Wnt Signalling in Acute Myeloid Leukaemia


Abstract
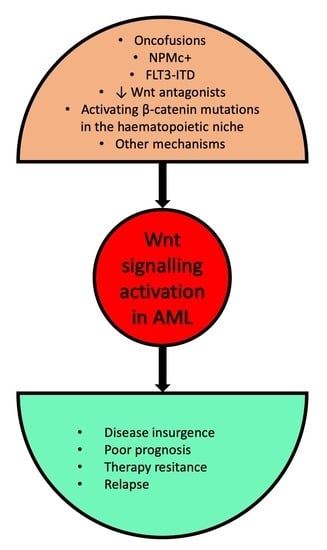
1. Introduction
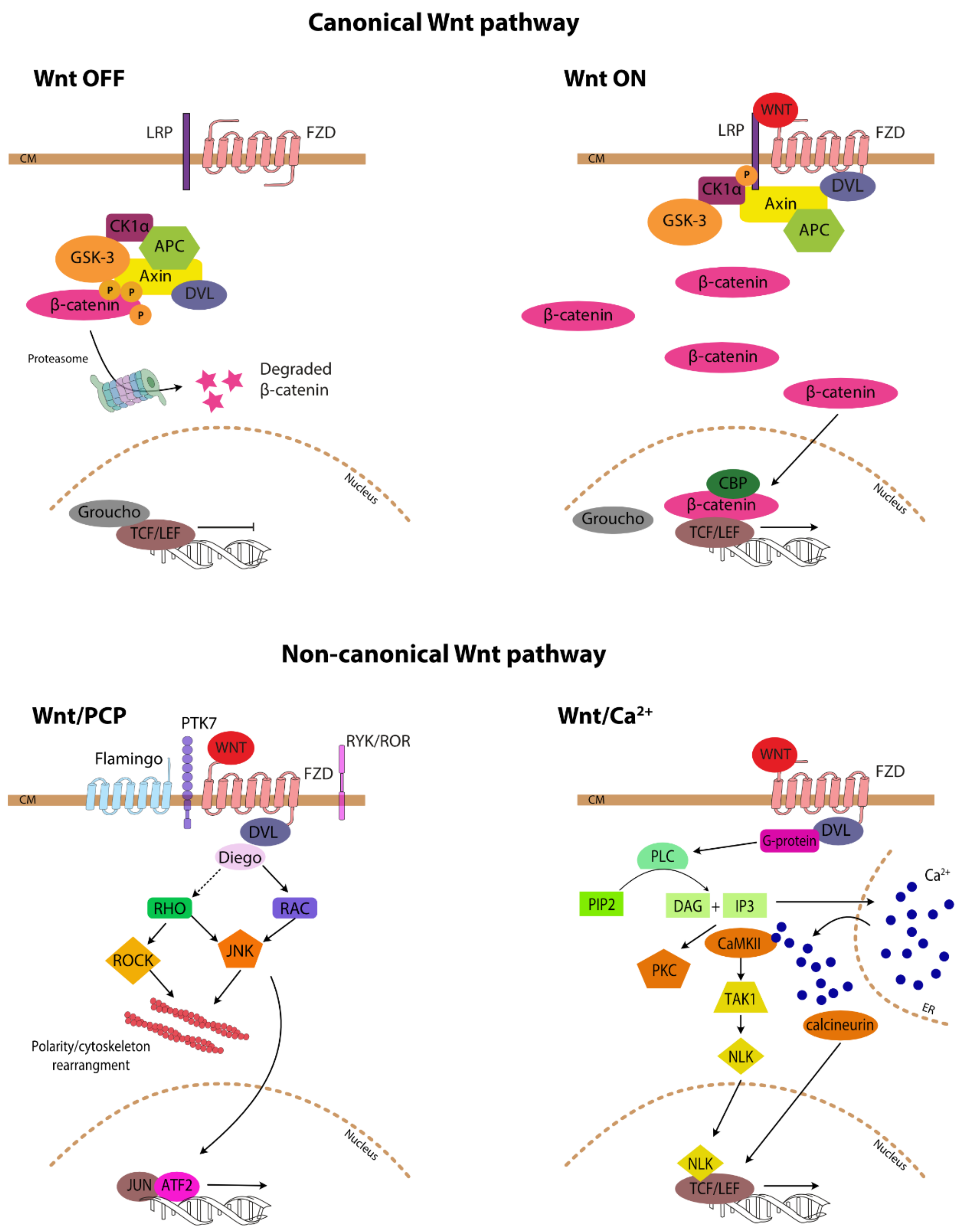
2. Wnt-Signalling Pathway
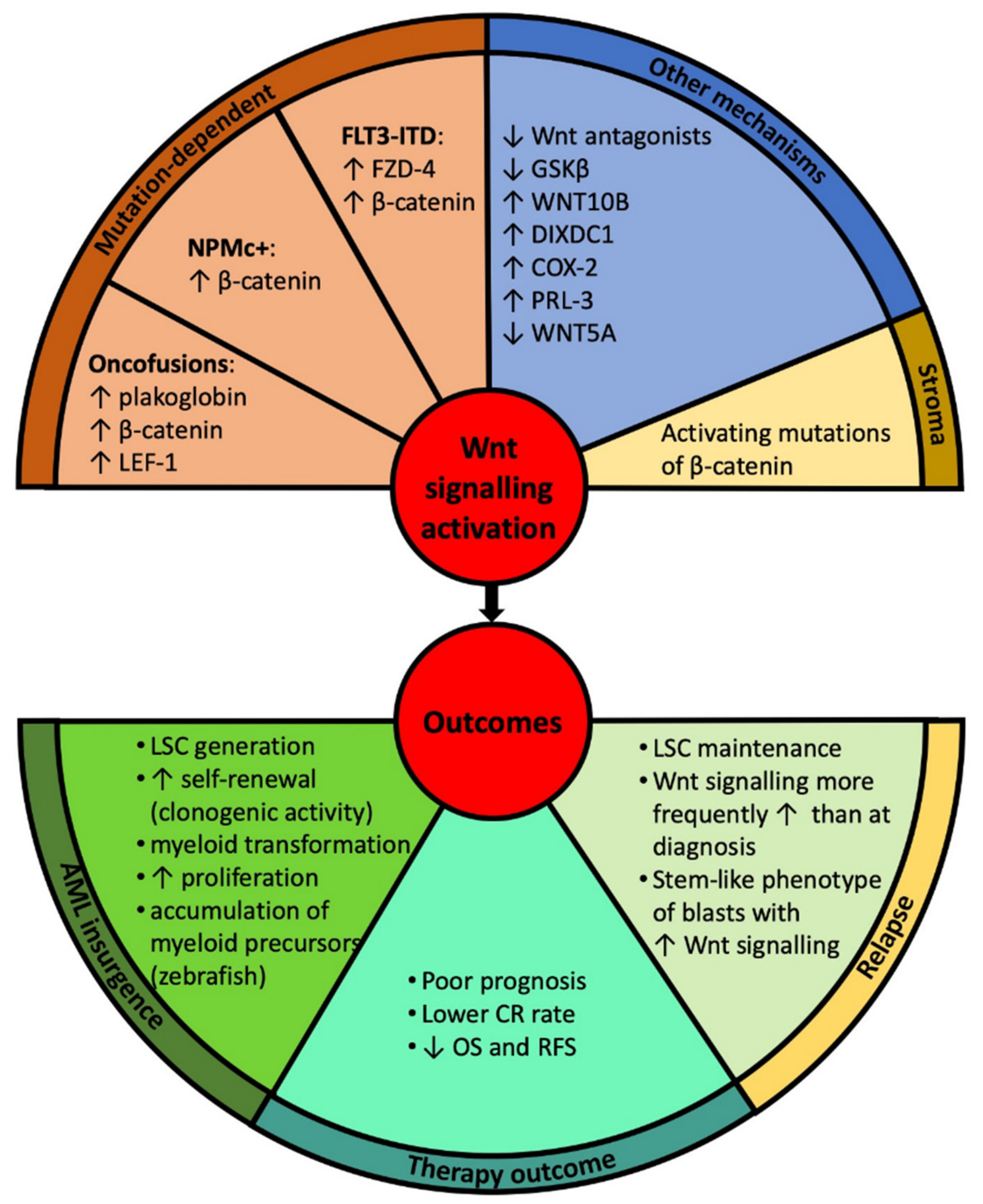
3. Wnt Signalling in the Pathogenesis of Acute Myeloid Leukaemia (AML)
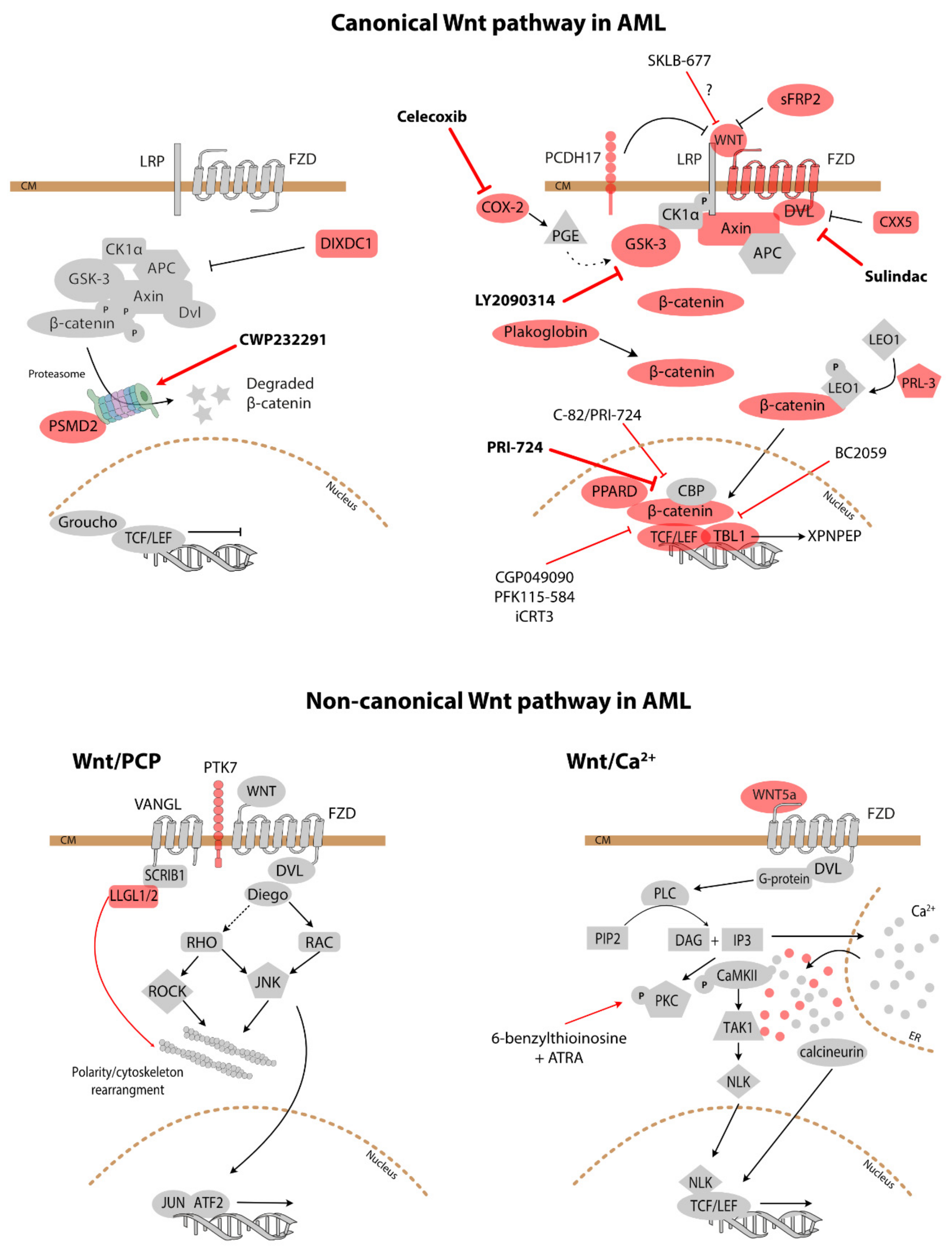
3.1. Canonical Wnt Signalling
3.2. Non-Canonical Wnt Signalling
4. Clinical Implications—Prognosis and Therapeutic Approaches
4.1. Prognostic Impact
4.2. Treatment
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.; Dohner, K. Prognostic implications of gene mutations in acute myeloid leukemia with normal cytogenetics. Semin. Oncol. 2008, 35, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.H.; Arreba-Tutusaus, P.; Armstrong, S.A.; Fischer, T. Evolutionarily conserved signaling pathways: Acting in the shadows of acute myelogenous leukemia’s genetic diversity. Clin. Cancer Res. 2015, 21, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Acute myeloid leukemia stem cells. Ann. N Y Acad. Sci. 2005, 1044, 1–5. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Ono, M.; Lai, K.K.Y.; Wu, K.; Nguyen, C.; Lin, D.P.; Murali, R.; Kahn, M. Nuclear receptor/Wnt beta-catenin interactions are regulated via differential CBP/p300 coactivator usage. PLoS ONE 2018, 13, e0200714. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Daulat, A.M.; Borg, J.P. Wnt/Planar Cell Polarity Signaling: New Opportunities for Cancer Treatment. Trends. Cancer 2017, 3, 113–125. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta. Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Muller-Tidow, C.; Steffen, B.; Cauvet, T.; Tickenbrock, L.; Ji, P.; Diederichs, S.; Sargin, B.; Kohler, G.; Stelljes, M.; Puccetti, E.; et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol. Cell Biol. 2004, 24, 2890–2904. [Google Scholar] [CrossRef]
- Zheng, X.; Beissert, T.; Kukoc-Zivojnov, N.; Puccetti, E.; Altschmied, J.; Strolz, C.; Boehrer, S.; Gul, H.; Schneider, O.; Ottmann, O.G.; et al. Gamma-catenin contributes to leukemogenesis induced by AML-associated translocation products by increasing the self-renewal of very primitive progenitor cells. Blood 2004, 103, 3535–3543. [Google Scholar] [CrossRef]
- Morgan, R.G.; Pearn, L.; Liddiard, K.; Pumford, S.L.; Burnett, A.K.; Tonks, A.; Darley, R.L. gamma-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of beta-catenin. Leukemia 2013, 27, 336–343. [Google Scholar] [CrossRef]
