A Matricryptic Conformation of the Integrin-Binding Domain of Fibronectin Regulates Platelet-Derived Growth Factor-Induced Intracellular Calcium Release


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
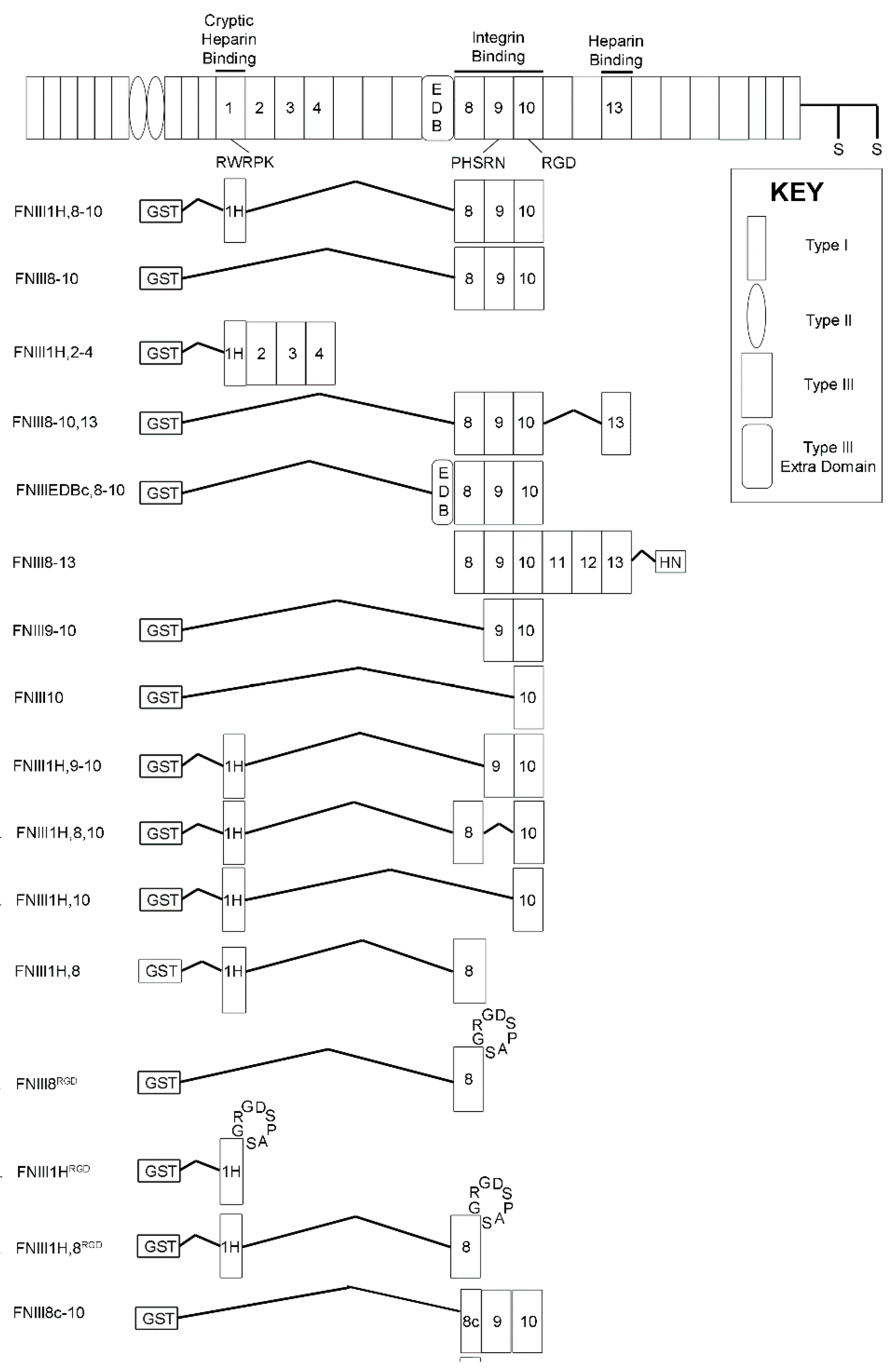
2.3. Recombinant Proteins
2.4. Intracellular Calcium Release
2.5. Immunoblotting
2.6. Molecular Dynamics Simulations
2.7. Cell Attachment Assays
2.8. Statistical Analyses
3. Results
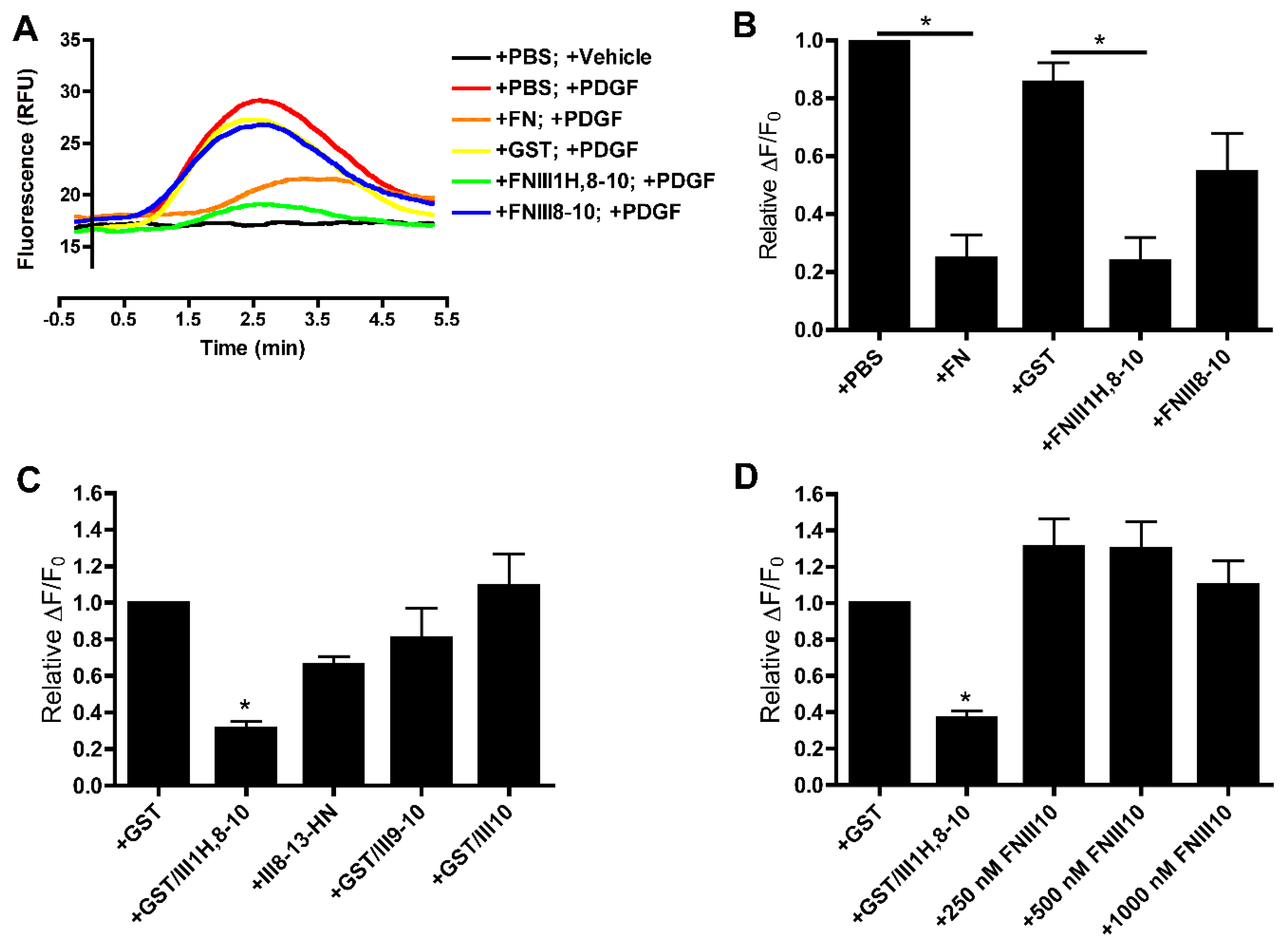
3.1. Effects of Cell-Binding Fibronectin Fragments on Platelet-Derived Growth Factor (PDGF)-Induced Intracellular Calcium Release
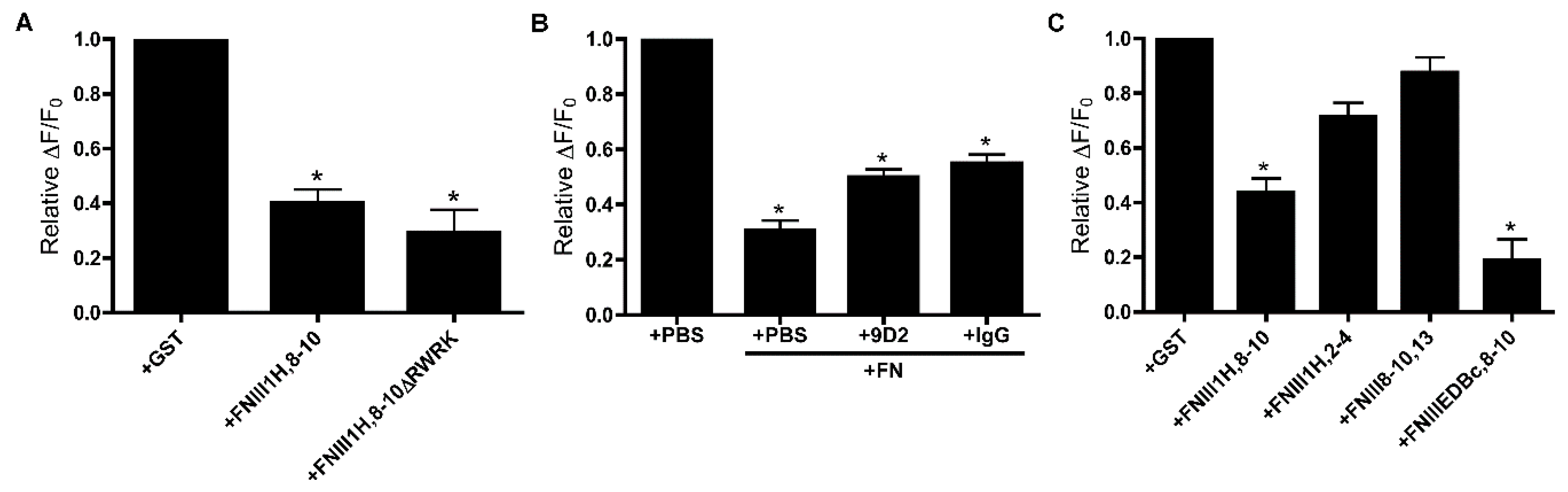
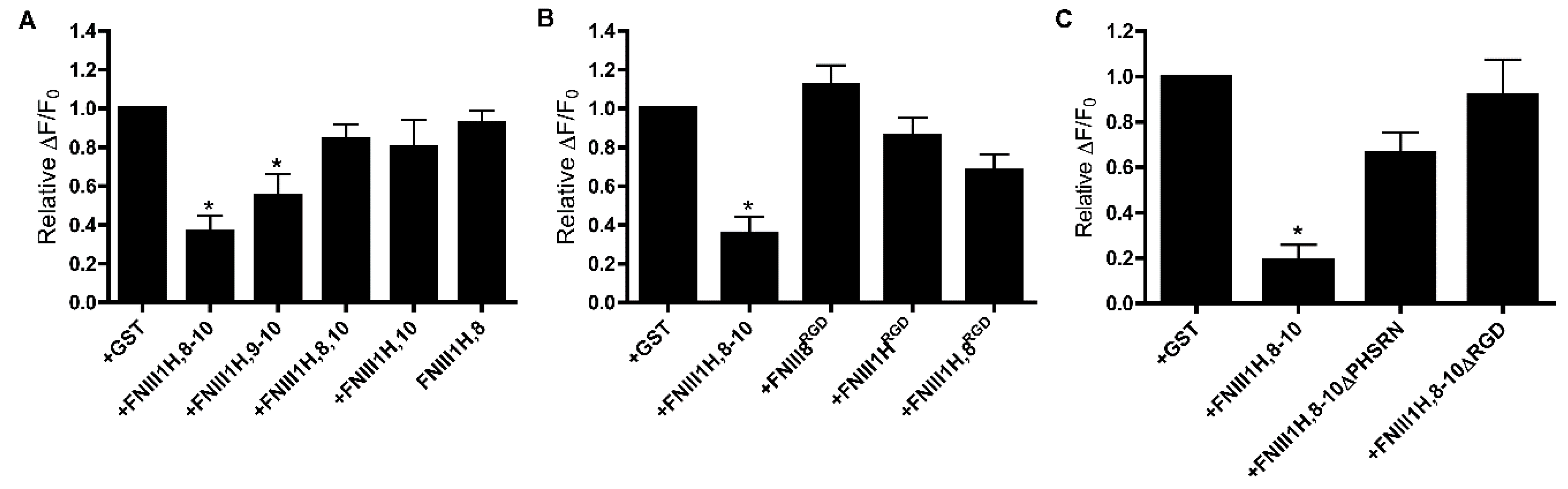
3.2. Role of FNIII1H in Attenuating PDGF-Induced Intracellular Calcium Release
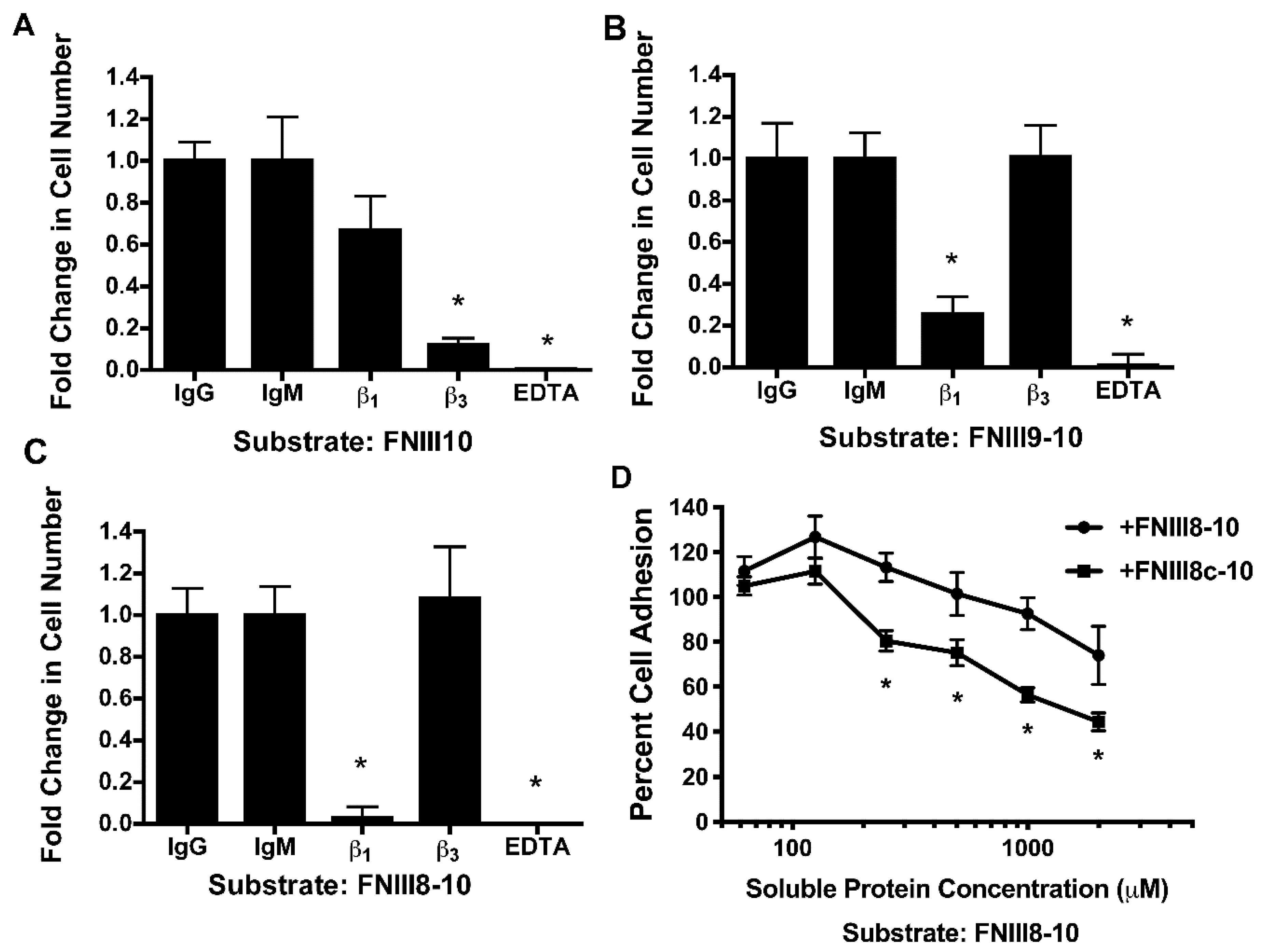
3.3. Role of FNIII9 and FNIII10 in Attenuating PDGF-Induced Intracellular Calcium Release
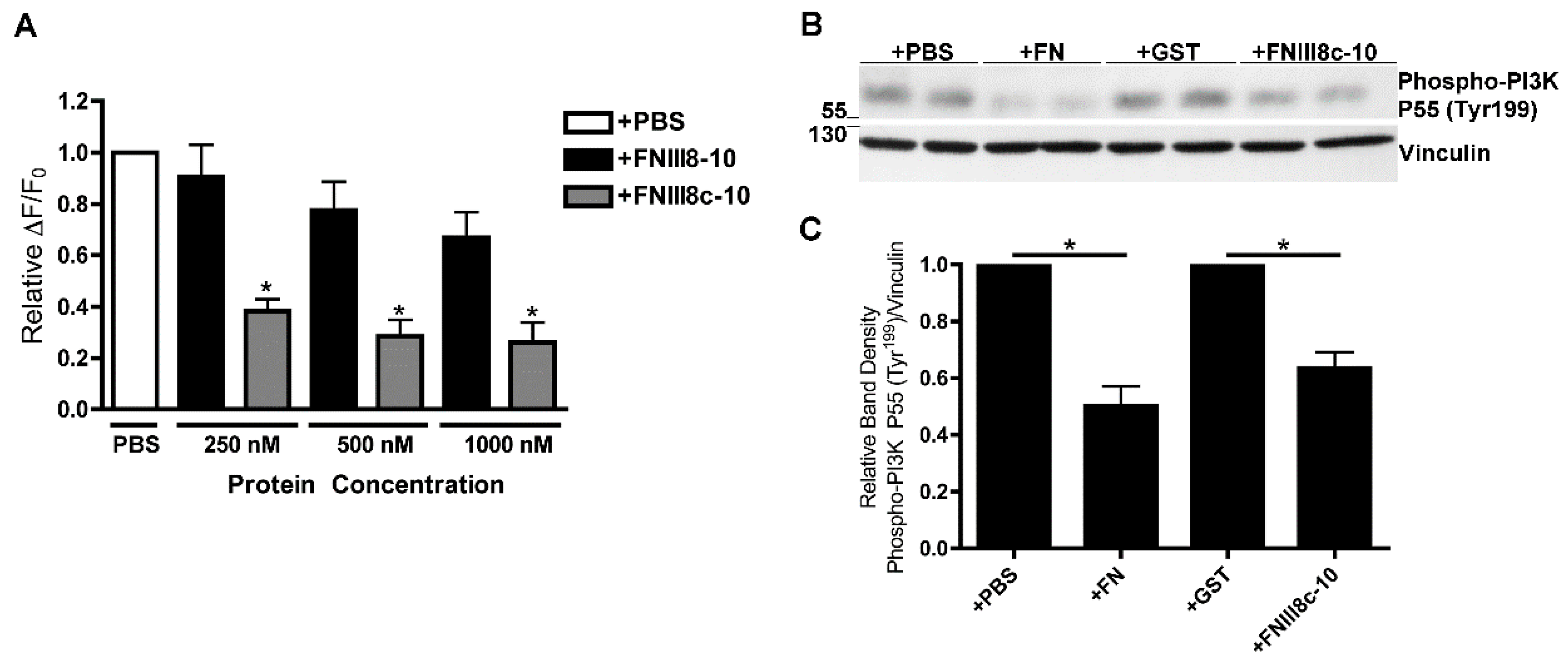
3.4. Removal of β Strand a from FNIII8 Confers Functional Activity
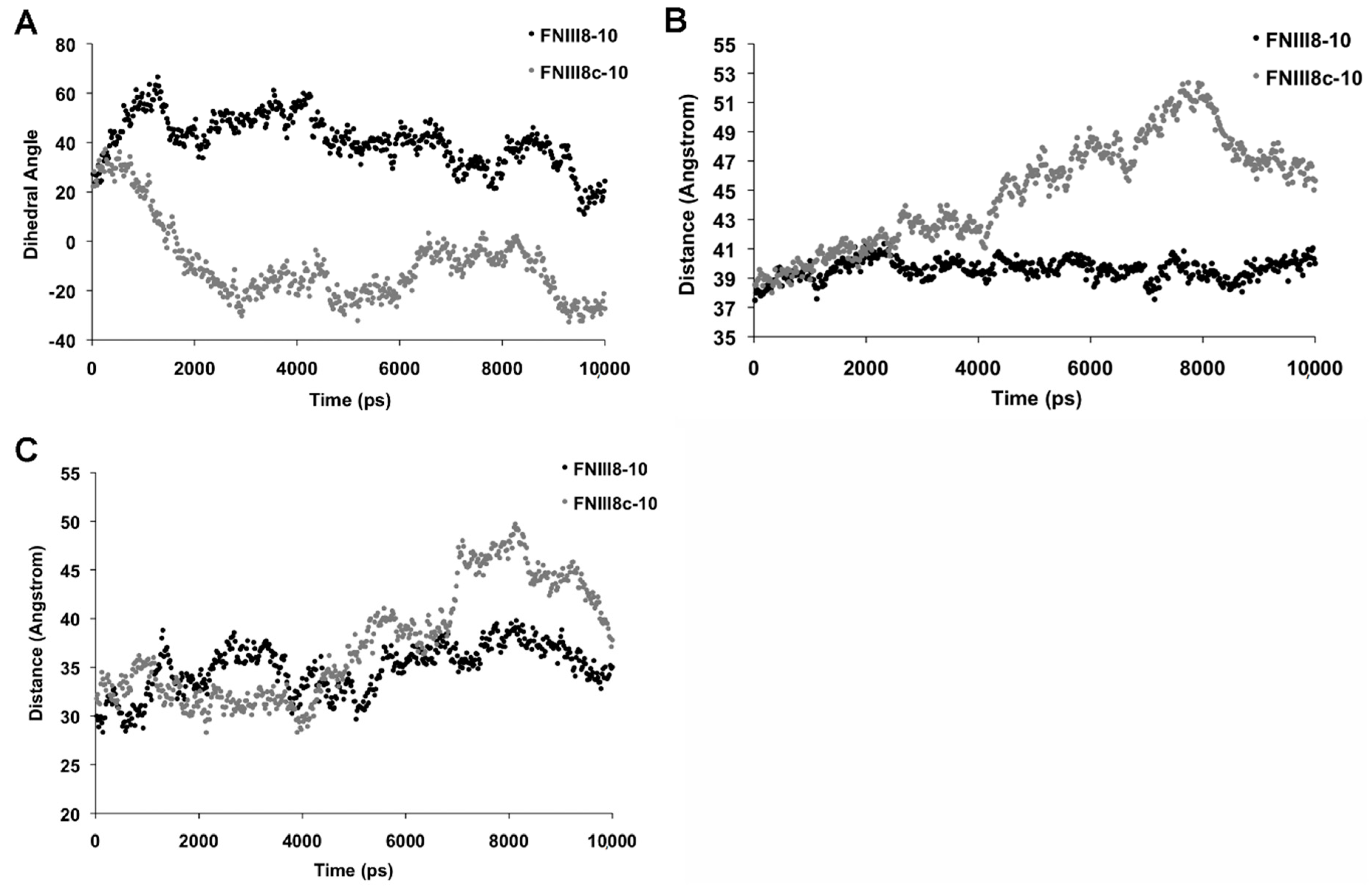
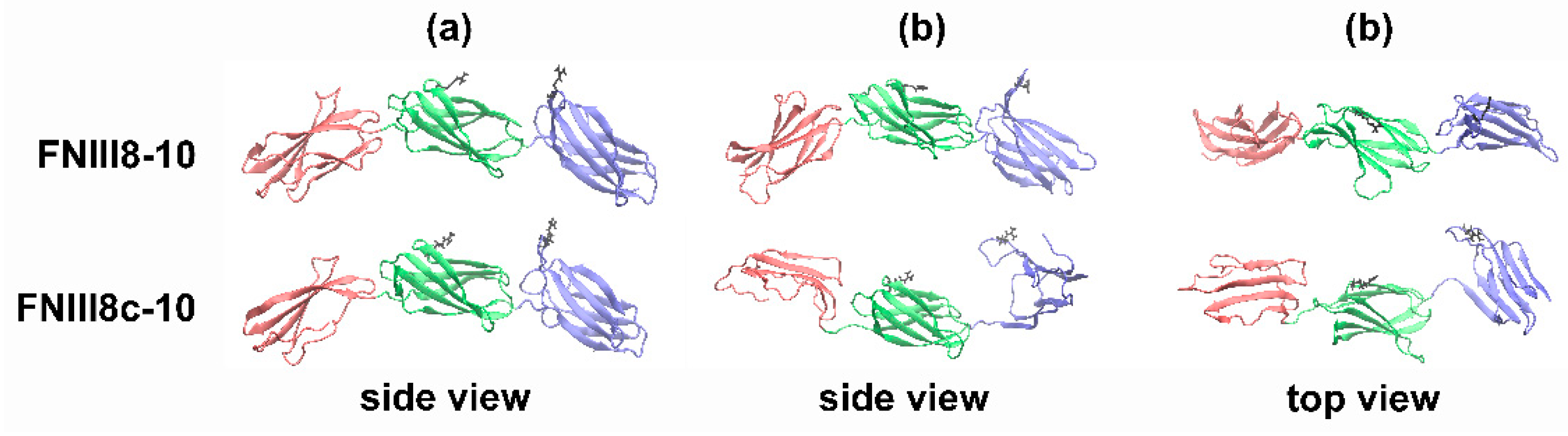
3.5. Molecular Modeling of FNIII8c-10
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Cooper, G.M. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, A.; Hammacher, A.; Westermark, B.; Heldin, C.H. Differential effects of the various isoforms of platelet-derived growth factor on chemotaxis of fibroblasts, monocytes, and granulocytes. J. Clin. Investig. 1990, 85, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Folkvord, J.M.; Hart, C.E.; Murray, M.J.; McPherson, J.M. Platelet isoforms of platelet-derived growth factor stimulate fibroblasts to contract collagen matrices. J. Clin. Investig. 1989, 84, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, D.; Tingstrom, A.; Thuresson, A.C.; Olsson, L.; Terracio, L.; Borg, T.K.; Rubin, K. Beta 1 integrin-mediated collagen gel contraction is stimulated by PDGF. Exp. Cell Res. 1990, 186, 264–272. [Google Scholar] [CrossRef]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Nagai, M.K.; Embil, J.M. Becaplermin: Recombinant platelet derived growth factor, a new treatment for healing diabetic foot ulcers. Expert Opin. Biol. 2002, 2, 211–218. