Biological Background of Resistance to Current Standards of Care in Multiple Myeloma

 , , , and
, , , and 
{kind=link}
Abstract
1. Introduction
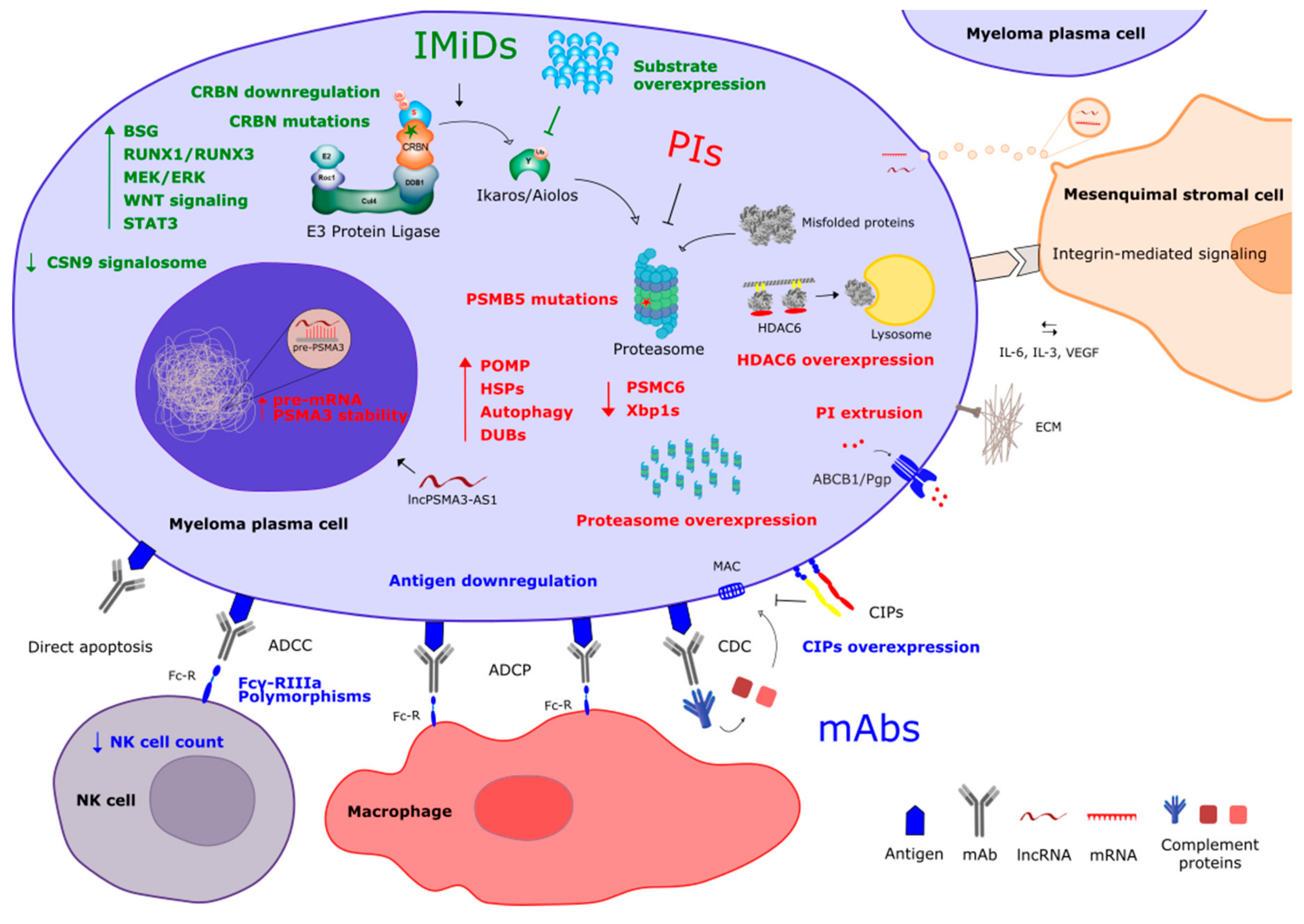
2. Resistance to Proteasome Inhibitors
2.1. Mechanisms of Action
2.2. Mechanisms of Resistance
2.3. Overcoming Drug Resistance
3. Resistance to Immunomodulatory Compounds
3.1. Mechanisms of Action
3.2. Mechanisms of Resistance
3.3. Overcoming Drug Resistance
4. Resistance to Monoclonal Antibodies
4.1. Mechanisms of Action
4.2. Mechanisms of Resistance
4.3. Overcoming Drug Resistance
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Costa, L.J.; Brill, I.K.; Omel, J.; Godby, K.; Kumar, S.K.; Brown, E.E. Recent trends in multiple myeloma incidence and survival by age, race, and ethnicity in the United States. Blood Adv. 2017, 1, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Callander, N.S.; Alsina, M.; Atanackovic, D.; Biermann, J.S.; Castillo, J.; Chandler, J.C.; Costello, C.; Faiman, M.; Fung, H.C.; et al. NCCN Guidelines Insights: Multiple Myeloma, Version 3.2018. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; San Miguel, J.; Sonneveld, P.; Mateos, M.V.; Zamagni, E.; Avet-Loiseau, H.; Hajek, R.; Dimopoulos, M.A.; Ludwig, H.; Einsele, H.; et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2017, 28, iv52–iv61. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Lee, J.H.; Lahuerta, J.J.; Morgan, G.; Richardson, P.G.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2012, 26, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Abeykoon, J.P.; Kumar, S.K.; Rajkumar, S.V.; Dingli, D.; Buadi, F.K.; Gonsalves, W.I.; Leung, N.; Dispenzieri, A.; Kourelis, T.V.; et al. Efficacy of daratumumab-based therapies in patients with relapsed, refractory multiple myeloma treated outside of clinical trials. Am. J. Hematol. 2017, 92, 1146–1155. [Google Scholar] [CrossRef]
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Baz, R.; Nooka, A.K.; Richter, J.; Cole, C.E.; et al. Selinexor and Low Dose Dexamethasone (Sd) in Patients with Lenalidomide, Pomalidomide, Bortezomib, Carfilzomib and Anti-CD38 Ab Refractory Multiple Myeloma (MM): STORM Study. Blood 2016, 128, 491. [Google Scholar] [CrossRef]
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J.; et al. Selective Inhibition of Nuclear Export With Oral Selinexor for Treatment of Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 859–866. [Google Scholar] [CrossRef]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Faict, S.; Maes, K.; Bruyne, E.D.; Valckenborgh, E.V.; Schots, R.; Vanderkerken, K.; Menu, E. Extracellular vesicle cross-talk in the bone marrow microenvironment: Implications in multiple myeloma. Oncotarget 2016, 7, 38927–38945. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; Bruyne, E.D.; Valckenborgh, E.V.; Lahoutte, T.; Wever, O.D.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Herrmann, M.; et al. Extensive Immunoglobulin Production Sensitizes Myeloma Cells for Proteasome Inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef]
- Ocio, E.M.; Mateos, M.-V.; San-Miguel, J.F. Novel agents derived from the currently approved treatments for MM: Novel proteasome inhibitors and novel IMIDs. Expert Opin. Investig. Drugs 2012, 21, 1075–1087. [Google Scholar] [CrossRef]
