Chaperone-Mediated Autophagy and Its Emerging Role in Hematological Malignancies




Abstract
1. General Introduction
2. Protein Renewal and Deciphering of the Main Cellular Catabolic Pathways
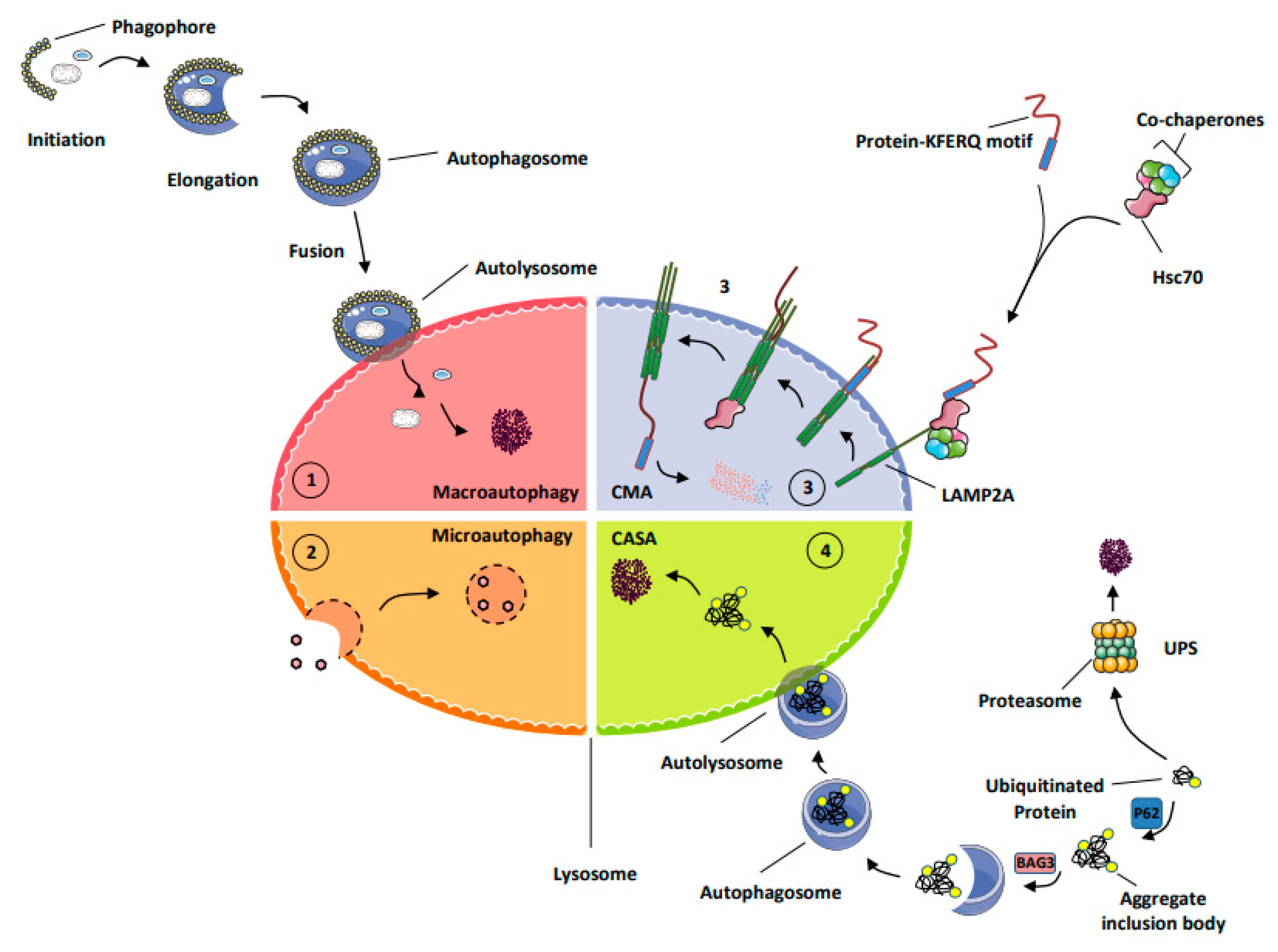
- MA (macro-autophagy): this process allows the degradation of long-lived proteins, protein aggregates, lipids, and carbohydrates but also damaged organelles, as well as intracellular micro-organisms into the lysosomes. During autophagy, the material to be degraded is engulfed in double-membrane vesicles, called autophagosomes, that fuse with lysosomes and are degraded by a large set of hydrolases and potentially recycled to sustain cell survival [9].
