al. Tumour necrosis factor-α inhibitors for ankylosing spondylitis and non-radiographic axial spondyloarthritis: A systematic review and economic evaluation. Health Technol. Assess. 2016, 20, 1–334. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Sanchez-Guerrero, J.; Merrill, J.T.; Furie, R.; Gladman, D.; Navarra, S.V.; Ginzler, E.M.; D’Cruz, D.P.; Doria, A.; Cooper, S.; et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: Combined results from two phase III trials. Ann. Rheum. Dis. 2012, 71, 1833–1838. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Koeller, M.; Weisman, M.H.; Emery, P. New therapies for treatment of rheumatoid arthritis. Lancet 2007, 370, 1861–1874. [Google Scholar] [CrossRef]
- Breedveld, F.C.; Combe, B. Understanding emerging treatment paradigms in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13 (Suppl. 1), S3. [Google Scholar]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Kuchroo, V.K. Th17 cell pathway in human immunity: Lessons from genetics and therapeutic interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef]
- Mahieu, M.A.; Strand, V.; Simon, L.S.; Lipsky, P.E.; Ramsey-Goldman, R. A critical review of clinical trials in systemic lupus erythematosus. Lupus 2016, 25, 1122–1140. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Bour-Jordan, H.; Cheng, M.; Anderson, M. T cells in the control of organ-specific autoimmunity. J. Clin. Invest. 2015, 125, 2250–2260. [Google Scholar] [CrossRef]
- Gorelik, G.; Richardson, B. Key role of ERK pathway signaling in lupus. Autoimmunity 2010, 43, 17–22. [Google Scholar] [CrossRef]
- Shui, J.W.; Boomer, J.S.; Han, J.; Xu, J.; Dement, G.A.; Zhou, G.; Tan, T.H. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat. Immunol. 2007, 8, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Lan, J.L.; Chen, D.Y.; Yang, C.Y.; Chen, Y.M.; Li, J.P.; Huang, C.Y.; Liu, P.E.; Wang, X.; Tan, T.H. The kinase GLK controls autoimmunity and NF-κB signaling by activating the kinase PKC-θ in T cells. Nat. Immunol. 2011, 12, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Yang, C.Y.; Chuang, H.C.; Lan, J.L.; Chen, D.Y.; Chen, Y.M.; Wang, X.; Chen, A.J.; Belmont, J.W.; Tan, T.H. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun. 2014, 5, 3618. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Overview of signal processing by the immune system in systemic lupus erythematosus. Autoimmun. Rev. 2009, 8, 177–178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molad, Y.; Amit-Vasina, M.; Bloch, O.; Yona, E.; Rapoport, M.J. Increased ERK and JNK activities correlate with disease activity in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2010, 69, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, A.; Orfanidou, T.; Liaskos, C.; Smyk, D.S.; Billinis, C.; Blank, M.; Rigopoulou, E.I.; Bogdanos, D.P. p38 mitogen-activated protein kinase (p38 MAPK)-mediated autoimmunity: Lessons to learn from ANCA vasculitis and pemphigus vulgaris. Autoimmun. Rev. 2013, 12, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Zwerina, J.; Firestein, G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 909–916. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Solit, D.B. Resistance to MEK inhibitors: Should we co-target upstream? Sci. Signal. 2011, 4, pe16. [Google Scholar] [CrossRef]
