Gγ and Gα Identity Dictate a G-Protein Heterotrimer Plasma Membrane Targeting



Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Protein Constructs
2.2. Cell Culture
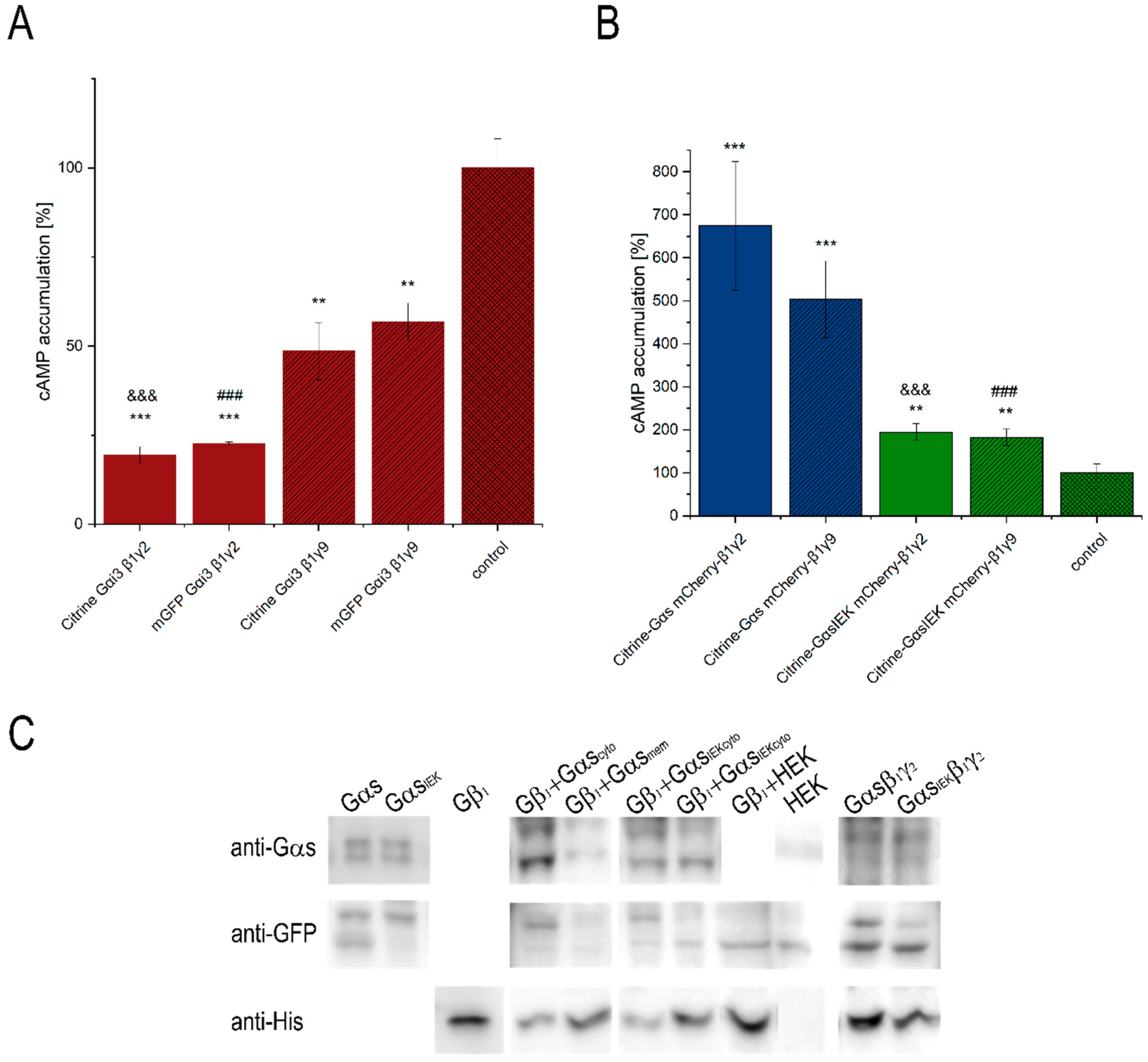
2.3. Determination of Intercellular cAMP Concentration
2.4. Pull-Down Assay
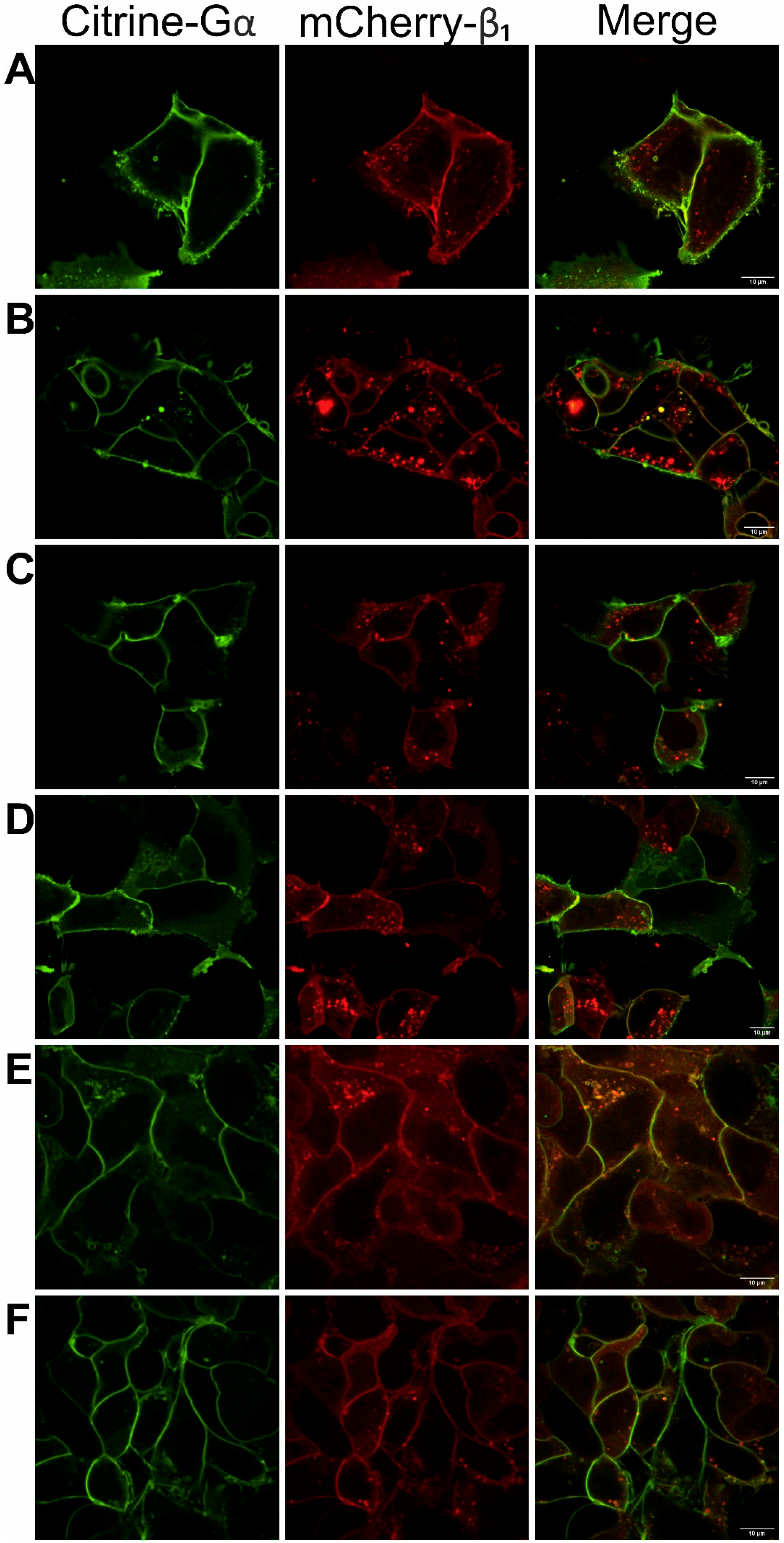
2.5. Confocal Microscopy
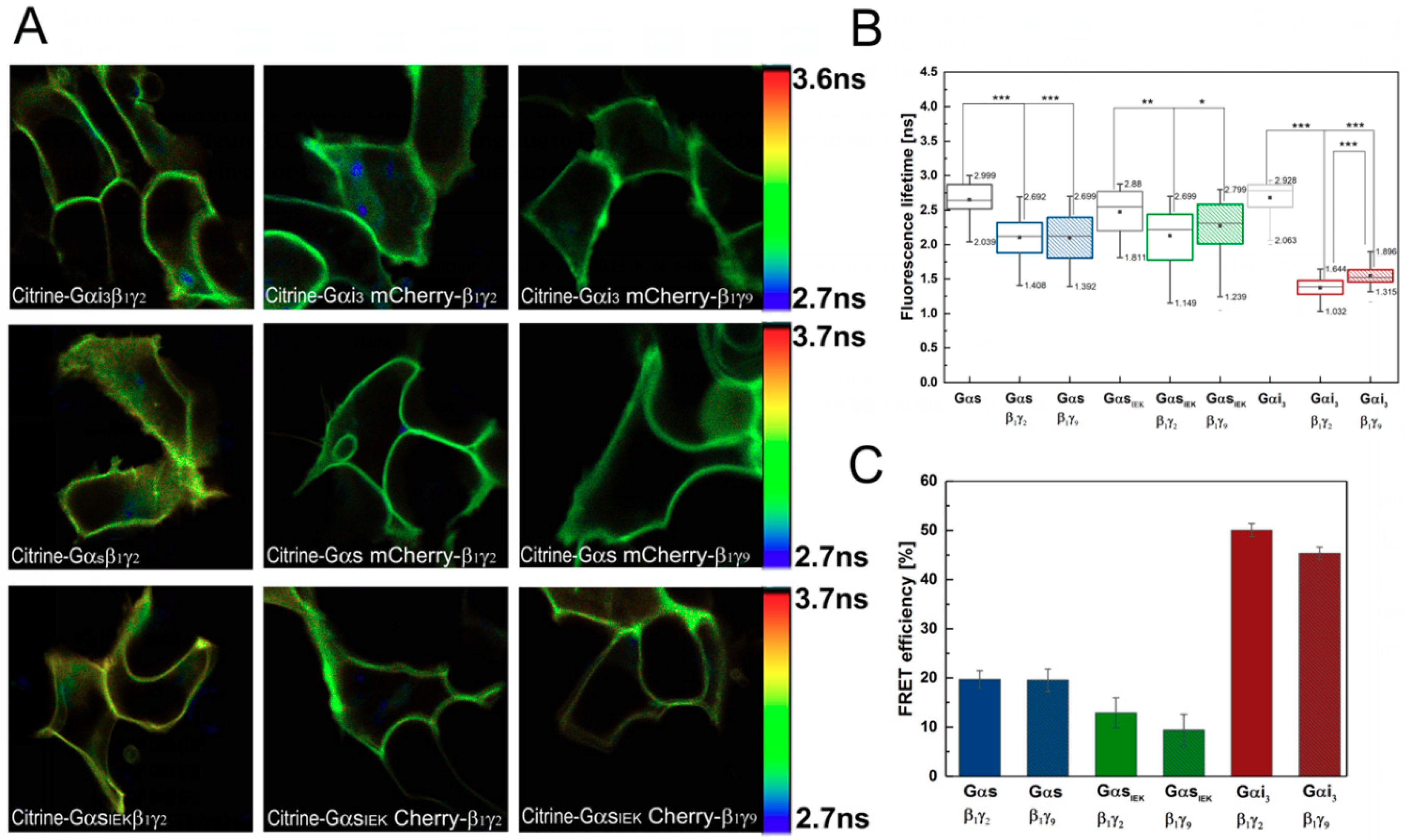
2.6. Fluorescence Lifetime Imaging
2.7. FRAP Measurements
2.8. Statistical Analyses
3. Results
3.1. Functionality of Fluorescently-Tagged G-Proteins
3.2. Analysis of G-Protein Interactions in Live Cells Using FLIM–FRET Microscopy
3.3. Effect of Gβγ Dimer on Gα Diffusion
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Vera, T.M.; Vanhauwe, J.; Thomas, T.O.; Medkova, M.; Preininger, A.; Mazzoni, M.R.; Hamm, H.E. Insights into G Protein Structure, Function, and Regulation. Endocr. Rev. 2003, 24, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hildebrandt, J.D. Genomic analysis of G protein γ subunits in human and mouse—The relationship between conserved gene structure and G protein βγ dimer formation. Cell. Signal. 2006, 18, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Degtyarev, M.Y.; Spiegel, A.M.; Jones, T.L.Z. Increased palmitoylation of the G(s) protein α subunit after activation by the β-adrenergic receptor or cholera toxin. J. Biol. Chem. 1993, 268, 23769–23772. [Google Scholar]
- Loisel, T.P.; Adam, L.; Hebert, T.E.; Bouvier, M. Agonist stimulation increases the turnover rate of β2AR-bound palmitate and promotes receptor depalmitoylation. Biochemistry 1996, 35, 15923–15932. [Google Scholar] [CrossRef] [PubMed]
- Kleuss, C.; Krause, E. Gαs is palmitoylated at the N-terminal glycine Christiane. EMBO J. 2003, 22, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. The Biology and Enzymology of Protein N-Myristoylation. J. Biol. Chem. 2001, 276, 39501–39504. [Google Scholar] [CrossRef] [PubMed]
- Kosloff, M.; Elia, N.; Selinger, Z. Structural homology discloses a bifunctional structural motif at the N-termini of Gα proteins. Biochemistry 2002, 41, 14518–14523. [Google Scholar] [CrossRef]