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsubara, H.; Akagi, S.; Sarashina, T.; Ejiri, K.; Kawakita, N.; Yoshida, M.; Miyoshi, T.; Watanabe, A.; Nishii, N.; et al. Nanoparticle-Mediated Drug Delivery System for Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Farrar, C.S.; Hocking, D.C. Assembly of fibronectin fibrils selectively attenuates platelet-derived growth factor-induced intracellular calcium release in fibroblasts. J. Biol. Chem. 2018, 293, 18655–18666. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, G.; Baugh, L.; Vogel, V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc. Natl. Acad. Sci. USA 2002, 99, 5139–5143. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Dysart, M.M.; Clarke, K.C.; Stabenfeldt, S.E.; Barker, T.H. Integrin alpha3beta1 Binding to Fibronectin Is Dependent on the Ninth Type III Repeat. J. Biol. Chem. 2015, 290, 25534–25547. [Google Scholar] [CrossRef] [PubMed]
- Krammer, A.; Craig, D.; Thomas, W.E.; Schulten, K.; Vogel, V. A structural model for force regulated integrin binding to fibronectin’s RGD-synergy site. Matrix Biol. 2002, 21, 139–147. [Google Scholar] [CrossRef]
- Zhong, C.; Chrzanowska-Wodnicka, M.; Brown, J.; Shaub, A.; Belkin, A.M.; Burridge, K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 1998, 141, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zeller, M.K.; Fiore, V.F.; Strane, P.; Bermudez, H.; Barker, T.H. Phage-based molecular probes that discriminate force-induced structural states of fibronectin in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 7251–7256. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Lemmon, C.A.; Erickson, H.P. Fibronectin Conformation and Assembly: Analysis of Fibronectin Deletion Mutants and Fibronectin Glomerulopathy (GFND) Mutants. Biochemistry 2017, 56, 4584–4591. [Google Scholar] [CrossRef]