- Hammaker, D.; Firestein, G.S. “Go upstream, young man”: Lessons learned from the p38 saga. Ann. Rheum. Dis. 2010, 69 (Suppl. 1), i77–i82. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Selectivity and therapeutic inhibition of kinases: To be or not to be? Nat. Immunol. 2009, 10, 356–360. [Google Scholar] [CrossRef]
- Chuang, H.C.; Wang, X.; Tan, T.H. MAP4K family kinases in immunity and inflammation. Adv. Immunol. 2016, 129, 277–314. [Google Scholar] [PubMed]
- Chen, Y.R.; Tan, T.H. The c-Jun N-terminal kinase pathway and apoptotic signaling. Int. J. Oncol. 2000, 16, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of dual-specificity phosphatase (DUSP) ubiquitination and protein stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Qiu, W.R.; Wang, X.; Meyer, C.F.; Tan, T.H. Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. 1996, 10, 2251–2264. [Google Scholar] [CrossRef]
- Boomer, J.S.; Tan, T.H. Functional interactions of HPK1 with adaptor proteins. J. Cell. Biochem. 2005, 95, 34–44. [Google Scholar] [CrossRef]
- Ling, P.; Meyer, C.F.; Redmond, L.P.; Shui, J.W.; Davis, B.; Rich, R.R.; Hu, M.C.; Wange, R.L.; Tan, T.H. Involvement of hematopoietic progenitor kinase 1 in T cell receptor signaling. J. Biol. Chem. 2001, 276, 18908–18914. [Google Scholar] [CrossRef]
- Pombo, C.M.; Kehrl, J.H.; Sanchez, I.; Katz, P.; Avruch, J.; Zon, L.I.; Woodgett, J.R.; Force, T.; Kyriakis, J.M. Activation of the SAPK pathway by the human STE20 homologue germinal centre kinase. Nature 1995, 377, 750–754. [Google Scholar] [CrossRef]
- Diener, K.; Wang, X.S.; Chen, C.; Meyer, C.F.; Keesler, G.; Zukowski, M.; Tan, T.H.; Yao, Z. Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 9687–9692. [Google Scholar] [CrossRef]
- Yao, Z.; Zhou, G.; Wang, X.S.; Brown, A.; Diener, K.; Gan, H.; Tan, T.H. A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 1999, 274, 2118–2125. [Google Scholar] [CrossRef]
- Fiedler, L.R.; Chapman, K.; Xie, M.; Maifoshie, E.; Jenkins, M.; Golforoush, P.A.; Bellahcene, M.; Noseda, M.; Faust, D.; Jarvis, A.; et al. MAP4K4 inhibition promotes survival of human stem cell-derived cardiomyocytes and reduces infarct size in vivo. Cell Stem Cell 2019, 24, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Tung, R.M.; Blenis, J. A novel human SPS1/STE20 homologue, KHS, activates Jun N-terminal kinase. Oncogene 1997, 14, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Dan, I.; Watanabe, N.M.; Kobayashi, T.; Yamashita-Suzuki, K.; Fukagaya, Y.; Kajikawa, E.; Kimura, W.K.; Nakashima, T.M.; Matsumoto, K.; Ninomiya-Tsuji, J.; et al. Molecular cloning of MINK, a novel member of mammalian GCK family kinases, which is up-regulated during postnatal mouse cerebral development. FEBS Lett 2000, 469, 19–23. [Google Scholar] [CrossRef]
- Chen, Y.R.; Meyer, C.F.; Tan, T.H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in γ radiation-induced apoptosis. J. Biol. Chem. 1996, 271, 631–634. [Google Scholar] [CrossRef]
- Chen, Y.R.; Wang, X.; Templeton, D.; Davis, R.J.; Tan, T.H. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation: Duration of JNK activation may determine cell death and proliferation. J. Biol. Chem. 1996, 271, 31929–31936. [Google Scholar] [CrossRef]