- Ugarte, G.D.; Vargas, M.F.; Medina, M.A.; Leon, P.; Necunir, D.; Elorza, A.A.; Gutierrez, S.E.; Moon, R.T.; Loyola, A.; De Ferrari, G.V. Wnt signaling induces transcription, spatial proximity, and translocation of fusion gene partners in human hematopoietic cells. Blood 2015, 126, 1785–1789. [Google Scholar] [CrossRef]
- Fu, Y.; Zhu, H.; Wu, W.; Xu, J.; Chen, T.; Xu, B.; Qian, S.; Li, J.; Liu, P. Clinical significance of lymphoid enhancer-binding factor 1 expression in acute myeloid leukemia. Leuk Lymphoma 2014, 55, 371–377. [Google Scholar] [CrossRef]
- Metzeler, K.H.; Heilmeier, B.; Edmaier, K.E.; Rawat, V.P.; Dufour, A.; Dohner, K.; Feuring-Buske, M.; Braess, J.; Spiekermann, K.; Buchner, T.; et al. High expression of lymphoid enhancer-binding factor-1 (LEF1) is a novel favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Blood 2012, 120, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Ridsdale, J.; Payne, M.; Heesom, K.J.; Wilson, M.C.; Davidson, A.; Greenhough, A.; Davies, S.; Williams, A.C.; Blair, A.; et al. LEF-1 drives aberrant beta-catenin nuclear localization in myeloid leukemia cells. Haematologica 2019, 104, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, K.; Arseni, N.; Schessl, C.; Stadler, C.R.; Rawat, V.P.; Deshpande, A.J.; Heilmeier, B.; Hiddemann, W.; Quintanilla-Martinez, L.; Bohlander, S.K.; et al. A novel role for Lef-1, a central transcription mediator of Wnt signaling, in leukemogenesis. J. Exp. Med. 2008, 205, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Becker, M.W.; Tian, Q.; Lee, T.L.; Yan, X.; Liu, R.; Chiang, J.H.; Hood, L.; Clarke, M.F.; Weissman, I.L. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3396–3401. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Deflorian, G.; Pezzimenti, F.; Valli, D.; Saia, M.; Meani, N.; Gruszka, A.M.; Alcalay, M. Nucleophosmin leukemogenic mutant activates Wnt signaling during zebrafish development. Oncotarget 2016, 7, 55302–55312. [Google Scholar] [CrossRef]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rucker, F.G.; Lux, S.; Blatte, T.J.; Skambraks, S.; Kronke, J.; Heidel, F.H.; Schnoder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 2031. [Google Scholar] [CrossRef]
- Reikvam, H.; Aasebo, E.; Brenner, A.K.; Bartaula-Brevik, S.; Gronningsaeter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, O. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 970. [Google Scholar] [CrossRef]
- Tsykunova, G.; Reikvam, H.; Hovland, R.; Bruserud, O. The surface molecule signature of primary human acute myeloid leukemia (AML) cells is highly associated with NPM1 mutation status. Leukemia 2012, 26, 557–559. [Google Scholar] [CrossRef]