- Hocking, D.C.; Kowalski, K. A cryptic fragment from fibronectin’s III1 module localizes to lipid rafts and stimulates cell growth and contractility. J. Cell Biol. 2002, 158, 175–184. [Google Scholar] [CrossRef]
- Hocking, D.C.; Titus, P.A.; Sumagin, R.; Sarelius, I.H. Extracellular matrix fibronectin mechanically couples skeletal muscle contraction with local vasodilation. Circ. Res. 2008, 102, 372–379. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Pierschbacher, M.D. New perspectives in cell adhesion: RGD and integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Altroff, H.; van der Walle, C.F.; Asselin, J.; Fairless, R.; Campbell, I.D.; Mardon, H.J. The eighth FIII domain of human fibronectin promotes integrin alpha5beta1 binding via stabilization of the ninth FIII domain. J. Biol. Chem. 2001, 276, 38885–38892. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Culp, L.A. Adhesion mediated by fibronectin’s alternatively spliced EDb (EIIIB) and its neighboring type III repeats. Exp. Cell Res. 1996, 223, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.Y.; Weng, Z.; Moll, S.; Kim, S.; Brown, C.T. Identification and validation of a novel cell-recognition site (KNEED) on the 8th type III domain of fibronectin. Biomaterials 2002, 23, 3865–3870. [Google Scholar] [CrossRef]
- Aota, S.; Nomizu, M.; Yamada, K.M. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 1994, 269, 24756–24761. [Google Scholar] [PubMed]
- Redick, S.D.; Settles, D.L.; Briscoe, G.; Erickson, H.P. Defining fibronectin’s cell adhesion synergy site by site-directed mutagenesis. J. Cell Biol. 2000, 149, 521–527. [Google Scholar] [CrossRef]
- Leahy, D.J.; Aukhil, I.; Erickson, H.P. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 1996, 84, 155–164. [Google Scholar] [CrossRef]
- Petrie, T.A.; Capadona, J.R.; Reyes, C.D.; Garcia, A.J. Integrin specificity and enhanced cellular activities associated with surfaces presenting a recombinant fibronectin fragment compared to RGD supports. Biomaterials 2006, 27, 5459–5470. [Google Scholar] [CrossRef]