- Kirk, C.J. Discovery and Development of Second-Generation Proteasome Inhibitors. Semin. Hematol. 2012, 49, 207–214. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal Structure of the Boronic Acid-Based Proteasome Inhibitor Bortezomib in Complex with the Yeast 20S Proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.B.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. [Google Scholar] [CrossRef]
- Piva, R.; Ruggeri, B.; Williams, M.; Costa, G.; Tamagno, I.; Ferrero, D.; Giai, V.; Coscia, M.; Peola, S.; Massaia, M.; et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 2008, 111, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A Highly Cytotoxic Proteasome Inhibitor from a Novel Microbial Source, a Marine Bacterium of the New Genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Anderson, K.C. Preclinical Studies of Novel Targeted Therapies. Hematol. Oncol. Clin. North Am. 2007, 21, 1071–1091. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McConkey, D.J.; Zhu, K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. Updates 2008, 11, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.P.; Zonder, J.A. Overview of Proteasome Inhibitor-Based Anti-cancer Therapies: Perspective on Bortezomib and Second Generation Proteasome Inhibitors versus Future Generation Inhibitors of Ubiquitin-Proteasome System. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef]
- Zangari, M.; Yaccoby, S.; Pappas, L.; Cavallo, F.; Kumar, N.S.; Ranganathan, S.; Suva, L.J.; Gruenwald, J.M.; Kern, S.; Zhan, F.; et al. A prospective evaluation of the biochemical, metabolic, hormonal and structural bone changes associated with bortezomib response in multiple myeloma patients. Haematologica 2011, 96, 333–336. [Google Scholar] [CrossRef]
- Von Metzler, I.; Krebbel, H.; Hecht, M.; Manz, R.A.; Fleissner, C.; Mieth, M.; Kaiser, M.; Jakob, C.; Sterz, J.; Kleeberg, L.; et al. Bortezomib inhibits human osteoclastogenesis. Leukemia 2007, 21, 2025–2034. [Google Scholar] [CrossRef]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A.; et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 2007, 110, 334–338. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Quwaider, D.; Canavese, M.; Ocio, E.M.; Tian, Z.; Blanco, J.F.; Berger, A.J.; Ortiz-de-Solorzano, C.; Hernández-Iglesias, T.; Martens, A.C.M.; et al. Preclinical Activity of the Oral Proteasome Inhibitor MLN9708 in Myeloma Bone Disease. Clin. Cancer Res. 2014, 20, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Hurchla, M.; Garcia-Gomez, A.; Hornick, M.; Ocio, E.; Li, A.; Blanco, J.; Collins, L.; Kirk, C.; Piwnica-Worms, D.; Vij, R.; et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia 2013, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: A role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447. [Google Scholar] [CrossRef]
- Balsas, P.; Galán-Malo, P.; Marzo, I.; Naval, J. Bortezomib resistance in a myeloma cell line is associated to PSMβ5 overexpression and polyploidy. Leuk. Res. 2012, 36, 212–218. [Google Scholar] [CrossRef]
- Niewerth, D.; Jansen, G.; Riethoff, L.F.V.; van Meerloo, J.; Kale, A.J.; Moore, B.S.; Assaraf, Y.G.; Anderl, J.L.; Zweegman, S.; Kaspers, G.J.L.; et al. Antileukemic Activity and Mechanism of Drug Resistance to the Marine Salinispora tropica Proteasome Inhibitor Salinosporamide A (Marizomib). Mol Pharm. 2014, 86, 12–19. [Google Scholar] [CrossRef]
- Rückrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin–proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef]
- Shuqing, L.; Jianmin, Y.; Chongmei, H.; Hui, C.; Wang, J. Upregulated expression of the PSMB5 gene may contribute to drug resistance in patient with multiple myeloma when treated with bortezomib-based regimen. Exp. Hematol. 2011, 39, 1117–1118. [Google Scholar] [CrossRef]
- Li, B.; Fu, J.; Chen, P.; Ge, X.; Li, Y.; Kuiatse, I.; Wang, H.; Wang, H.; Zhang, X.; Orlowski, R.Z. The Nuclear Factor (Erythroid-derived 2)-like 2 and Proteasome Maturation Protein Axis Mediate Bortezomib Resistance in Multiple Myeloma. J. Biol. Chem. 2015, 290, 29854–29868. [Google Scholar] [CrossRef]
- Shi, C.-X.; Kortüm, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198. [Google Scholar] [CrossRef] [PubMed]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Zhang, L.; Fok, J.H.L.; Davies, F.E. Heat shock proteins in multiple myeloma. Oncotarget 2014, 5, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzezniakiewicz, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736. [Google Scholar] [CrossRef]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.M.; López-Pérez, R.; Mateo, G.; Gutiérrez, N.; Atadja, P.; Pandiella, A.; Miguel, J.F.S. The Histone Deacetylase Inhibitor LBH589 Is a Potent Antimyeloma Agent that Overcomes Drug Resistance. Cancer Res. 2006, 66, 5781–5789. [Google Scholar] [CrossRef]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Peterson, L.F.; Sun, H.; Liu, Y.; Potu, H.; Kandarpa, M.; Ermann, M.; Courtney, S.M.; Young, M.; Showalter, H.D.; Sun, D.; et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 2015, 125, 3588–3597. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Assaraf, Y.G.; Dijkmans, B.A.C.; Scheffer, G.L.; Al, M.; den Uyl, D.; Oerlemans, R.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; et al. Inactivating PSMB5 Mutations and P-Glycoprotein (Multidrug Resistance-Associated Protein/ATP-Binding Cassette B1) Mediate Resistance to Proteasome Inhibitors: Ex Vivo Efficacy of (Immuno)Proteasome Inhibitors in Mononuclear Blood Cells from Patients with Rheumatoid Arthritis. J. Pharm. Exp. 2012, 341, 174–182. [Google Scholar]
- Hao, M.; Zhang, L.; An, G.; Sui, W.; Yu, Z.; Zou, D.; Xu, Y.; Chang, H.; Qiu, L. Suppressing miRNA-15a/-16 expression by interleukin-6 enhances drug-resistance in myeloma cells. J. Hematol. Oncol. 2011, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Hazlehurst, L.; Lynch, C.C. Dissecting the multiple myeloma-bone microenvironment reveals new therapeutic opportunities. J. Mol. Med. 2016, 94, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Franke, N.E.; Kaspers, G.L.; Assaraf, Y.G.; van Meerloo, J.; Niewerth, D.; Kessler, F.L.; Poddighe, P.J.; Kole, J.; Smeets, S.J.; Ylstra, B.; et al. Exocytosis of polyubiquitinated proteins in bortezomib-resistant leukemia cells: A role for MARCKS in acquired resistance to proteasome inhibitors. Oncotarget 2016, 7, 74779–74796. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 Promotes Proteasome Inhibitors Resistance in Multiple Myeloma. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Gu, J.; Li, J.; Zhou, Z.; Liu, J.; Huang, B.; Zheng, D.; Su, C. Differentiation induction enhances bortezomib efficacy and overcomes drug resistance in multiple myeloma. Biochem. Biophys. Res. Commun. 2012, 420, 644–650. [Google Scholar] [CrossRef]