- Chaperone-mediated autophagy (CMA) represents a highly selective process of degradation of cytosolic proteins endowed with a KFERQ or KFERQ-like motif in their amino-acid sequences. During CMA, the KFERQ motif present in protein substrates is recognized by the cytosolic chaperone heat-shock protein cognate protein Hsc70c, also called HSPA8, responsible for their unfolding and subsequent transport to LAMP2A (lysosomal-associated membrane protein 2A), which serves as the specific receptor for CMA. Proteins transported to the lysosomal lumen are ultimately degraded by lysosomal proteases, and the products of degradation (amino acids) are potentially recycled to maintain cellular homeostasis and/or promote survival [9].
- Chaperone-assisted selective autophagy (CASA) ensures cellular protein quality control and, as such, allows the selective ubiquitin-dependent degradation of dysfunctional chaperone-bound proteins in lysosomes. The ubiquitinated proteins are next engulfed in autophagosomes and delivered to lysosomes for degradation [12].
- The ubiquitin proteasome system (UPS) is the mechanism by which short-lived proteins and dysfunctional or unfolded proteins are addressed to the proteasome for degradation and potential recycling [13].
3. Chaperone-Mediated Autophagy (CMA)
3.1. Introduction
3.2. Mechanisms of CMA
3.3. Modulation of CMA
3.4. CMA Substrates
3.5. Crosstalk Regulation between MA and CMA
3.6. Crosstalk between UPS and CMA
4. Physiological and Pathological Functions of CMA
4.1. Function of CMA in Cancer Initiation and Progression
4.2. Implication of CMA in Hematological Malignancies
4.3. CMA Substrates with a Special Relevance to Hematopoietic Malignancies
4.3.1. AF1Q/MLLT11
4.3.2. Bcl2-L10 (Bcl2-Like Protein 10/Bcl-2L10)
4.3.3. c-Myc
4.3.4. TP53
4.3.5. TFEB
4.3.6. IκB
4.3.7. PKM2
4.3.8. HK2
4.3.9. Elimination of Fusion Protein by MA
5. Targeting CMA in Hematological Malignancies
5.1. Small Molecules Compounds that Affect CMA
5.2. Potential Role of CMA in APL Cell Differentiation and Treatment
6. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy: Many paths to the same end. Mol. Cell Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells 2019, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef]
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Cuervo, A.M. Chaperone-mediated autophagy: Dice’s ’wild’ idea about lysosomal selectivity. Nat. Rev. Mol. Cell Biol. 2011, 12, 535–541. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Santambrogio, L.; Cuervo, A.M. Chasing the elusive mammalian microautophagy. Autophagy 2011, 7, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Zhang, C.; Cuervo, A.M. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006, 73, 205–235. [Google Scholar] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S.; Chiang, H.L.; Goldberg, A.L.; Dice, J.F. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem. J. 1991, 275 Pt 1, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [PubMed]
- Agarraberes, F.A.; Dice, J.F. Protein translocation across membranes. Biochim. Biophys. Acta 2001, 1513, 1–24. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615. [Google Scholar] [CrossRef]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Arias, E.; Diaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef]
- Wang, D.W.; Peng, Z.J.; Ren, G.F.; Wang, G.X. The different roles of selective autophagic protein degradation in mammalian cells. Oncotarget 2015, 6, 37098–37116. [Google Scholar] [CrossRef]
- Li, P.; Ji, M.; Lu, F.; Zhang, J.; Li, H.; Cui, T.; Li Wang, X.; Tang, D.; Ji, C. Degradation of AF1Q by chaperone-mediated autophagy. Exp. Cell Res. 2014, 327, 48–56. [Google Scholar] [CrossRef]