- Altroff, H.; Schlinkert, R.; van der Walle, C.F.; Bernini, A.; Campbell, I.D.; Werner, J.M.; Mardon, H.J. Interdomain tilt angle determines integrin-dependent function of the ninth and tenth FIII domains of human fibronectin. J. Biol. Chem. 2004, 279, 55995–56003. [Google Scholar] [CrossRef]
- Martino, M.M.; Mochizuki, M.; Rothenfluh, D.A.; Rempel, S.A.; Hubbell, J.A.; Barker, T.H. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials 2009, 30, 1089–1097. [Google Scholar] [CrossRef]
- Ambesi, A.; McKeown-Longo, P.J. Conformational remodeling of the fibronectin matrix selectively regulates VEGF signaling. J. Cell Sci. 2014, 127, 3805–3816. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Nicosia, J.; Larouche, J.; Zhang, Y.; Bachman, H.; Brown, A.C.; Holmgren, L.; Barker, T.H. Detection of an Integrin-Binding Mechanoswitch within Fibronectin during Tissue Formation and Fibrosis. ACS Nano 2017, 11, 7110–7117. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Andresen Eguiluz, R.C.; Wu, F.; Seo, B.R.; Fischbach, C.; Gourdon, D. Stiffening and unfolding of early deposited-fibronectin increase proangiogenic factor secretion by breast cancer-associated stromal cells. Biomaterials 2015, 54, 63–71. [Google Scholar] [CrossRef]
- Liang, X.; Garcia, B.L.; Visai, L.; Prabhakaran, S.; Meenan, N.A.; Potts, J.R.; Humphries, M.J.; Hook, M. Allosteric Regulation of Fibronectin/alpha5beta1 Interaction by Fibronectin-Binding MSCRAMMs. PLoS ONE 2016, 11, e0159118. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.C.; Hocking, D.C. Recombinant fibronectin matrix mimetics specify integrin adhesion and extracellular matrix assembly. Tissue Eng. Part. A 2013, 19, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.K.; Yamada, S.S.; Chen, W.T.; Yamada, K.M. Analysis of fibronectin receptor function with monoclonal antibodies: Roles in cell adhesion, migration, matrix