- Crouthamel, M.; Abankwa, D.; Zhang, L.; DiLizio, C.; Manning, D.R.; Hancock, J.F.; Wedegaertner, P.B. An N-Terminal Polybasic Motif of G q Is Required for Signaling and Influences Membrane Nanodomain Distribution. Mol. Pharmacol. 2010, 78, 767–777. [Google Scholar] [CrossRef]
- Pedone, K.H.; Hepler, J.R. The Importance of N-terminal polycysteine and polybasic sequences for G14αand G16α palmitoylation, plasma membrane localization, and signaling function. J. Biol. Chem. 2007, 282, 25199–25212. [Google Scholar] [CrossRef]
- Crouthamela, M.; Thiyagarajan, M.M.; Evanko, D.S.; Wedegaertner, P.B. N-terminal polybasic motifs are required for plasma membrane localization of Gαs AND Gαq. Cell Signal. 2008, 20, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Sondek, J.; Bohm, A.; Lambright, D.G.; Hamm, H.E.; Sigler, P.B. Crystal structure of a GA protein βγdimer at 2.1 Å resolution. Nature 1996, 379, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.B.; Casey, P.J. The role of prenylation in G-protein assembly and function. Cell. Signal. 1996, 8, 433–437. [Google Scholar] [CrossRef]
- Dunphy, J.T.; Linder, M.E. Signalling functions of protein palmitoylation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1998, 1436, 245–261. [Google Scholar] [CrossRef]
- McLaughlin, S.; Aderem, A. The myristoyl-electrostatic switch: A modulator of reversible protein-membrane interactions. Trends Biochem. Sci. 1995, 20, 272–276. [Google Scholar] [CrossRef]
- Resh, M.D. Fatty acylation of proteins: New insights into membrane targeting of myristylated and palmitoylated proteins. Biochim. Biophys. Acta Mol. Cell Res. 1999, 1451, 1–16. [Google Scholar] [CrossRef]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Morales, J.; Fishburn, C.S.; Wilson, P.T.; Bourne, H.R. Plasma Membrane Localization of Galpha z Requires Two Signals. Mol. Biol. Cell 1998, 9, 1–14. [Google Scholar] [CrossRef]
- Karunarathne, W.K.A.; O’Neill, P.R.; Martinez-Espinosa, P.L.; Kalyanaraman, V.; Gautam, N. All G protein βγ complexes are capable of translocation on receptor activation. Biochem. Biophys Res. Commun. 2012, 421, 605–611. [Google Scholar] [CrossRef]