- Jia, L.; Gopinathan, G.; Sukumar, J.T.; Gribben, J.G. Blocking Autophagy Prevents Bortezomib-Induced NF-κB Activation by Reducing I-κBα Degradation in Lymphoma Cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A Small Molecule Inhibitor of Ubiquitin-Specific Protease-7 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Bortezomib Resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.-S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. New Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Spencer, A.; Attal, M.; Prince, H.M.; Harousseau, J.-L.; Dmoszynska, A.; San Miguel, J.; Hellmann, A.; Facon, T.; Foà, R.; et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2007, 357, 2123–2132. [Google Scholar] [CrossRef]
- Schey, S.a.; Fields, P.; Bartlett, J.B.; Clarke, I.A.; Ashan, G.; Knight, R.D.; Streetly, M.; Dalgleish, A.G. Phase I Study of an Immunomodulatory Thalidomide Analog, CC-4047, in Relapsed or Refractory Multiple Myeloma. JCO 2004, 22, 3269–3276. [Google Scholar] [CrossRef]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) Plus Low-Dose Dexamethasone As Therapy for Relapsed Multiple Myeloma. JCO 2009, 27, 5008–5014. [Google Scholar] [CrossRef]
- Lepper, E.; Smith, N.; Cox, M.; Scripture, C.; Figg, W. Thalidomide Metabolism and Hydrolysis: Mechanisms and Implications. CDM 2006, 7, 677–685. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kasserra, C.; Reyes, J.; Schafer, P.; Kosek, J.; Capone, L.; Parton, A.; Kim-Kang, H.; Surapaneni, S.; Kumar, G. Absorption, metabolism and excretion of [14C]pomalidomide in humans following oral administration. Cancer Chemother. Pharm. 2013, 71, 489–501. [Google Scholar] [CrossRef]
- Chen, N.; Wen, L.; Lau, H.; Surapaneni, S.; Kumar, G. Pharmacokinetics, metabolism and excretion of [14C]-lenalidomide following oral administration in healthy male subjects. Cancer Chemother. Pharm. 2012, 69, 789–797. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Shima, Y.; Raje, N.; Davies, F.E.; Tai, Y.-T.; Treon, S.P.; Lin, B.; Schlossman, R.L.; Richardson, P.; et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood 2000, 96, 2943–2950. [Google Scholar] [CrossRef]
- Offidani, M.; Corvatta, L.; Caraffa, P.; Leoni, P.; Pautasso, C.; Larocca, A.; Palumbo, A. Pomalidomide for the treatment of relapsed–refractory multiple myeloma: A review of biological and clinical data. Expert Rev. Anticancer Ther. 2014, 14, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2019, 25, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; van de Donk, N.W.C.J.; Popat, R.; Zonder, J.A.; Minnema, M.C.; Larsen, J.; Nguyen, T.V.; Chen, M.S.; Bensmaine, A.; Cota, M.; et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). JCO 2019, 37, 8006. [Google Scholar]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; de Campos, C.B.; Braggio, E.; Bergsagel, P.L.; et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9, 19. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Richardson, P.G.; Hideshima, T.; Munshi, N.C.; Treon, S.P.; Anderson, K.C. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: Therapeutic implications. Blood 2002, 99, 4525–4530. [Google Scholar] [CrossRef]