- Tickenbrock, L.; Schwable, J.; Wiedehage, M.; Steffen, B.; Sargin, B.; Choudhary, C.; Brandts, C.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood 2005, 105, 3699–3706. [Google Scholar] [CrossRef]
- Valencia, A.; Roman-Gomez, J.; Cervera, J.; Such, E.; Barragan, E.; Bolufer, P.; Moscardo, F.; Sanz, G.F.; Sanz, M.A. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia 2009, 23, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Li, L.; Cheng, S.H.; Ng, K.; Chan, N.P.; Ip, R.K.; Wong, R.S.; Shing, M.M.; Li, C.K.; Ng, M.H. Secreted-frizzled related protein 1 is a transcriptional repression target of the t(8;21) fusion protein in acute myeloid leukemia. Blood 2011, 118, 6638–6648. [Google Scholar] [CrossRef] [PubMed]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Lazzaroni, F.; Del Giacco, L.; Biasci, D.; Turrini, M.; Prosperi, L.; Brusamolino, R.; Cairoli, R.; Beghini, A. Intronless WNT10B-short variant underlies new recurrent allele-specific rearrangement in acute myeloid leukaemia. Sci. Rep. 2016, 6, 37201. [Google Scholar] [CrossRef] [PubMed]
- Beghini, A.; Corlazzoli, F.; Del Giacco, L.; Re, M.; Lazzaroni, F.; Brioschi, M.; Valentini, G.; Ferrazzi, F.; Ghilardi, A.; Righi, M.; et al. Regeneration-associated WNT signaling is activated in long-term reconstituting AC133bright acute myeloid leukemia cells. Neoplasia 2012, 14, 1236–1248. [Google Scholar] [CrossRef]
- Xin, H.; Li, C.; Wang, M. DIXDC1 promotes the growth of acute myeloid leukemia cells by upregulating the Wnt/beta-catenin signaling pathway. Biomed. Pharmacother 2018, 107, 1548–1555. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Lilly, M.B.; Drapiza, L.; Sheth, M.; Zemskova, M.; Bashkirova, S.; Morris, J. Expression of Cyclooxygenase-2 (COX-2) in Human Leukemias and Hematopoietic Cells. Blood 2004, 104, 4336. [Google Scholar]
- Duciel, L.; Monraz Gomez, L.C.; Kondratova, M.; Kuperstein, I.; Saule, S. The Phosphatase PRL-3 Is Involved in Key Steps of Cancer Metastasis. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Zhou, J.; Bi, C.; Chng, W.J.; Cheong, L.L.; Liu, S.C.; Mahara, S.; Tay, K.G.; Zeng, Q.; Li, J.; Guo, K.; et al. PRL-3, a metastasis associated tyrosine phosphatase, is involved in FLT3-ITD signaling and implicated in anti-AML therapy. PLoS ONE 2011, 6, e19798. [Google Scholar] [CrossRef]
