Retroelement-Linked H3K4me1 Histone Tags Uncover Regulatory Evolution Trends of Gene Enhancers and Feature Quickly Evolving Molecular Processes in Human Physiology

 , , ,
, , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of RE-Linked H3K4me1 Modification Tags
2.2. Gene Expression Data
2.3. Measuring Gene Enrichment by RE–Linked H3K4me1 Histone Modification Tags
2.4. Measuring Molecular Pathway Enrichment by RE– H3K4me1 Tags
2.5. Measuring Enrichment of Gene Sets by Non-Coding RNA Genes
2.6. Gene Ontology Enrichment Analysis
2.7. Significance of Correlations
2.8. Significance of Correlations
3. Results
3.1. Genes and Molecular Pathways Impacted by RE-Linked Histone Modification Marks
3.2. Functional Characteristics of Top RRE-Enriched and Deficient Genes
3.2.1. RRE-Enriched Genes
3.2.2. RRE-Deficient Genes
3.2.3. Alternative GO Annotation Analysis
3.2.4. Enrichment by Non-Coding RNA Genes
3.2.5. Significance of RRE-Based Functional Gene Annotations
3.3. Functional Analysis of Top RRE-Enriched and Deficient Molecular Pathways
3.3.1. RRE-Enriched Pathways
3.3.2. RRE-Deficient Pathways
3.4. Comparison of Gene- and Pathway-Based RRE Data
3.4.1. RRE-Enriched Processes
- (1)
- The “Posttranscriptional silencing by small RNAs” group was identified using Gorilla functional annotation of RRE-enriched genes and included microRNA-mediated gene silencing.
- (2)
- The “DNA Metabolism and Chromatin Structure” group included double strand break DNA repair, transcription-coupled DNA repair, DNA strand displacement and chromatin remodeling processes.
- (3)
- The “Sensory Perception and Neurotransmission” group consisted of olfactory receptors activity (more specifically, class C3 metabotropic glutamate pheromonic receptors activity) and retinoid cycle in cones, which is responsible for color vision in primates.
- (4)
- The “Lipids Metabolism” group contained fatty acid biosynthesis, beta-oxidation and desaturation of fatty acids as well as phospholipase A2 activity and modification of sterols via cytochrome P450.
3.4.2. RRE-Deficient Processes
- (1)
- “Immune System” was a heterogenous group of molecular processes such as dendritic cell chemotaxis and cytokine production, IL-1, IL-3, IL-4, TLR and PD-1 signaling, asthma-related signaling and activation of RAS GTPase in B-cells.
- (2)
- The “Protein Ubiquitination and Degradation” group included ubiquitin-ligase activity, K63 polyubiquitin binding (non-degradative signal), protein degradation in proteasomes and autodegradation of E3 ubiquitin-ligase.
- (3)
- The “Cell Adhesion, Migration and Interaction” group included tight junction formation, E-cadherin binding, cell interaction with extracellular matrix (hyaluronanglucosaminidase activity, laminin binding and inhibition of matrix metalloproteinases), MMIF-mediated angiogenesis and platelet aggregation processes.
- (4)
- The “Metals Metabolism and Ion Transport” group consisted of zinc and calcium ion binding, chloride and potassium channels activity.
- (5)
- The “Cell Death” group contained various signaling pathways responsible for activation of apoptosis (p53, MEF2D-mediated apoptosis in T cells and BAD translocation to mitochondria), PTEN-mediated cell cycle arrest leading to apoptosis, caspase cascade and permeabilization of mitochondrial outer membrane.
- (6)
- The “General Signaling Pathways” group included wide variety of signaling pathways such as NK-κB, VEGF, EGF, IGF and mTOR signaling.