- O’Neill, P.R.; Karunarathne, W.K.A.; Kalyanaraman, V.; Silvius, J.R.; Gautam, N. G-protein signaling leverages subunit-dependent membrane affinity to differentially control translocation to intracellular membranes. Proc. Natl. Acad. Sci. USA 2012, 109, E3568–E3577. [Google Scholar]
- Chisari, M.; Saini, D.K.; Kalyanaraman, V.; Gautam, N. Shuttling of G protein subunits between the plasma membrane and intracellular membranes. J. Biol. Chem. 2007, 282, 24092–24098. [Google Scholar] [CrossRef] [PubMed]
- Mystek, P.; Tworzydło, M.; Dziedzicka-Wasylewska, M.; Polit, A. New insights into the model of dopamine D1 receptor and G-proteins interactions. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 594–603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Evanko, D.S.; Thiyagarajan, M.M.; Wedegaertner, P.B. Interaction with Gbetagamma is required for membrane targeting and palmitoylation of Galpha(s) and Galpha(q). J. Biol. Chem. 2000, 275, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Bond, S.R.; Naus, C.C. RF-Cloning.org: An online tool for the design of restriction-free cloning projects. Nucleic Acids Res. 2012, 40, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Mystek, P.; Dutka, P.; Tworzydło, M.; Dziedzicka-Wasylewska, M.; Polit, A. The role of cholesterol and sphingolipids in the dopamine D1 receptor and G protein distribution in the plasma membrane. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescent Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Gibson, S.K.; Gilman, A.G. Gialpha and Gbeta subunits both define selectivity of G protein activation by alpha2-adrenergic receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Hillenbrand, M.; Schori, C.; Schöppe, J.; Plückthun, A. Comprehensive analysis of heterotrimeric G-protein complex diversity and their interactions with GPCRs in solution. Proc. Natl. Acad. Sci. USA 2015, 112, E1181–E1190. [Google Scholar] [CrossRef] [PubMed]