- Zhou, J.; Chan, Z.L.; Bi, C.; Lu, X.; Chong, P.S.; Chooi, J.Y.; Cheong, L.L.; Liu, S.C.; Ching, Y.Q.; Zhou, Y.; et al. LIN28B Activation by PRL-3 Promotes Leukemogenesis and a Stem Cell-like Transcriptional Program in AML. Mol. Cancer Res. 2017, 15, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.S.Y.; Zhou, J.; Chooi, J.Y.; Chan, Z.L.; Toh, S.H.M.; Tan, T.Z.; Wee, S.; Gunaratne, J.; Zeng, Q.; Chng, W.J. Non-canonical activation of beta-catenin by PRL-3 phosphatase in acute myeloid leukemia. Oncogene 2019, 38, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, L.; Wellbrock, J.; Fiedler, W. Acute Myeloid Leukemia and the Bone Marrow Niche-Take a Closer Look. Front. Oncol. 2018, 8, 444. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Kunisada, T.; Tsukamoto, H.; Yamazaki, H.; Niwa, H.; Takada, S.; Hayashi, S.I. Wnt signaling regulates hemopoiesis through stromal cells. J. Immunol. 2001, 167, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Wang, Y.J.; Lo Celso, C.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G.; et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Mosialou, I.; Manavalan, S.J.; Rathinam, C.V.; Friedman, R.A.; Teruya-Feldstein, J.; Bhagat, G.; Berman, E.; Kousteni, S. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 2016, 30, 1–13. [Google Scholar] [CrossRef]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/beta-catenin signaling pathway. J. Hematol. Oncol. 2015, 8, 1. [Google Scholar] [CrossRef]
- Chien, S.; Haq, S.U.; Pawlus, M.; Moon, R.T.; Estey, E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L.; Becker, P.S. Adhesion of Acute Myeloid Leukemia Blasts to E-Selectin In The Vascular Niche Enhances Their Survival By Mechanisms Such As Wnt Activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef]
- Heidel, F.H.; Bullinger, L.; Arreba-Tutusaus, P.; Wang, Z.; Gaebel, J.; Hirt, C.; Niederwieser, D.; Lane, S.W.; Dohner, K.; Vasioukhin, V.; et al. The cell fate determinant Llgl1 influences HSC fitness and prognosis in AML. J. Exp. Med. 2013, 210, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Beekman, R.; Valkhof, M.G.; Sanders, M.A.; van Strien, P.M.; Haanstra, J.R.; Broeders, L.; Geertsma-Kleinekoort, W.M.; Veerman, A.J.; Valk, P.J.; Verhaak, R.G.; et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood 2012, 119, 5071–5077. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Lhoumeau, A.C.; Arnoulet, C.; Aulas, A.; Marchetto, S.; Audebert, S.; Puppo, F.; Chabannon, C.; Sainty, D.; Santoni, M.J.; et al. The cell polarity PTK7 receptor acts as a modulator of the chemotherapeutic response in acute myeloid leukemia and impairs clinical outcome. Blood 2010, 116, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Lhoumeau, A.C.; Arcangeli, M.L.; De Grandis, M.; Giordano, M.; Orsoni, J.C.; Lembo, F.; Bardin, F.; Marchetto, S.; Aurrand-Lions, M.; Borg, J.P. Ptk7-Deficient Mice Have Decreased Hematopoietic Stem Cell Pools as a Result of Deregulated Proliferation and Migration. J. Immunol. 2016, 196, 4367–4377. [Google Scholar] [CrossRef] [PubMed]