- Quintavalle, C.; Di Costanzo, S.; Zanca, C.; Tasset, I.; Fraldi, A.; Incoronato, M.; Mirabelli, P.; Monti, M.; Ballabio, A.; Pucci, P.; et al. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J. Cell Physiol. 2014, 229, 1359–1368. [Google Scholar] [CrossRef]
- Ali, A.B.; Nin, D.S.; Tam, J.; Khan, M. Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS ONE 2011, 6, e25268. [Google Scholar] [CrossRef]
- Razidlo, G.L.; Wang, Y.; Chen, J.; Krueger, E.W.; Billadeau, D.D.; McNiven, M.A. Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev. Cell 2013, 24, 573–585. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Aniento, F.; Roche, E.; Cuervo, A.M.; Knecht, E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 1993, 268, 10463–10470. [Google Scholar] [PubMed]
- Welsch, T.; Younsi, A.; Disanza, A.; Rodriguez, J.A.; Cuervo, A.M.; Scita, G.; Schmidt, J. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp. Cell Res. 2010, 316, 1914–1924. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, Y.; Fei, M.; Tan, C.; Wu, J.; Zheng, J.; Tang, J.; Sun, W.; Lv, Z.; Bao, J.; et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 2014, 10, 1015–1035. [Google Scholar] [CrossRef]
- Yang, Q.; She, H.; Gearing, M.; Colla, E.; Lee, M.; Shacka, J.J.; Mao, Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Kruger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 2010, 6, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Zhang, X.D.; Wu, J.C.; Lin, F.; Wang, J.; DiFiglia, M.; Qin, Z.H. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 2012, 7, e46834. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, L.; Jiao, H.; Guan, L.; Zha, J.; Li, X.; Wu, M.; Wang, Z.; Han, J.; You, H. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy 2015, 11, 1623–1635. [Google Scholar] [CrossRef]
- Dubois, A.; Furstoss, N.; Calleja, A.; Zerhouni, M.; Cluzeau, T.; Savy, C.; Marchetti, S.; Hamouda, M.A.; Boulakirba, S.; Orange, F.; et al. LAMP2 expression dictates azacytidine response and prognosis in MDS/AML. Leukemia 2019, 33, 1501–1513. [Google Scholar] [CrossRef]
- Hao, Y.; Kacal, M.; Ouchida, A.T.; Zhang, B.; Norberg, E.; Vakifahmetoglu-Norberg, H. Targetome analysis of chaperone-mediated autophagy in cancer cells. Autophagy 2019, 1–14. [Google Scholar] [CrossRef]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of chaperone-mediated autophagy. Int J. Biochem. Cell Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Cuervo, A.M. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin Cell Dev. Biol. 2010, 21, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Jiang, H.; Cheng, D.; Liu, W.; Peng, J.; Feng, J. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem. Biophys. Res. Commun. 2010, 395, 471–476. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef]
- Huang, J.; Xu, J.; Pang, S.; Bai, B.; Yan, B. Age-related decrease of the LAMP-2 gene expression in human leukocytes. Clin. Biochem. 2012, 45, 1229–1232. [Google Scholar] [CrossRef]
- Mufti, G.J. Pathobiology, classification, and diagnosis of myelodysplastic syndrome. Best Pract Res. Clin. Haematol. 2004, 17, 543–557. [Google Scholar] [CrossRef]
- Fenaux, P. Myelodysplastic syndromes: From pathogenesis and prognosis to treatment. Semin Hematol 2004, 41, 6–12. [Google Scholar] [CrossRef]
- Cai, Z.; Zeng, W.; Tao, K.; E, Z.; Wang, B.; Yang, Q. Chaperone-mediated autophagy: Roles in neuroprotection. Neurosci. Bull. 2015, 31, 452–458. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis 2013, 4, e545. [Google Scholar] [CrossRef]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome membrane lipid microdomains: Novel regulators of chaperone-mediated autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Mann, L.; Bonten, E.J.; d’Azzo, A.; Dice, J.F. Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 2003, 22, 47–59. [Google Scholar] [CrossRef]