- Li, S.; Pal, R.; Monaghan, S.A.; Schafer, P.; Ouyang, H.; Mapara, M.; Galson, D.L.; Lentzsch, S. IMiD immunomodulatory compounds block C/EBPβ translation through eIF4E down-regulation resulting in inhibition of MM. Blood 2011, 117, 5157–5165. [Google Scholar] [CrossRef]
- Raje, N.; Kumar, S.; Hideshima, T.; Ishitsuka, K.; Chauhan, D.; Mitsiades, C.; Podar, K.; Gouill, S.L.; Richardson, P.; Munshi, N.C.; et al. Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood 2004, 104, 4188–4193. [Google Scholar] [CrossRef]
- Corral, L.G.; Haslett, P.A.J.; Muller, G.W.; Chen, R.; Wong, L.-M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential Cytokine Modulation and T Cell Activation by Two Distinct Classes of Thalidomide Analogues That Are Potent Inhibitors of TNF-α. J. Immunol. 1999, 163, 380–386. [Google Scholar] [PubMed]
- LeBlanc, R.; Hideshima, T.; Catley, L.P.; Shringarpure, R.; Burger, R.; Mitsiades, N.; Mitsiades, C.; Cheema, P.; Chauhan, D.; Richardson, P.G.; et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004, 103, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Corral, L.G.; Fleming, Y.W.; Stein, B. Immunomodulatory drugs Revlimid® (lenalidomide) and CC-4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol. Immunother. 2008, 57, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Manni, S.; Carrino, M.; Manzoni, M.; Gianesin, K.; Nunes, S.C.; Costacurta, M.; Tubi, L.Q.; Macaccaro, P.; Taiana, E.; Cabrelle, A.; et al. Inactivation of CK1α in multiple myeloma empowers drug cytotoxicity by affecting AKT and β-catenin survival signaling pathways. Oncotarget 2017, 8, 14604–14619. [Google Scholar] [CrossRef]
- Hu, Y.; Song, W.; Cirstea, D.; Lu, D.; Munshi, N.C.; Anderson, K.C. CSNK1α1 mediates malignant plasma cell survival. Leukemia 2015, 29, 474–482. [Google Scholar] [CrossRef][Green Version]
- Carrino, M.; Quotti Tubi, L.; Fregnani, A.; Canovas Nunes, S.; Barilà, G.; Trentin, L.; Zambello, R.; Semenzato, G.; Manni, S.; Piazza, F. Prosurvival autophagy is regulated by protein kinase CK1 alpha in multiple myeloma. Cell Death Discov. 2019, 5, 98. [Google Scholar] [CrossRef]
- Manni, S.; Carrino, M.; Piazza, F. Role of protein kinases CK1α and CK2 in multiple myeloma: Regulation of pivotal survival and stress-managing pathways. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef]
- Eichner, R.; Heider, M.; Fernández-Sáiz, V.; van Bebber, F.; Garz, A.-K.; Lemeer, S.; Rudelius, M.; Targosz, B.-S.; Jacobs, L.; Knorn, A.-M.; et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Wier, S.V.; Chang, X.-B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Fernández-Lázaro, D.; San-Segundo, L.; López-Corral, L.; Corchete, L.A.; Gutiérrez, N.C.; Garayoa, M.; Paíno, T.; García-Gómez, A.; Delgado, M.; et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia 2015, 29, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Heintel, D.; Rocci, A.; Ludwig, H.; Bolomsky, A.; Caltagirone, S.; Schreder, M.; Pfeifer, S.; Gisslinger, H.; Zojer, N.; Jäger, U.; et al. High expression of cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Br. J. Haematol. 