assembly, and cytoskeletal organization. J. Cell Biol. 1989, 109, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Chernousov, M.A.; Fogerty, F.J.; Koteliansky, V.E.; Mosher, D.F. Role of the I-9 and III-1 modules of fibronectin in formation of an extracellular fibronectin matrix. J. Biol. Chem. 1991, 266, 10851–10858. [Google Scholar]
- Sottile, J.; Hocking, D.C.; Swiatek, P.J. Fibronectin matrix assembly enhances adhesion-dependent cell growth. J. Cell Sci. 1998, 111 Pt 19, 2933–2943. [Google Scholar]
- Hocking, D.C.; Smith, R.K.; McKeown-Longo, P.J. A novel role for the integrin-binding III-10 module in fibronectin matrix assembly. J. Cell Biol. 1996, 133, 431–444. [Google Scholar] [CrossRef]
- Hocking, D.C.; Sottile, J.; Reho, T.; Fassler, R.; McKeown-Longo, P.J. Inhibition of fibronectin matrix assembly by the heparin-binding domain of vitronectin. J. Biol. Chem. 1999, 274, 27257–27264. [Google Scholar] [CrossRef]
- Gui, L.; Wojciechowski, K.; Gildner, C.D.; Nedelkovska, H.; Hocking, D.C. Identification of the heparin-binding determinants within fibronectin repeat III1: Role in cell spreading and growth. J. Biol. Chem. 2006, 281, 34816–34825. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.C.; Wilke-Mounts, S.J.; Hocking, D.C. Chimeric fibronectin matrix mimetic as a functional growth- and migration-promoting adhesive substrate. Biomaterials 2011, 32, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.R.; Hocking, D.C. Cooperative effects of fibronectin matrix assembly and initial cell-substrate adhesion strength in cellular self-assembly. Acta Biomater. 2016, 32, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Hocking, D.C.; Sottile, J.; McKeown-Longo, P.J. Fibronectin’s III-1 module contains a conformation-dependent binding site for the amino-terminal region of fibronectin. J. Biol. Chem. 1994, 269, 19183–19187. [Google Scholar] [PubMed]