- Hamouda, M.A.; Jacquel, A.; Robert, G.; Puissant, A.; Richez, V.; Cassel, R.; Fenouille, N.; Roulland, S.; Gilleron, J.; Griessinger, E.; et al. BCL-B (BCL2L10) is overexpressed in patients suffering from multiple myeloma (MM) and drives an MM-like disease in transgenic mice. J. Exp. Med. 2016, 213, 1705–1722. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.P.; et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Vidal, V.; Robert, G.; Goursaud, L.; Durand, L.; Ginet, C.; Karsenti, J.M.; Luciano, F.; Gastaud, L.; Garnier, G.; Braun, T.; et al. BCL2L10 positive cells in bone marrow are an independent prognostic factor of azacitidine outcome in myelodysplastic syndrome and acute myeloid leukemia. Oncotarget 2017, 8, 47103–47109. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rooswinkel, R.W.; van de Kooij, B.; de Vries, E.; Paauwe, M.; Braster, R.; Verheij, M.; Borst, J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 2014, 123, 2806–2815. [Google Scholar] [CrossRef]
- van de Kooij, B.; Rooswinkel, R.W.; Kok, F.; Herrebout, M.; de Vries, E.; Paauwe, M.; Janssen, G.M.; van Veelen, P.A.; Borst, J. Polyubiquitination and proteasomal turnover controls the anti-apoptotic activity of Bcl-B. Oncogene 2013, 32, 5439–5448. [Google Scholar] [CrossRef]
- Robert, G.; Gastaldi, C.; Puissant, A.; Hamouda, A.; Jacquel, A.; Dufies, M.; Belhacene, N.; Colosetti, P.; Reed, J.C.; Auberger, P.; et al. The anti-apoptotic Bcl-B protein inhibits BECN1-dependent autophagic cell death. Autophagy 2012, 8, 637–649. [Google Scholar] [CrossRef]
- Godoi, P.H.; Wilkie-Grantham, R.P.; Hishiki, A.; Sano, R.; Matsuzawa, Y.; Yanagi, H.; Munte, C.E.; Chen, Y.; Yao, Y.; Marassi, F.M.; et al. Orphan Nuclear Receptor NR4A1 Binds a Novel Protein Interaction Site on Anti-apoptotic B Cell Lymphoma Gene 2 Family Proteins. J. Biol. Chem. 2016, 291, 14072–14084. [Google Scholar] [CrossRef]
- Ding, Q.; Xie, X.L.; Wang, M.M.; Yin, J.; Tian, J.M.; Jiang, X.Y.; Zhang, D.; Han, J.; Bai, Y.; Cui, Z.J.; et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp. Mol. Med. 2019, 51, 6. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. TFEB regulates autophagy: An integrated coordination of cellular degradation and recycling processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef]
- Scott, M.L.; Fujita, T.; Liou, H.C.; Nolan, G.P.; Baltimore, D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev. 1993, 7, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef] [PubMed]
- Koss, K.; Harrison, R.F.; Gregory, J.; Darnton, S.J.; Anderson, M.R.; Jankowski, J.A. The metabolic marker tumour pyruvate kinase type M2 (tumour M2-PK) shows increased expression along the metaplasia-dysplasia-adenocarcinoma sequence in Barrett’s oesophagus. J. Clin. Pathol. 2004, 57, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.S.; Dew, T.; Lawton, F.G.; Papadopoulos, A.J.; Devaja, O.; Raju, K.S.; Sherwood, R.A. M2-PK as a novel marker in ovarian cancer. A prospective cohort study. Eur J. Gynaecol Oncol. 2007, 28, 83–88. [Google Scholar] [PubMed]