- (7)
- The “Hormone Signaling Pathways” group included steroid hormone mediated signaling pathways mediating response to estrogen and testosterone and PELP1 modulation of estrogen receptor activity.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rodić, N.; Burns, K.H. Long interspersed element-1 (LINE-1): Passenger or driver in human neoplasms? PLoS Genet. 2013, 9, e1003402. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Policarpi, C.; Crepaldi, L.; Brookes, E.; Nitarska, J.; French, S.M.; Coatti, A.; Riccio, A. Enhancer SINEs Link Pol III to Pol II Transcription in Neurons. Cell Rep. 2017, 21, 2879–2894. [Google Scholar] [CrossRef]
- Suntsova, M.; Gogvadze, E.V.; Salozhin, S.; Gaifullin, N.; Eroshkin, F.; Dmitriev, S.E.; Martynova, N.; Kulikov, K.; Malakhova, G.; Tukhbatova, G.; et al. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc. Natl. Acad. Sci. USA 2013, 110, 19472–19477. [Google Scholar] [CrossRef]
- Pan, G.; Ameur, A.; Enroth, S.; Bysani, M.; Nord, H.; Cavalli, M.; Essand, M.; Gyllensten, U.; Wadelius, C. PATZ1 down-regulates FADS1 by binding to rs174557 and is opposed by SP1/SREBP1c. Nucleic Acids Res. 2017, 45, 2408–2422. [Google Scholar] [CrossRef]
- Suntsova, M.; Garazha, A.; Ivanova, A.; Kaminsky, D.; Zhavoronkov, A.; Buzdin, A. Molecular functions of human endogenous retroviruses in health and disease. Cell. Mol. Life Sci. 2015, 72, 3653–3675. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Dunn-fletcher, C.E.; Muglia, L.M.; Pavlicev, M.; Wolf, G.; Sun, M.A.; Hu, Y.C.; Huffman, E.; Tumukuntala, S.; Thiele, K.; Mukherjee, A.; et al. Anthropoid primate-specific retroviral element THE1B controls expression of CRH in placenta and alters gestation length. PLoS Biol. 2018, 16, e2006337. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, E.; Buzdin, A. Retroelements and their impact on genome evolution and functioning. Cell. Mol. Life Sci. 2009, 66, 3727–3742. [Google Scholar] [CrossRef] [PubMed]
- Mita, P.; Boeke, J.D. How retrotransposons shape genome regulation. Curr. Opin. Genet. Dev. 2016, 37, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Marnetto, D.; Mantica, F.; Molineris, I.; Grassi, E.; Pesando, I.; Provero, P. Evolutionary Rewiring of Human Regulatory Networks by Waves of Genome Expansion. Am. J. Hum. Genet. 2018, 102, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Blinka, S.; Reimer, M.H.; Pulakanti, K.; Pinello, L.; Yuan, G.C.; Rao, S. Identification of Transcribed Enhancers by Genome-Wide Chromatin Immunoprecipitation Sequencing. Methods Mol. Biol. 2017, 1468, 91–109. [Google Scholar] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell. 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Siggens, L.; Ekwall, K. Epigenetics, chromatin and genome organization: Recent advances from the ENCODE project. J. Intern. Med. 2014, 276, 201–214. [Google Scholar] [CrossRef]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef]
- Rubinstein, M.; De souza, F.S. Evolution of transcriptional enhancers and animal diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20130017. [Google Scholar] [CrossRef]
- Carelli, F.N.; Liechti, A.; Halbert, J.; Warnefors, M.; Kaessmann, H. Repurposing of promoters and enhancers during mammalian evolution. Nat. Commun. 2018, 9, 4066. [Google Scholar] [CrossRef]
- Emera, D.; Yin, J.; Reilly, S.K.; Gockley, J.; Noonan, J.P. Origin and evolution of developmental enhancers in the mammalian neocortex. Proc. Natl. Acad. Sci. USA 2016, 113, E2617–E2626. [Google Scholar] [CrossRef]
- Nikitin, D.; Penzar, D.; Garazha, A.; Sorokin, M.; Tkachev, V.; Borisov, N.; Poltorak, A.; Prassolov, V.; Buzdin, A.A. Profiling of Human Molecular Pathways Affected by Retrotransposons at the Level of Regulation by Transcription Factor Proteins. Front. Immunol. 2018, 9, 30. [Google Scholar] [CrossRef]
- Nikitin, D.; Garazha, A.; Sorokin, M.; Penzar, D.; Tkachev, V.; Markov, A.; Gaifullin, N.; Borger, P.; Poltorak, A.; Buzdin, A. Retroelement-Linked Transcription Factor Binding Patterns Point to Quickly Developing Molecular Pathways in Human Evolution. Cells 2019, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Pontarotti, P. Evolution, Origin of Life, Concepts and Methods, 1st ed.; Springer International Publishing: New York, NY, USA, 2019. [Google Scholar]
- Danino, Y.M.; Even, D.; Ideses, D.; Juven-gershon, T. The core promoter: At the heart of gene expression. Biochim. Biophys. Acta 2015, 1849, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.D.; Paccanaro, A.; Fu, Y.; Weissman, S.; Weng, Z.; Chang, J.; Snyder, M.; Gerstein, M.B. Statistical analysis of the genomic distribution and correlation of regulatory elements in the ENCODE regions. Genome Res. 2007, 17, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation—New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin Cancer Biol. 2018, 53, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Mooney, M.A.; Wilmot, B. Gene set analysis: A step-by-step guide. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2015, 168, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Aliper, A.M.; Korzinkin, M.B.; Kuzmina, N.B.; Zenin, A.A.; Venkova, L.S.; Smirnov, P.Y.; Zhavoronkov, A.A.; Buzdin, A.A.; Borisov, N.M. Mathematical Justification of Expression-Based Pathway Activation Scoring (PAS). Methods Mol. Biol. 2017, 1613, 31–51. [Google Scholar] [PubMed]
- Zolotovskaia, M.A.; Sorokin, M.I.; Roumiantsev, S.A.; Borisov, N.M.; Buzdin, A.A. Pathway Instability Is an Effective New Mutation-Based Type of Cancer Biomarkers. Front. Oncol. 2019, 8, 658. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef]
- Gaudet, P.; Dessimoz, C. Gene Ontology: Pitfalls, Biases, and Remedies. Methods Mol. Biol. 2017, 1446, 189–205. [Google Scholar]
- Miletić, A.; Krmpotić, A.; Jonjić, S. The evolutionary arms race between NK cells and viruses: Who gets the short end of the stick? Eur. J. Immunol. 2013, 43, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S. Color vision diversity and significance in primates inferred from genetic and field studies. Genes Genomics 2016, 38, 779–791. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Database. Available online: https://www.encodeproject.org/ (accessed on 16 August 2019).