- Straub, S.V.; Giovannucci, D.R.; Yule, D.I. Calcium wave propagation in pancreatic acinar cells: Functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J. Gen. Physiol. 2000, 116, 547–560. [Google Scholar] [CrossRef]
- Johenning, F.W.; Zochowski, M.; Conway, S.J.; Holmes, A.B.; Koulen, P.; Ehrlich, B.E. Distinct intracellular calcium transients in neurites and somata integrate neuronal signals. J. Neurosci. 2002, 22, 5344–5353. [Google Scholar] [CrossRef]
- Qui, D.; Shenkin, P.S.; Hollinger, F.P.; Still, W.C. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate born radii. J. Phys. Chem. A 1997, 101, 3005–3014. [Google Scholar]
- Moolenaar, W.H.; Tertoolen, L.G.; de Laat, S.W. Growth factors immediately raise cytoplasmic free Ca2+ in human fibroblasts. J. Biol. Chem. 1984, 259, 8066–8069. [Google Scholar]
- Sarelius, I.H.; Titus, P.A.; Maimon, N.; Okech, W.; Wilke-Mounts, S.J.; Brennan, J.R.; Hocking, D.C. Extracellular matrix fibronectin initiates endothelium-dependent arteriolar dilatation via the heparin-binding, matricryptic RWRPK sequence of the first type III repeat of fibrillar fibronectin. J. Physiol. 2016, 594, 687–697. [Google Scholar] [CrossRef]
- Okech, W.; Abberton, K.M.; Kuebel, J.M.; Hocking, D.C.; Sarelius, I.H. Extracellular matrix fibronectin mediates an endothelial cell response to shear stress via the heparin-binding, matricryptic RWRPK sequence of FNIII1H. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1063–H1071. [Google Scholar] [CrossRef]
- Garcia, A.J.; Schwarzbauer, J.E.; Boettiger, D. Distinct activation states of alpha5beta1 integrin show differential binding to RGD and synergy domains of fibronectin. Biochemistry 2002, 41, 9063–9069. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.; Krammer, A.; Schulten, K.; Vogel, V. Comparison of the early stages of forced unfolding for fibronectin type III modules. Proc. Natl. Acad. Sci. USA 2001, 98, 5590–5595. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Ostman, A.; Ronnstrand, L. Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta 1998, 1378, F79–F113. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Craig, D.; Lequin, O.; Campbell, I.D.; Vogel, V.; Schulten, K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc. Natl. Acad. Sci. USA 2003, 100, 14784–14789. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Craig, D.; Vogel, V.; Schulten, K. Identifying unfolding intermediates of FN-III(10) by steered molecular dynamics. J. Mol. Biol. 2002, 323, 939–950. [Google Scholar] [CrossRef]
- Pan, D.; Song, Y. Role of altered sialylation of the I-like domain of beta1 integrin in the binding of fibronectin to beta1 integrin: Thermodynamics and conformational analyses. Biophys. J. 2001, 99, 208–217. [Google Scholar] [CrossRef]
- Smith, M.L.; Gourdon, D.; Little, W.C.; Kubow, K.E.; Eguiluz, R.A.; Luna-Morris, S.; Vogel, V. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 2007, 5, e268. [Google Scholar] [CrossRef]
- Bencharit, S.; Cui, C.B.; Siddiqui, A.; Howard-Williams, E.L.; Sondek, J.; Zuobi-Hasona, K.; Aukhil, I. Structural insights into fibronectin type III domain-mediated signaling. J. Mol. Biol. 2007, 367, 303–309. [Google Scholar] [CrossRef]
- Ugarova, T.P.; Zamarron, C.; Veklich, Y.; Bowditch, R.D.; Ginsberg, M.H.; Weisel, J.W.; Plow, E.F. Conformational transitions in the cell binding domain of fibronectin. Biochemistry 1995, 34, 4457–4466. [Google Scholar] [CrossRef]
- Craig, J.A.; Rexeisen, E.L.; Mardilovich, A.; Shroff, K.; Kokkoli, E. Effect of linker and spacer on the design of a fibronectin-mimetic peptide evaluated via cell studies and AFM adhesion forces. Langmuir 2008, 24, 10282–10292. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Mukherjee, S.; Duan, F.; Kolb, M.R.; Janssen, L.J. Platelet derived growth factor-evoked Ca2+ wave and matrix gene expression through phospholipase C in human pulmonary fibroblast. Int. J. Biochem. Cell Biol. 2013, 45, 1516–1524. [Google Scholar] [CrossRef]