2013, 161, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.R.; Kortuem, K.M.; Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Ahmann, G.; Kumar, S.; Rajkumar, S.V.; et al. The clinical significance of cereblon expression in multiple myeloma. Leuk. Res. 2014, 38, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, T.; Zhou, W.; Xing, L.; Wang, S.; Ho, M.; Peng, Z.; Tai, Y.-T.; Hideshima, T.; Anderson, K.C.; et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia 2019, 33, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, A.; Gandhi, A.K.; Waldman, M.F.; Bjorklund, C.; Ning, Y.; Mendy, D.; Schafer, P.; Lopez-Girona, A.; Lentzsch, S.; Schey, S.A.; et al. Absence of mutations in cereblon (CRBN) and DNA damage-binding protein 1 (DDB1) genes and significance for IMiD therapy. Leukemia 2014, 28, 1129–1131. [Google Scholar] [CrossRef] [PubMed]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef]
- Sperling, A.S.; Burgess, M.; Keshishian, H.; Gasser, J.; Jan, M.; Sharma, R.; Fulciniti, M.; Munshi, N.; Carr, S.A.; Ebert, B.L. Discovery of a Novel Mechanism of Resistance to Thalidomide Derivatives. Blood 2018, 132, 949. [Google Scholar] [CrossRef]
- Zhou, N.; Gutierrez-Uzquiza, A.; Zheng, X.Y.; Chang, R.; Vogl, D.T.; Garfall, A.L.; Bernabei, L.; Saraf, A.; Florens, L.; Washburn, M.P.; et al. RUNX proteins desensitize multiple myeloma to lenalidomide via protecting IKZFs from degradation. Leukemia 2019, 33, 2006–2021. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Ma, W.; Wang, Z.Q.; Davis, R.E.; Kuhn, D.J.; Kornblau, S.M.; Wang, M.; Shah, J.J.; Orlowski, R.Z. Evidence of a role for activation of Wnt/beta-catenin signaling in the resistance of plasma cells to lenalidomide. J. Biol. Chem. 2011, 286, 11009–11020. [Google Scholar] [CrossRef]
- Sebastian, S.; Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Panchabhai, S.C.; Van Wier, S.A.; Ahmann, G.J.; Chesi, M.; Bergsagel, P.L.; Stewart, A.K.; et al. Multiple myeloma cells’ capacity to decompose H2O2 determines lenalidomide sensitivity. Blood 2017, 129, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Helbo, A.S.; Munch-Petersen, H.F.; Sjö, L.; Christensen, J.; Kristensen, L.S.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Zamagni, E.; Tacchetti, P.; Pantani, L.; Cavo, M. Anti-CD38 and anti-SLAMF7: The future of myeloma immunotherapy. Expert Rev. Hematol. 2018, 11, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.-V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Palumbo, A.; San-Miguel, J.; Shpilberg, O.; Anderson, K.; Grosicki, S.; Spicka, I.; et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br. J. Haematol. 2017, 178, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.-F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef]
- Nooka, A.K.; Kaufman, J.L.; Hofmeister, C.C.; Joseph, N.S.; Heffner, T.L.; Gupta, V.A.; Sullivan, H.C.; Neish, A.S.; Dhodapkar, M.V.; Lonial, S. Daratumumab in multiple myeloma. Cancer 2019, 125, 2364–2382. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.J.; Breij, E.; Martens, A.C.M.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 2015, 7, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Jansen, J.H.M.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.J.; Parren, P.W.H.I.; Leusen, J.H.W.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Wetzel, M.-C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallee, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, A Novel Humanized CD38-Targeting Antibody, Demonstrates Potent Antitumor Activity in Models of Multiple Myeloma and Other CD38+ Hematologic Malignancies. Clin.Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.-T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef]