- García-Nafría, J.; Lee, Y.; Bai, X.; Carpenter, B.; Tate, C.G. Cryo-EM structure of the adenosine A 2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7, 267674. [Google Scholar] [CrossRef]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the μ-opioid receptor-G i protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Hou, Y.; Azpiazu, I.; Smrcka, A.; Gautam, N. Selective role of G protein γ subunits in receptor interaction. J. Biol. Chem. 2000, 275, 38961–38964. [Google Scholar] [CrossRef] [PubMed]
- Myung, C.S.; Lim, W.K.; DeFilippo, J.M.; Yasuda, H.; Neubig, R.R.; Garrison, J.C. Regions in the G Protein Subunit Important for Interaction with Receptors and Effectors. Mol. Pharmacol. 2005, 69, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Sheets, E.D.; Wagenknecht-Wiesner, A.; Heikal, A.A. Quantitative analysis of the fluorescence properties of intrinsically fluorescent proteins in living cells. Biophys. J. 2003, 85, 2566–2580. [Google Scholar] [CrossRef]
- Ghosh, A.; Isbaner, S.; Veiga-Gutiérrez, M.; Gregor, I.; Enderlein, J.; Karedla, N. Quantifying Microsecond Transition Times Using Fluorescence Lifetime Correlation Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 6022–6028. [Google Scholar] [CrossRef]
- Heikal, A.A.; Hess, S.T.; Baird, G.S.; Tsien, R.Y.; Webb, W.W. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: Coral red (dsRed) and yellow (Citrine). Proc. Natl. Acad. Sci. USA 2000, 97, 11996–12001. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Akrap, N.; Seidel, T.; Barisas, G.B. Förster distances for FRET between mCherry and other Visible Fluorescent Proteins. Anal. Biochem. 2010, 402, 105–106. [Google Scholar] [CrossRef]
- Vogel, S.S.; van der Meer, B.W.; Blank, P.S. Estimating the distance separating fluorescent protein FRET pairs. Methods 2014, 66, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Lambright, D.G.; Sondek, J.; Bohm, A.; Skiba, N.P.; Hamm, H.E.; Sigler, P.B. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature 1996, 379, 311–319. [Google Scholar] [CrossRef]
- Lim, W.K.; Myung, C.; Garrison, J.C.; Neubig, R.R. Receptor-G Protein γ Specificity: γ11 Shows Unique Potency for A1 Adenosine and 5-HT1A Receptors. Biochemistry 2001, 40, 10532–10541. [Google Scholar] [CrossRef]