- Ferguson, E.C.; Rathmell, J.C. New roles for pyruvate kinase M2: Working out the Warburg effect. Trends Biochem. Sci. 2008, 33, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Ouchida, A.T.; Li, Y.; Geng, J.; Najafov, A.; Ofengeim, D.; Sun, X.; Yu, Q.; Yuan, J. Synergistic effect of a novel autophagy inhibitor and Quizartinib enhances cancer cell death. Cell Death Dis. 2018, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Platanias, L.C. BCR-ABL1-induced leukemogenesis and autophagic targeting by arsenic trioxide. Autophagy 2013, 9, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Auberger, P. BCR-ABL/p62/SQSTM1: A cannibal embrace. Blood 2012, 120, 3389–3390. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nasr, R.; Guillemin, M.C.; Ferhi, O.; Soilihi, H.; Peres, L.; Berthier, C.; Rousselot, P.; Robledo-Sarmiento, M.; Lallemand-Breitenbach, V.; Gourmel, B.; et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008, 14, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Vitaliano-Prunier, A.; Halftermeyer, J.; Ablain, J.; de Reynies, A.; Peres, L.; Le Bras, M.; Metzger, D.; de The, H. Clearance of PML/RARA-bound promoters suffice to initiate APL differentiation. Blood 2014, 124, 3772–3780. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef]
- Allende-Vega, N.; Villalba, M. Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci. Rep. 2019, 9, 5637. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Niederst, M.; Fecteau, J.F.; Nguyen, V.M.; Kipps, T.J.; Messmer, D.; Newton, A.C.; Handel, T.M. Mechanisms and consequences of the loss of PHLPP1 phosphatase in chronic lymphocytic leukemia (CLL). Leukemia 2012, 26, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Suljagic, M.; Laurenti, L.; Tarnani, M.; Alam, M.; Malek, S.N.; Efremov, D.G. Reduced expression of the tumor suppressor PHLPP1 enhances the antiapoptotic B-cell receptor signal in chronic lymphocytic leukemia B-cells. Leukemia 2010, 24, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Shao, K.; Zhou, Q.; Sun, J.; Wang, Z.; Yan, F.; Liu, T.; Wu, X.; Ye, B.; Huang, H.; et al. Autophagy and Ubiquitin-Mediated Proteolytic Degradation of PML/Raralpha Fusion Protein in Matrine-Induced Differentiation Sensitivity Recovery of ATRA-Resistant APL (NB4-LR1) Cells: In Vitro and in Vivo Studies. Cell Physiol. Biochem. 2018, 48, 2286–2301. [Google Scholar] [CrossRef]
- Humbert, M.; Federzoni, E.A.; Tschan, M.P. Distinct TP73-DAPK2-ATG5 pathway involvement in ATO-mediated cell death versus ATRA-mediated autophagy responses in APL. J. Leukoc Biol. 2017, 102, 1357–1370. [Google Scholar] [CrossRef]
- Dembitz, V.; Lalic, H.; Visnjic, D. 5-Aminoimidazole-4-carboxamide ribonucleoside-induced autophagy flux during differentiation of monocytic leukemia cells. Cell Death Discov. 2017, 3, 17066. [Google Scholar] [CrossRef]
- Segal-Bendirdjian, E.; Tschan, M.P.; Reiffers, J.; Djavaheri-Mergny, M. Pro-survival role of p62 during granulocytic differentiation of acute myeloid leukemia cells. Mol. Cell Oncol. 2014, 1, e970066. [Google Scholar] [CrossRef]
- Trocoli, A.; Mathieu, J.; Priault, M.; Reiffers, J.; Souquere, S.; Pierron, G.; Besancon, F.; Djavaheri-Mergny, M. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy 2011, 7, 1108–1114. [Google Scholar] [CrossRef]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Riffelmacher, T.; Simon, A.K. Mechanistic roles of autophagy in hematopoietic differentiation. FEBS J. 2017, 284, 1008–1020. [Google Scholar] [CrossRef] [PubMed]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Nicastri, M.C.; McLaughlin, N.; Fennelly, C.; McAfee, Q.; Ronghe, A.; Nofal, M.; Lim, C.Y.; Witze, E.; Chude, C.I.; et al. A Unified Approach to Targeting the Lysosome’s Degradative and Growth Signaling Roles. Cancer Discov. 2017, 7, 1266–1283. [Google Scholar] [CrossRef]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Symbol | Protein Full Name | Function | Deregulated in: | CMA Substrat Ref: |
|---|---|---|---|---|
| GAPDH | Glyceraldehyde 3-phosphate deshydrogenase | Carbohydrate Metabolism | Non hodgkin’s B lymphoma | [34] |
| HK-2 | Hexokinase-2 | Carbohydrate Metabolism | Ovarian cancer | [32] |
| PKM2 | Pyruvate Kinase M2 | Carbohydrate Metabolism | AML, Melanoma | [31] |