- Hsu, C.L.; Lee, E.X.; Gordon, K.L.; Paz, E.A.; Shen, W.C.; Ohnishi, K.; Meisenhelder, J.; Hunter, T.; La Spada, A.R. MAP4K3 mediates amino acid-dependent regulation of autophagy via phosphorylation of TFEB. Nat. Commun. 2018, 9, 942. [Google Scholar] [CrossRef]
- Chuang, H.C.; Sheu, W.H.; Lin, Y.T.; Tsai, C.Y.; Yang, C.Y.; Cheng, Y.J.; Huang, P.Y.; Li, J.P.; Chiu, L.L.; Wang, X.; et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat. Commun. 2014, 5, 4602. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.P.; Kuo, H.K.; Chiu, L.L.; Dement, G.A.; Lan, J.L.; Chen, D.Y.; Yang, C.Y.; Hu, H.; Tan, T.H. Down-regulation of B cell receptor signaling by hematopoietic progenitor kinase 1 (HPK1)-mediated phosphorylation and ubiquitination of activated B cell linker protein (BLNK). J. Biol. Chem. 2012, 287, 11037–11048. [Google Scholar] [CrossRef]
- Zhang, Q.; Long, H.; Liao, J.; Zhao, M.; Liang, G.; Wu, X.; Zhang, P.; Ding, S.; Luo, S.; Lu, Q. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J. Autoimmun. 2011, 37, 180–189. [Google Scholar] [CrossRef]
- Stoeckman, A.K.; Baechler, E.C.; Ortmann, W.A.; Behrens, T.W.; Michet, C.J.; Peterson, E.J. A distinct inflammatory gene expression profile in patients with psoriatic arthritis. Genes Immun. 2006, 7, 583–591. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chuang, H.C.; Lan, J.L.; Chen, Y.M.; Hung, W.T.; Lai, K.L.; Tan, T.H. Germinal center kinase-like kinase (GLK/MAP4K3) expression is increased in adult-onset Still’s disease and may act as an activity marker. BMC Med. 2012, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Chuang, H.C.; Lin, W.C.; Tsai, C.Y.; Wu, C.W.; Gong, N.R.; Hung, W.T.; Lan, T.H.; Lan, J.L.; Tan, T.H.; et al. Germinal center kinase-like kinase overexpression in T cells as a novel biomarker in rheumatoid arthritis. Arthritis Rheum. 2013, 65, 2573–2582. [Google Scholar] [PubMed]
- Chen, Y.R.; Meyer, C.F.; Ahmed, B.; Yao, Z.; Tan, T.H. Caspase-mediated cleavage and functional changes of hematopoietic progenitor kinase 1 (HPK1). Oncogene 1999, 18, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Shui, J.W.; Zhang, X.; Zheng, B.; Han, S.; Tan, T.H. HIP-55 is important for T-cell proliferation, cytokine production, and immune responses. Mol. Cell. Biol. 2005, 25, 6869–6878. [Google Scholar] [CrossRef]
- Han, J.; Kori, R.; Shui, J.W.; Chen, Y.R.; Yao, Z.; Tan, T.H. The SH3 domain-containing adaptor HIP-55 mediates c-Jun N-terminal kinase activation in T cell receptor signaling. J. Biol. Chem. 2003, 278, 52195–52202. [Google Scholar] [CrossRef]
- Ma, W.; Xia, C.; Ling, P.; Qiu, M.; Luo, Y.; Tan, T.H.; Liu, M. Leukocyte-specific adaptor protein Grap2 interacts with hematopoietic progenitor kinase 1 (HPK1) to activate JNK signaling pathway in T lymphocytes. Oncogene 2001, 20, 1703–1714. [Google Scholar] [CrossRef]
- Ling, P.; Yao, Z.; Meyer, C.F.; Wang, X.S.; Oehrl, W.; Feller, S.M.; Tan, T.H. Interaction of hematopoietic progenitor kinase 1 with adapter proteins Crk and CrkL leads to synergistic activation of c-Jun N-terminal kinase. Mol. Cell. Biol. 1999, 19, 1359–1368. [Google Scholar] [CrossRef]