- Senarath, K.; Payton, J.L.; Kankanamge, D.; Siripurapu, P.; Tennakoon, M.; Karunarathne, A. Gγ identity dictates efficacy of Gβγ signaling and macrophage migration. J. Biol. Chem. 2018, 293, 2974–2989. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, O.; Pronin, A.; Ermolaeva, M.; Gautam, N. Receptor-G protein coupling is established by a potential conformational switch in the beta gamma complex. Proc. Natl. Acad. Sci. USA 2006, 92, 9102–9106. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.M.; Sleno, R.; Gora, S.; Zylbergold, P.; Laverdure, J.-P.; Labbe, J.-C.; Miller, G.J.; Hebert, T.E. The Expanding Roles of G Subunits in G Protein-Coupled Receptor Signaling and Drug Action. Pharmacol. Rev. 2013, 65, 545–577. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.E.; Skiba, N.P.; Bae, H.; Daaka, Y.; Reuveny, E.; Shekter, L.R.; Rosal, R.; Weng, G.; Yang, C.S.; Iyengar, R.; et al. Molecular basis for interactions of G protein βΓ subunits with effectors. Science 1998, 280, 1271–1274. [Google Scholar] [CrossRef]
- Cook, L.A.; Schey, K.L.; Cleator, J.H.; Wilcox, M.D.; Dingus, J.; Hildebrandt, J.D. Identification of a region in G protein γ subunits conserved across species but hypervariable among subunit isoforms. Protein Sci. 2009, 10, 2548–2555. [Google Scholar] [CrossRef]
- McCormick, P.; Dumaresq-Doiron, K.; Pluviose, A.-S.; Pichette, V.; Tosato, G.; Lefrancois, S. Palmitoylation Controls Recycling in Lysosomal Sorting and Trafficking. Traffic 2008, 9, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2006, 2, 584–590. [Google Scholar] [CrossRef]
- Chen, C.A.; Manning, D.R. Regulation of g proteins by covalent modification. Oncogene 2001, 20, 1643–1652. [Google Scholar] [CrossRef]
- Zhang, Z.; Melia, T.J.; He, F.; Yuan, C.; McGough, A.; Schmid, M.F.; Wensel, T.G. How a G protein binds a membrane. J. Biol. Chem. 2004, 279, 33937–33945. [Google Scholar] [CrossRef]
- Vogler, O.; Casas, J.; Capo, D.; Nagy, T.; Borchert, G.; Martorell, G.; Escriba, P.V. The G Dimer Drives the Interaction of Heterotrimeric Gi Proteins with Nonlamellar Membrane Structures. J. Biol. Chem. 2004, 279, 36540–36545. [Google Scholar] [CrossRef]