- Zang, S.; Liu, N.; Wang, H.; Wald, D.N.; Shao, N.; Zhang, J.; Ma, D.; Ji, C.; Tse, W. Wnt signaling is involved in 6-benzylthioinosine-induced AML cell differentiation. BMC Cancer 2014, 14, 886. [Google Scholar] [CrossRef] [PubMed]
- Ysebaert, L.; Chicanne, G.; Demur, C.; De Toni, F.; Prade-Houdellier, N.; Ruidavets, J.B.; Mansat-De Mas, V.; Rigal-Huguet, F.; Laurent, G.; Payrastre, B.; et al. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006, 20, 1211–1216. [Google Scholar] [CrossRef]
- Xu, J.; Suzuki, M.; Niwa, Y.; Hiraga, J.; Nagasaka, T.; Ito, M.; Nakamura, S.; Tomita, A.; Abe, A.; Kiyoi, H.; et al. Clinical significance of nuclear non-phosphorylated beta-catenin in acute myeloid leukaemia and myelodysplastic syndrome. Br. J. Haematol. 2008, 140, 394–401. [Google Scholar] [CrossRef]
- Chen, C.C.; Gau, J.P.; You, J.Y.; Lee, K.D.; Yu, Y.B.; Lu, C.H.; Lin, J.T.; Lan, C.; Lo, W.H.; Liu, J.M.; et al. Prognostic significance of beta-catenin and topoisomerase IIalpha in de novo acute myeloid leukemia. Am. J. Hematol. 2009, 84, 87–92. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Golding, M.C.; Srivastava, P.; Povinelli, B.J.; James, S.R.; Ford, L.A.; Wetzler, M.; Wang, E.S.; Nemeth, M.J. Pharmacological targeting of beta-catenin in normal karyotype acute myeloid leukemia blasts. Haematologica 2015, 100, e49–e52. [Google Scholar] [CrossRef]
- Xu, J.; Wu, W.; Shen, W.; Liu, P. The clinical significance of gamma-catenin in acute myeloid leukemia. Onco. Targets Ther. 2016, 9, 3861–3871. [Google Scholar] [CrossRef] [PubMed]
- Skokowa, J.; Cario, G.; Uenalan, M.; Schambach, A.; Germeshausen, M.; Battmer, K.; Zeidler, C.; Lehmann, U.; Eder, M.; Baum, C.; et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat. Med. 2006, 12, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P.; Qiu, Y.; Coombes, K.R.; Zhang, N.; Neeley, E.S.; Ruvolo, V.R.; Hail, N., Jr.; Borthakur, G.; Konopleva, M.; Andreeff, M.; et al. Phosphorylation of GSK3alpha/beta correlates with activation of AKT and is prognostic for poor overall survival in acute myeloid leukemia patients. BBA Clin. 2015, 4, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Taskesen, E.; Staal, F.J.; Reinders, M.J. An integrated approach of gene expression and DNA-methylation profiles of WNT signaling genes uncovers novel prognostic markers in acute myeloid leukemia. BMC Bioinformatics 2015, 16 (Suppl. 4), S4. [Google Scholar] [CrossRef]