- Ensenat, D.; Yao, Z.; Wang, X.S.; Kori, R.; Zhou, G.; Lee, S.C.; Tan, T.H. A novel src homology 3 domain-containing adaptor protein, HIP-55, that interacts with hematopoietic progenitor kinase 1. J. Biol. Chem. 1999, 274, 33945–33950. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.P.; Chiu, L.L.; Lan, J.L.; Chen, D.Y.; Boomer, J.; Tan, T.H. Attenuation of T cell receptor signaling by serine phosphorylation-mediated lysine 30 ubiquitination of SLP-76 protein. J. Biol. Chem. 2012, 287, 34091–34100. [Google Scholar] [CrossRef]
- Chuang, H.C.; Tan, T.H. MAP4K4 and IL-6+ Th17 cells play important roles in non-obese type 2 diabetes. J. Biomed. Sci. 2017, 24, 4. [Google Scholar] [CrossRef]
- Chuang, H.C.; Wang, J.S.; Lee, I.T.; Sheu, W.H.; Tan, T.H. Epigenetic regulation of HGK/MAP4K4 in T cells of type 2 diabetes patients. Oncotarget 2016, 7, 10976–10989. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Carlsson Almlof, J.; Leonard, D.; Alexsson, A.; Nordmark, G.; Eloranta, M.L.; Rantapaa-Dahlqvist, S.; Bengtsson, A.A.; Jonsen, A.; Padyukov, L.; et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.H.; Chuang, H.C. MAP Kinase Kinase Kinase Kinase 3 (MAP4K3) as a Biomarker and Therapeutic Target for Autoimmune Disease, Cancer, Inflammation and IL-17-Associated Disease. U.S. Patent 8,846,311 B2, 2014. [Google Scholar]
- Hsu, C.P.; Chuang, H.C.; Lee, M.C.; Tsou, H.H.; Lee, L.W.; Li, J.P.; Tan, T.H. GLK/MAP4K3 overexpression associates with recurrence risk for non-small cell lung cancer. Oncotarget 2016, 7, 41748–41757. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.H.; Chuang, H.C.; Wu, I.C.; Tsai, H.W.; Lin, Y.J.; Sun, H.Y.; Young, K.C.; Chiu, Y.C.; Cheng, P.N.; Liu, W.C.; et al. Prediction of early hepatocellular carcinoma recurrence using germinal center kinase-like kinase. Oncotarget 2016, 7, 49765–49776. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chuang, H.C.; Chang, C.C.; Teng, C.F.; Hsueh, C.H.; Chiu, L.L.; Hsu, P.M.; Lee, M.C.; Hsu, C.P.; Chen, Y.R.; Liu, Y.C.; et al. MAP4K3/GLK promotes lung cancer metastasis by phosphorylating and activating IQGAP1. Cancer Res. 2019, 79, 4978–4993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wu, L.; Qian, J.; Qu, B.; Xia, S.; La, T.; Wu, Y.; Ma, J.; Zeng, J.; Guo, Q.; et al. Identification of the long noncoding RNA NEAT1 as a novel inflammatory regulator acting through MAPK pathway in human lupus. J. Autoimmun. 2016, 75, 96–104. [Google Scholar] [CrossRef]
- Farooq, A.; Zhou, M.M. Structure and regulation of MAPK phosphatases. Cell. Signal. 2004, 16, 769–779. [Google Scholar] [CrossRef]
- MacCorkle, R.A.; Tan, T.H. Mitogen-activated protein kinases in cell-cycle control. Cell Biochem. Biophys. 2005, 43, 451–461. [Google Scholar] [CrossRef]
- Chen, A.J.; Zhou, G.; Juan, T.; Colicos, S.M.; Cannon, J.P.; Cabriera-Hansen, M.; Meyer, C.F.; Jurecic, R.; Copeland, N.G.; Gilbert, D.J.; et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2002, 277, 36592–36601. [Google Scholar] [CrossRef]
- Lin, H.P.; Ho, H.M.; Chang, C.W.; Yeh, S.D.; Su, Y.W.; Tan, T.H.; Lin, W.J. DUSP22 suppresses prostate cancer proliferation by targeting the EGFR-AR axis. FASEB J. 2019. [Google Scholar] [CrossRef]