- Abankwa, D.; Vogel, H. A FRET map of membrane anchors suggests distinct microdomains of heterotrimeric G proteins. J. Cell Sci. 2007, 120, 2953–2962. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Johannes, L.; Pezeshkian, W.; Ipsen, J.H.; Shillcock, J.C. Clustering on Membranes: Fluctuations and More. Trends Cell Biol. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Parajo, M.F.; Cambi, A.; Torreno-Pina, J.A.; Thompson, N.; Jacobson, K. Nanoclustering as a dominant feature of plasma membrane organization. J. Cell Sci. 2014, 127, 4995–5005. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dapp (µm2/s) | Mf (%) | n | |
|---|---|---|---|
| Gαs † | 0.130 ± 0.004 | 84.5 ± 1.5 | 49 |
| Gαs Gβ1γ2 | 0.246 ± 0.009 | 92.4 ± 0.8 | 143 |
| Gαs Gβ1γ9 | 0.202 ± 0.007 | 89.5 ± 1.1 | 92 |
| Gαi3 † | 0.338 ± 0.022 | 94.2 ± 1.7 | 34 |
| Gαi3 Gβ1γ2 | 0.424 ± 0.014 | 93.5 ± 0.9 | 66 |
| Gαi3 Gβ1γ9 | 0.475 ± 0.021 | 92.8 ± 1.2 | 60 |
| GαsIEK Gβ1γ2 | 0.214 ± 0.010 | 87.8 ± 1.5 | 61 |
| GαsIEK Gβ1γ9 | 0.214 ± 0.005 | 89.7 ± 0.9 | 108 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mystek, P.; Rysiewicz, B.; Gregrowicz, J.; Dziedzicka-Wasylewska, M.; Polit, A. Gγ and Gα Identity Dictate a G-Protein Heterotrimer Plasma Membrane Targeting. Cells 2019, 8, 1246. https://doi.org/10.3390/cells8101246
Mystek P, Rysiewicz B, Gregrowicz J, Dziedzicka-Wasylewska M, Polit A. Gγ and Gα Identity Dictate a G-Protein Heterotrimer Plasma Membrane Targeting. Cells. 2019; 8(10):1246. https://doi.org/10.3390/cells8101246
Chicago/Turabian StyleMystek, Paweł, Beata Rysiewicz, Jan Gregrowicz, Marta Dziedzicka-Wasylewska, and Agnieszka Polit. 2019. "Gγ and Gα Identity Dictate a G-Protein Heterotrimer Plasma Membrane Targeting" Cells 8, no. 10: 1246. https://doi.org/10.3390/cells8101246
APA StyleMystek, P., Rysiewicz, B., Gregrowicz, J., Dziedzicka-Wasylewska, M., & Polit, A. (2019). Gγ and Gα Identity Dictate a G-Protein Heterotrimer Plasma Membrane Targeting. Cells, 8(10), 1246. https://doi.org/10.3390/cells8101246