| TP53 | Tumor Protein P53 | Tumor suppressor protein | Most of cancers | [26] |
| Mutant TP53 | Mutant Tumor Protein P53 | Oncogene | Most of cancers | [37] |
| MDM2 | Mouse Double Minute 2 homolog | E3 ubiquitin Ligase | Glioma, ALL, Melanoma | |
| PUMA | P53 upregulated modulator of apoptosis | BH3-only Pro-Apoptotic protein | Breast, Colon cancers | [46] |
| AF1Q (MLLT11) | MLLT11 Transcription Factor 7 Cofactor | Oncogene | AML | [27] |
| c-Myc | MYC Proto-Oncogene, BHLH Transcription Factor | Oncogene | Most of cancers | [44] |
| IκΒ | NFKB Inhibitor Alpha | NF-κB Inhibitor | B-cell lymphoma | |
| CHK1 | Checkpoint Kinase 1 | Cell cycle arrest | Breast, Ovarian Cancers | |
| Vav1 | Vav Guanine Nucleotide Exchange Factor 1 | (GEFs) for Rho family GTPases | Pancreatic cancer | [30] |
| HIF-1α | hypoxia Inducible Factor 1 alpha | Transcriptional regulator of the adaptive response to hypoxia | Lymphoma, colorectal cancers | [39] |
| NCOR1 | Nuclear Receptor Corepressor 1 | Promotes histone deacetylation and the formation of repressive chromatin structures | NSCLC, Gastric cancer | [29] |
| PED | Phosphoprotein Enriched in diabetes | Facilitate glucose transport | Gastric cancer | [28] |
| EPS8 | Epidermal Growth Factor Receptor Pathway Substrate 8 | Signaling adaptapter | Pancreatic cancer | [35] |
| RND3 | Rho Family GTPase 3 | Negative regulator of cytoskeletal organization | Gastric cancer | [36] |
| ANXs | Annexins | membrane scaffold, linking Ca2+ signalling to membrane dynamics | Breast Cancer | |
| TFEB | Transcription Factor EB | Transcription factor of lysosomal genes | Pancreatic, Renal cancers | [45] |
| EGFR | Epidermal Growth Factor receptor | Receptor tyrosine kinase binding ligands of the EGF family | Head and neck squamous cell carcinoma (HNSCC) | |
| GAL3 | Galectine-3 | Numerous cellular function: cell growth, adhesion, mitosis, proliferation and apoptosis | Diffuse large B-cell lymphoma (DLBCL), Prostate, liver cancer | |
| RKIP | Raf Kinase Inhibitor Protein | Raf Kinase Inhibitor | Prostate cancer | |
| UBQLN1 | Ubiquilin 1 | Ubiquitin like protein | Gastric cancer | |
| Bcl2-L10 | Bcl2 Like 10 | Anti-apoptotic protein of BCL2 family members | MM, MDS and AML | [47] |
| Compounds | Target | Effect on CMA | Refs |
|---|---|---|---|
| Cycloheximide | Protein synthesis inhibitor | Inhibition | [99] |
| Anisomycin | Protein synthesis inhibitor | Inhibition | [99] |
| SB230580 | P38 MAPK inhibitor | Inhibition | [99] |
| Geldanamycin | HSP90 inhibitor | Activation | [99] |
| 17-AAG/DCA | HSP90 inhibitor + PDK1 inhibitor | Activation | [100] |
| 6-aminonicotinamide | G6PDH inhibitor | Activation | [99] |
| synthetic ATRA derivatives | RAR-alpha inhibitor | Activation | [101] |
| torin | TORC2 inhibitor | Activation | [24] |
| TAK165/AC220 | MA inhibitor + FLT3 Inhibitor | Activation | [91] |
| Spautin/AC220 | MA inhibitor + FLT3 Inhibitor | Activation | [32] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robert, G.; Jacquel, A.; Auberger, P. Chaperone-Mediated Autophagy and Its Emerging Role in Hematological Malignancies. Cells 2019, 8, 1260. https://doi.org/10.3390/cells8101260
Robert G, Jacquel A, Auberger P. Chaperone-Mediated Autophagy and Its Emerging Role in Hematological Malignancies. Cells. 2019; 8(10):1260. https://doi.org/10.3390/cells8101260
Chicago/Turabian StyleRobert, Guillaume, Arnaud Jacquel, and Patrick Auberger. 2019. "Chaperone-Mediated Autophagy and Its Emerging Role in Hematological Malignancies" Cells 8, no. 10: 1260. https://doi.org/10.3390/cells8101260
APA StyleRobert, G., Jacquel, A., & Auberger, P. (2019). Chaperone-Mediated Autophagy and Its Emerging Role in Hematological Malignancies. Cells, 8(10), 1260. https://doi.org/10.3390/cells8101260