- Martin, T.; Baz, R.; Benson, D.M.; Lendvai, N.; Wolf, J.; Munster, P.; Lesokhin, A.M.; Wack, C.; Charpentier, E.; Campana, F.; et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 129, 3294–3303. [Google Scholar] [CrossRef]
- Mikhael, J.; Richardson, P.; Usmani, S.Z.; Raje, N.; Bensinger, W.; Karanes, C.; Campana, F.; Kanagavel, D.; Dubin, F.; Liu, Q.; et al. A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood 2019, 134, 123–133. [Google Scholar] [CrossRef]
- Raab, M.S.; Chatterjee, M.; Goldschmidt, H.; Agis, H.; Blau, I.; Einsele, H.; Engelhardt, M.; Ferstl, B.; Gramatzki, M.; Röllig, C.; et al. A Phase I/IIa Study of the CD38 Antibody MOR202 Alone and in Combination with Pomalidomide or Lenalidomide in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2016, 128, 1152. [Google Scholar] [CrossRef]
- Busch, L.; Mougiakakos, D.; Büttner-Herold, M.; Müller, M.J.; Volmer, D.A.; Bach, C.; Fabri, M.; Bittenbring, J.T.; Neumann, F.; Boxhammer, R.; et al. Lenalidomide enhances MOR202-dependent macrophage-mediated effector functions via the vitamin D pathway. Leukemia 2018, 32, 2445–2458. [Google Scholar] [CrossRef] [PubMed]
- van Bueren, J.L.; Jakobs, D.; Kaldenhoven, N.; Roza, M.; Hiddingh, S.; Meesters, J.; Voorhorst, M.; Gresnigt, E.; Wiegman, L.; Buijsse, A.O.; et al. Direct in Vitro Comparison of Daratumumab with Surrogate Analogs of CD38 Antibodies MOR03087, SAR650984 and Ab79. Blood 2014, 124, 3474. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J. Reprint of “Immunomodulatory effects of CD38-targeting antibodies”. Immunol. Lett. 2019, 205, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Roepcke, S.; Plock, N.; Yuan, J.; Fedyk, E.R.; Lahu, G.; Zhao, L.; Smithson, G. Pharmacokinetics and pharmacodynamics of the cytolytic anti-CD38 human monoclonal antibody TAK-079 in monkey - model assisted preparation for the first in human trial. Pharm. Res. Perspect. 2018, 6, e00402. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Groen, R.W.J.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Kitadate, A.; Kobayashi, H.; Abe, Y.; Narita, K.; Miura, D.; Takeuchi, M.; Matsue, K. CD38 Expression Levels on Myeloma Cells and the Frequency of Circulating CD38-Positive Treg Cells Are Associated with the Response to Daratumumab in Multiple Myeloma. Blood 2018, 132, 1883. [Google Scholar] [CrossRef]
- Pick, M.; Vainstein, V.; Goldschmidt, N.; Lavie, D.; Libster, D.; Gural, A.; Grisariu, S.; Avni, B.; Ben Yehuda, D.; Gatt, M.E. Daratumumab resistance is frequent in advanced-stage multiple myeloma patients irrespective of CD38 expression and is related to dismal prognosis. Eur. J. Haematol. 2018, 100, 494–501. [Google Scholar] [CrossRef]
- Viola, D.; Dona, A.; Gunes, E.G.; Troadec, E.; Wu, X.; Branciamore, S.; McDonald, T.; Ghoda, L.; Streatfield, A.; Sanchez, J.F.; et al. Immune Mediated Mechanisms of Resistance to Daratumumab. Blood 2018, 132, 3201. [Google Scholar] [CrossRef]