- Martin, V.; Valencia, A.; Agirre, X.; Cervera, J.; San Jose-Eneriz, E.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Sanz, M.A.; Herrera, C.; Torres, A.; et al. Epigenetic regulation of the non-canonical Wnt pathway in acute myeloid leukemia. Cancer Sci. 2010, 101, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Xiang, T.; Mu, J.; Mao, H.; Li, L.; Huang, X.; Li, C.; Feng, Y.; Luo, X.; Wei, Y.; et al. Protocadherin 17 functions as a tumor suppressor suppressing Wnt/beta-catenin signaling and cell metastasis and is frequently methylated in breast cancer. Oncotarget 2016, 7, 51720–51732. [Google Scholar] [CrossRef]
- Xu, Z.J.; Ma, J.C.; Zhou, J.D.; Wen, X.M.; Yao, D.M.; Zhang, W.; Ji, R.B.; Wu, D.H.; Tang, L.J.; Deng, Z.Q.; et al. Reduced protocadherin17 expression in leukemia stem cells: The clinical and biological effect in acute myeloid leukemia. J. Transl. Med. 2019, 17, 102. [Google Scholar] [CrossRef]
- Astori, A.; Fredly, H.; Aloysius, T.A.; Bullinger, L.; Mansat-De Mas, V.; de la Grange, P.; Delhommeau, F.; Hagen, K.M.; Recher, C.; Dusanter-Fourt, I.; et al. CXXC5 (retinoid-inducible nuclear factor, RINF) is a potential therapeutic target in high-risk human acute myeloid leukemia. Oncotarget 2013, 4, 1438–1448. [Google Scholar] [CrossRef]
- Bruserud, O.; Reikvam, H.; Fredly, H.; Skavland, J.; Hagen, K.M.; van Hoang, T.T.; Brenner, A.K.; Kadi, A.; Astori, A.; Gjertsen, B.T.; et al. Expression of the potential therapeutic target CXXC5 in primary acute myeloid leukemia cells - high expression is associated with adverse prognosis as well as altered intracellular signaling and transcriptional regulation. Oncotarget 2015, 6, 2794–2811. [Google Scholar] [CrossRef]
- Kuhnl, A.; Valk, P.J.; Sanders, M.A.; Ivey, A.; Hills, R.K.; Mills, K.I.; Gale, R.E.; Kaiser, M.F.; Dillon, R.; Joannides, M.; et al. Downregulation of the Wnt inhibitor CXXC5 predicts a better prognosis in acute myeloid leukemia. Blood 2015, 125, 2985–2994. [Google Scholar] [CrossRef]
- Minke, K.S.; Staib, P.; Puetter, A.; Gehrke, I.; Gandhirajan, R.K.; Schlosser, A.; Schmitt, E.K.; Hallek, M.; Kreuzer, K.A. Small molecule inhibitors of WNT signaling effectively induce apoptosis in acute myeloid leukemia cells. Eur. J. Haematol. 2009, 82, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Hu, C.; Mei, C.; Ren, Z.; Vera, J.C.; Zhuang, Z.; Jin, J.; Tong, H. Sequential combination of decitabine and idarubicin synergistically enhances anti-leukemia effect followed by demethylating Wnt pathway inhibitor promoters and downregulating Wnt pathway nuclear target. J. Transl. Med. 2014, 12, 167. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yang, L.L.; Niu, T.; Cheng, C.; Zhong, L.; Zheng, M.W.; Xiong, Y.; Li, L.L.; Xiang, R.; Chen, L.J.; et al. SKLB-677, an FLT3 and Wnt/beta-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci. Rep. 2015, 5, 15646. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Mak, D.H.; Kornblau, S.; Zhang, Q.; Ruvolo, P.; Burks, J.K.; Zhang, W.; et al. Disruption of Wnt/beta-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2417–2429. [Google Scholar] [CrossRef]
- Chen, C.; Xu, W.; Wang, C.M. Combination of celecoxib and doxorubicin increases growth inhibition and apoptosis in acute myeloid leukemia cells. Leuk Lymphoma 2013, 54, 2517–2522. [Google Scholar] [CrossRef]
- Casanova, I.; Bosch, R.; Lasa, A.; Parreno, M.; Cespedes, M.V.; Brunet, S.; Nomdedeu, J.F.; Mangues, M.A.; Sierra, J.; Mangues, R. A celecoxib derivative inhibits focal adhesion signaling and induces caspase-8-dependent apoptosis in human acute myeloid leukemia cells. Int. J. Cancer 2008, 123, 217–226. [Google Scholar] [CrossRef]
- Winer, E.S.; Stone, R.M. Novel therapy in Acute myeloid leukemia (AML): Moving toward targeted approaches. Ther. Adv. Hematol. 2019, 10, 2040620719860645. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wnt Antagonist | Name | Target | Mechanism of Action | Canonical/Non-Canonical Signalling Inhibition |
|---|---|---|---|---|
| Secreted | sFRP | Wnt | Sequestration of Wnt | Both |
| FZD | Binding to FZD receptor | |||
| DKK 1, 2 and 4 | LRP6 | Disruption of FZD-LRP6 complex | Canonical | |
| Induction of LRP6 endocytosis | ||||
| WIF | Wnt | Sequestration of Wnt | Both | |
| Wise/SOST | LRP6 | Disruption of FZD-LRP6 complex | Canonical | |
| Cerberus | Wnt | Sequestration of Wnt | Both | |
| IGFBP | LRP6 and FZD | Disruption of FZD-LRP6 complex | Canonical | |
| Trans-membrane | Waif1 | LRP6 | Disruption of FZD-LRP6 complex | Canonical |
| APCDD1 | Wnt and LRP5 | Sequestration of Wnt | Canonical | |
| Tiki1 | Wnt | Removal of eight amino-terminal residues from Wnt | Both |
| Prognostic Factor | Number of Cases | Age (Median) | AML Type | Therapy | Technology Used | Prognosis | Reference |
|---|---|---|---|---|---|---|---|
| High β-catenin | 82 | 57 | De novo | Induction + consolidation | WB | Poor | [57] |
| 54 | 53 | De novo | Not specified | IHC | [58] | ||
| 59 | 59.6 | De novo | Induction + anthracycline or low dose cytarabine | IHC | [59] | ||
| 21 | 65 | De novo/relapse | Not specified | Flow cytometry | [60] | ||