- Chen, Y.R.; Chou, H.C.; Yang, C.H.; Chen, H.Y.; Liu, Y.W.; Lin, T.Y.; Yeh, C.L.; Chao, W.T.; Tsou, H.H.; Chuang, H.C.; et al. Deficiency in VHR/DUSP3, a suppressor of focal adhesion kinase, reveals its role in regulating cell adhesion and migration. Oncogene 2017, 36, 6509–6517. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Lin, Y.C.; Hsiao, W.Y.; Liao, F.H.; Huang, P.Y.; Tan, T.H. DUSP4 deficiency enhances CD25 expression and CD4+ T-cell proliferation without impeding T-cell development. Eur. J. Immunol. 2012, 42, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.C.; Chen, M.Y.; Hsu, S.C.; Huang, L.R.; Kao, C.Y.; Cheng, W.H.; Pan, C.H.; Wu, M.S.; Yu, G.Y.; Hung, M.S.; et al. DUSP6 mediates T cell receptor-engaged glycolysis and restrains TFH cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, E8027–E8036. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Liu, L.; Ji, X.; Gao, Y.; Chen, X.; Liu, Y.; Liu, Y.; Zhao, X.; Li, Y.; Li, Y.; et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat. Immunol. 2015, 16, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Raffi, F.A.M. Dual-specificity phosphatases in immunity and infection: An update. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Chuang, H.C.; Chen, Y.M.; Hung, W.T.; Li, J.P.; Chen, D.Y.; Lan, J.L.; Tan, T.H. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget 2016, 7, 57593–57605. [Google Scholar] [CrossRef]
- Johar, A.S.; Mastronardi, C.; Rojas-Villarraga, A.; Patel, H.R.; Chuah, A.; Peng, K.; Higgins, A.; Milburn, P.; Palmer, S.; Silva-Lara, M.F.; et al. Novel and rare functional genomic variants in multiple autoimmune syndrome and Sjogren’s syndrome. J. Transl. Med. 2015, 13, 173. [Google Scholar] [CrossRef]
- Hofmann, S.R.; Mabert, K.; Kapplusch, F.; Russ, S.; Northey, S.; Beresford, M.W.; Tsokos, G.C.; Hedrich, C.M. cAMP response element modulator α induces dual specificity protein phosphatase 4 to promote effector T cells in juvenile-onset lupus. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Castro-Sanchez, P.; Ramirez-Munoz, R.; Lamana, A.; Ortiz, A.; Gonzalez-Alvaro, I.; Roda-Navarro, P. mRNA profiling identifies low levels of phosphatases dual-specific phosphatase-7 (DUSP7) and cell division cycle-25B (CDC25B) in patients with early arthritis. Clin. Exp. Immunol. 2017, 189, 113–119. [Google Scholar] [CrossRef]
- Balada, E.; Felip, L.; Ordi-Ros, J.; Vilardell-Tarres, M. DUSP23 is over-expressed and linked to the expression of DNMTs in CD4+ T cells from systemic lupus erythematosus patients. Clin. Exp. Immunol. 2017, 187, 242–250. [Google Scholar] [CrossRef]
- Li, J.P.; Fu, Y.N.; Chen, Y.R.; Tan, T.H. JNK pathway-associated phosphatase dephosphorylates focal adhesion kinase and suppresses cell migration. J. Biol. Chem. 2010, 285, 5472–5478. [Google Scholar] [CrossRef] [PubMed]
- Matache, C.; Stefanescu, M.; Onu, A.; Tanaseanu, S.; Matei, I.; Frade, R.; Szegli, G. p56lck activity and expression in peripheral blood lymphocytes from patients with systemic lupus erythematosus. Autoimmunity 1999, 29, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Jury, E.C.; Isenberg, D.A.; Mauri, C.; Ehrenstein, M.R. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J. Immunol. 2006, 177, 7416–7422. [Google Scholar] [CrossRef]