- Danhof, S.; Strifler, S.; Hose, D.; Kortüm, M.; Bittrich, M.; Hefner, J.; Einsele, H.; Knop, S.; Schreder, M. Clinical and biological characteristics of myeloma patients influence response to elotuzumab combination therapy. J. Cancer Res. Clin. Oncol. 2019, 145, 561–571. [Google Scholar] [CrossRef]
- Jakubowiak, A.; Offidani, M.; Pegourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A.; et al. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Poulart, V.; Ying-Ming, J.; Delmonte, T.; Robbins, M. Fc-Gamma Receptor Polymorphisms And Progression-Free Survival; EHA Library: Copenhagen, Denmark, 2016. [Google Scholar]
- Van de Donk, N.W.C.J.; Casneuf, T.; Di Cara, A.; Parren, P.W.; Zweegman, S.; van Kessel, B.; Lokhorst, H.M.; Usmani, S.Z.; Lonial, S.; Richardson, P.G.; et al. Impact of Fc gamma receptor polymorphisms on efficacy and safety of daratumumab in relapsed/refractory multiple myeloma. Br. J. Haematol. 2019, 184, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, A.; Quarona, V.; Zito, A.; Morandi, F.; Marimpietri, D.; Cuccioloni, M.; Robert, O.J.; Mark, C.S.; Bolzoni, M.; Toscani, D.; et al. Generation and Characterization of Microvesicles after Daratumumab Interaction with Myeloma Cells. Blood 2015, 126, 1849. [Google Scholar] [CrossRef]
- Horenstein, A.; Chillemi, A.; Quarona, V.; Zito, A.; Roato, I.; Morandi, F.; Marimpietri, D.; Bolzoni, M.; Toscani, D.; Oldham, R.; et al. NAD+-Metabolizing Ectoenzymes in Remodeling Tumor–Host Interactions: The Human Myeloma Model. Cells 2015, 4, 520–537. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.C.; Zweegman, S.; van Meerloo, J.; Musters, R.J.P.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef]
- Ghose, J.; Viola, D.; Terrazas, C.; Caserta, E.; Troadec, E.; Khalife, J.; Gunes, E.G.; Sanchez, J.; McDonald, T.; Marcucci, G.; et al. Daratumumab induces CD38 internalization and impairs myeloma cell adhesion. Oncoimmunology 2018, 7, e1486948. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Gogishvili, T.; Danhof, S.; Schreder, M.; Pallaud, C.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017, 129, 3386–3388. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mogollón, P.; Díaz-Tejedor, A.; Algarín, E.M.; Paíno, T.; Garayoa, M.; Ocio, E.M. Biological Background of Resistance to Current Standards of Care in Multiple Myeloma. Cells 2019, 8, 1432. https://doi.org/10.3390/cells8111432
Mogollón P, Díaz-Tejedor A, Algarín EM, Paíno T, Garayoa M, Ocio EM. Biological Background of Resistance to Current Standards of Care in Multiple Myeloma. Cells. 2019; 8(11):1432. https://doi.org/10.3390/cells8111432
Chicago/Turabian StyleMogollón, Pedro, Andrea Díaz-Tejedor, Esperanza M. Algarín, Teresa Paíno, Mercedes Garayoa, and Enrique M. Ocio. 2019. "Biological Background of Resistance to Current Standards of Care in Multiple Myeloma" Cells 8, no. 11: 1432. https://doi.org/10.3390/cells8111432
APA StyleMogollón, P., Díaz-Tejedor, A., Algarín, E. M., Paíno, T., Garayoa, M., & Ocio, E. M. (2019). Biological Background of Resistance to Current Standards of Care in Multiple Myeloma. Cells, 8(11), 1432. https://doi.org/10.3390/cells8111432