- Hollenbeck, S.T.; Nelson, P.R.; Yamamura, S.; Faries, P.L.; Liu, B.; Kent, K.C. Intracellular calcium transients are necessary for platelet-derived growth factor but not extracellular matrix protein-induced vascular smooth muscle cell migration. J. Vasc. Surg. 2004, 40, 351–358. [Google Scholar] [CrossRef]
- Martino, M.M.; Tortelli, F.; Mochizuki, M.; Traub, S.; Ben-David, D.; Kuhn, G.A.; Muller, R.; Livne, E.; Eming, S.A.; Hubbell, J.A. Engineering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Sci. Transl. Med. 2011, 3, 100ra189. [Google Scholar] [CrossRef]
- Lin, F.; Zhu, J.; Tonnesen, M.G.; Taira, B.R.; McClain, S.A.; Singer, A.J.; Clark, R.A. Fibronectin peptides that bind PDGF-BB enhance survival of cells and tissue under stress. J. Invest. Derm. 2014, 134, 1119–1127. [Google Scholar] [CrossRef]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farrar, C.S.; Rouin, G.T.; Miller, B.L.; Raeman, C.H.; Mooney, N.A.; Hocking, D.C. A Matricryptic Conformation of the Integrin-Binding Domain of Fibronectin Regulates Platelet-Derived Growth Factor-Induced Intracellular Calcium Release. Cells 2019, 8, 1351. https://doi.org/10.3390/cells8111351
Farrar CS, Rouin GT, Miller BL, Raeman CH, Mooney NA, Hocking DC. A Matricryptic Conformation of the Integrin-Binding Domain of Fibronectin Regulates Platelet-Derived Growth Factor-Induced Intracellular Calcium Release. Cells. 2019; 8(11):1351. https://doi.org/10.3390/cells8111351
Chicago/Turabian StyleFarrar, Christopher S., Geoffrey T. Rouin, Benjamin L. Miller, Carol H. Raeman, Nancie A. Mooney, and Denise C. Hocking. 2019. "A Matricryptic Conformation of the Integrin-Binding Domain of Fibronectin Regulates Platelet-Derived Growth Factor-Induced Intracellular Calcium Release" Cells 8, no. 11: 1351. https://doi.org/10.3390/cells8111351
APA StyleFarrar, C. S., Rouin, G. T., Miller, B. L., Raeman, C. H., Mooney, N. A., & Hocking, D. C. (2019). A Matricryptic Conformation of the Integrin-Binding Domain of Fibronectin Regulates Platelet-Derived Growth Factor-Induced Intracellular Calcium Release. Cells, 8(11), 1351. https://doi.org/10.3390/cells8111351