- Ostensson, M.; Monten, C.; Bacelis, J.; Gudjonsdottir, A.H.; Adamovic, S.; Ek, J.; Ascher, H.; Pollak, E.; Arnell, H.; Browaldh, L.; et al. A possible mechanism behind autoimmune disorders discovered by genome-wide linkage and association analysis in celiac disease. PLoS ONE 2013, 8, e70174. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Solski, P.A.; Khosravi-Far, R.; Der, C.J.; Kelly, K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J. Biol. Chem. 1996, 271, 6497–6501. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairi, M.; Al-Harthi, S.; Cadalbert, L.; Plevin, R. Over-expression of mitogen-activated protein kinase phosphatase-2 enhances adhesion molecule expression and protects against apoptosis in human endothelial cells. Br. J. Pharmacol. 2010, 161, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.Y.; Lin, Y.C.; Liao, F.H.; Chan, Y.C.; Huang, C.Y. Dual-specificity phosphatase 4 regulates STAT5 protein stability and helper T cell polarization. PLoS ONE 2015, 10, e0145880. [Google Scholar] [CrossRef]
- Moser, K.L.; Neas, B.R.; Salmon, J.E.; Yu, H.; Gray-McGuire, C.; Asundi, N.; Bruner, G.R.; Fox, J.; Kelly, J.; Henshall, S.; et al. Genome scan of human systemic lupus erythematosus: Evidence for linkage on chromosome 1q in African-American pedigrees. Proc. Natl. Acad. Sci. USA 1998, 95, 14869–14874. [Google Scholar] [CrossRef]
- Zhang, Y.; Reynolds, J.M.; Chang, S.H.; Martin-Orozco, N.; Chung, Y.; Nurieva, R.I.; Dong, C. MKP-1 is necessary for T cell activation and function. J. Biol. Chem. 2009, 284, 30815–30824. [Google Scholar] [CrossRef]
- Moon, S.J.; Lim, M.A.; Park, J.S.; Byun, J.K.; Kim, S.M.; Park, M.K.; Kim, E.K.; Moon, Y.M.; Min, J.K.; Ahn, S.M.; et al. Dual-specificity phosphatase 5 attenuates autoimmune arthritis in mice via reciprocal regulation of the Th17/Treg cell balance and inhibition of osteoclastogenesis. Arthritis Rheumatol. 2014, 66, 3083–3095. [Google Scholar] [CrossRef]
- Yang, C.Y.; Li, J.P.; Chiu, L.L.; Lan, J.L.; Chen, D.Y.; Chuang, H.C.; Huang, C.Y.; Tan, T.H. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J. Immunol. 2014, 192, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chiu, L.L.; Tan, T.H. TRAF2-mediated Lys63-linked ubiquitination of DUSP14/MKP6 is essential for its phosphatase activity. Cell. Signal. 2016, 28, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chiu, L.L.; Chang, C.C.; Chuang, H.C.; Tan, T.H. Induction of DUSP14 ubiquitination by PRMT5-mediated arginine methylation. FASEB J. 2018, 32, 6760–6770. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, H.-C.; Tan, T.-H. MAP4K Family Kinases and DUSP Family Phosphatases in T-Cell Signaling and Systemic Lupus Erythematosus. Cells 2019, 8, 1433. https://doi.org/10.3390/cells8111433
Chuang H-C, Tan T-H. MAP4K Family Kinases and DUSP Family Phosphatases in T-Cell Signaling and Systemic Lupus Erythematosus. Cells. 2019; 8(11):1433. https://doi.org/10.3390/cells8111433
Chicago/Turabian StyleChuang, Huai-Chia, and Tse-Hua Tan. 2019. "MAP4K Family Kinases and DUSP Family Phosphatases in T-Cell Signaling and Systemic Lupus Erythematosus" Cells 8, no. 11: 1433. https://doi.org/10.3390/cells8111433
APA StyleChuang, H.-C., & Tan, T.-H. (2019). MAP4K Family Kinases and DUSP Family Phosphatases in T-Cell Signaling and Systemic Lupus Erythematosus. Cells, 8(11), 1433. https://doi.org/10.3390/cells8111433