| High LEF-1 | 101 | 47 | De novo | Untreated | RT-qPCR | Good | [20] |
| 406 | <60 | De novo | Induction + consolidation | Microarrays and RT-qPCR | [21] | ||
| 344 | 48 | De novo/relapse/secondary | Induction + consolidation ± transplantation | Genome-wide mRNA and DNA-methylation profiling data * | [64] | ||
| High phosphorylated-GSKβ | 511 | 65.7 | De novo | Not specified | Reverse-phase protein analysis | Poor | [63] |
| Low PSMD2 | 344 | 48 | De novo/relapse/secondary | Induction + consolidation ± transplantation | Genome-wide mRNA and DNA-methylation profiling data * | Good | [64] |
| High PPARD | Good | ||||||
| High XPNPEP | Poor | ||||||
| Low sFRP2 | Poor | ||||||
| Hypermethylated RUNX1 | Poor | ||||||
| Low AXIN2 | Poor | ||||||
| Low WNT5A | 252 | 57 | De novo | Induction + consolidation ± transplantation | Methylation-specific PCR | Poor | [65] |
| Low PCDH17 | 626 | 52 | De novo | Induction + consolidation ± transplantation | RT-qPCR | Poor | [67] |
| High CXXC5 | 27 | 64 | De novo $# | Induction + consolidation | RT-qPCR | Poor | [68] |
| 67 | 64 | De novo/secondary $ | Not specified | RT-qPCR | [69] | ||
| Low CXXC5 | 529 | 46 | De novo | Induction + consolidation ± transplantation | Microarray and RT-qPCR | Good | [70] |
| Low LLGL1 | 83 | <60 | Normal karyotype | Not specified | RT-qPCR | Poor | [52] |
| High PTK7 | 184 | 63 | De novo/secondary $ | Induction + consolidation ± transplantation | Flow cytometry | Poor | [54] |
| Compound | Target | Clinical Trial Phase (Number) | Status | Results |
|---|---|---|---|---|
| Celecoxib | COX-2 | Phase Ib (NCT03878524) | Not yet recruiting | NA |
| CWP232291 | β-catenin degradation | Phase I (NCT01398462) | Completed | No results posted |
| LY2090314 | GSK3β | Phase II (NCT01214603) | Completed | Well tolerated No CR or PR |
| PRI-724 | β-catenin/CBP | Phase I (NCT01606579) | Completed | No results posted |
| Sulindac | PDZ domain of DVL | Phase II (NCT01843179) | Withdrawn (lack of funding) | No results posted |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403. https://doi.org/10.3390/cells8111403
Gruszka AM, Valli D, Alcalay M. Wnt Signalling in Acute Myeloid Leukaemia. Cells. 2019; 8(11):1403. https://doi.org/10.3390/cells8111403
Chicago/Turabian StyleGruszka, Alicja M., Debora Valli, and Myriam Alcalay. 2019. "Wnt Signalling in Acute Myeloid Leukaemia" Cells 8, no. 11: 1403. https://doi.org/10.3390/cells8111403
APA StyleGruszka, A. M., Valli, D., & Alcalay, M. (2019). Wnt Signalling in Acute Myeloid Leukaemia. Cells, 8(11), 1403. https://doi.org/10.3390/cells8111403