- ENCODE Database. Histone ChIP-seq protocol. Available online: https://www.encodeproject.org/chip-seq/histone/ (accessed on 16 August 2019).
- ENCODE Database, ChIP-seq Read Mapping. Available online: https://www.encodeproject.org/pipelines/ENCPL220NBH/ (accessed on 16 August 2019).
- ENCODE Database, ChIP-seq Peak Calling. Available online: https://www.encodeproject.org/pipelines/ENCPL272XAE/ (accessed on 16 August 2019).
- RepeatMasker. Available online: http://www.repeatmasker.org (accessed on 16 August 2019).
- UCSC Browser, Human Genome. Available online: https://genome.ucsc.edu/cgi-bin/hgs (accessed on 16 August 2019).
- BioCarta. Available online: https://cgap.nci.nih.gov/Pathways/BioCarta_Pathways (accessed on 16 August 2019).
- KEGG. Available online: http://www.genome.jp/kegg/ (accessed on 16 August 2019).
- National Cancer Institute. Available online: https://cactus.nci.nih.gov/ncicadd/about.htm (accessed on 16 August 2019).
- Reactome. Available online: http://reactome.org (accessed on 16 August 2019).
- Pathway Central. Available online: http://www.sabiosciences.com/pathwaycentral.php (accessed on 16 August 2019).
- Buzdin, A.; Sorokin, M.; Poddubskaya, E.; Borisov, N. High-Throughput Mutation Data Now Complement Transcriptomic Profiling: Advances in Molecular Pathway Activation Analysis Approach in Cancer Biology. Cancer Inform. 2019, 18, 838–844. [Google Scholar] [CrossRef] [PubMed]
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov/ (accessed on 16 August 2019).
- USCS Genome Browser. Available online: https://genome.ucsc.edu/ (accessed on 16 August 2019).
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Python Seaborn package. Available online: http://seaborn.pydata.org/ (accessed on 16 August 2019).
- Python Sklearn package. Available online: https://scikit-learn.org/stable/documentation.html (accessed on 16 August 2019).
- Gene Ontology documentation. Available online: http://geneontology.org/docs/ontology-documentation/ (accessed on 16 August 2019).
- GOrilla-A tool for Identifying Enriched GO Terms. Available online: http://cbl-gorilla.cs.technion.ac.il (accessed on 16 August 2019).
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat Rev Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Gross, D.S.; Garrard, W.T. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988, 57, 159–197. [Google Scholar] [CrossRef]
- Natoli, G.; Andrau, J.C. Noncoding transcription at enhancers: General principles and functional models. Annu. Rev. Genet. 2012, 46, 1–19. [Google Scholar] [CrossRef]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor-DNA binding: Beyond binding site motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef]
- Bogdanovic, O.; Fernandez-miñán, A.; Tena, J.J.; de la Calle-Mustienes, E.; Hidalgo, C.; van Kruysbergen, I.; van Heeringen, S.J.; Veenstra, G.J.; Gómez-Skarmeta, J.L. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res. 2012, 22, 2043–2053. [Google Scholar] [CrossRef]
- Bonn, S.; Zinzen, R.P.; Girardot, C.; Gustafson, E.H.; Perez-Gonzalez, A.; Delhomme, N.; Ghavi-Helm, Y.; Wilczyński, B.; Riddell, A.; Furlong, E.E. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef]
- Ganesh, S.; Svoboda, P. Retrotransposon-associated long non-coding RNAs in mice and men. Pflugers Arch. 2016, 468, 1049–1060. [Google Scholar] [CrossRef]
- Gaiti, F.; Calcino, A.D.; Tanurdžić, M.; Degnan, B.M. Origin and evolution of the metazoan non-coding regulatory genome. Dev. Biol. 2017, 427, 193–202. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar]
- Kaas, J.H. The evolution of complex sensory systems in mammals. J. Exp. Biol. 1989, 146, 165–176. [Google Scholar]
- Khrameeva, E.; Kurochkin, I.; Bozek, K.; Giavalisco, P.; Khaitovich, P. Lipidome Evolution in Mammalian Tissues. Mol. Biol. Evol. 2018, 35, 1947–1957. [Google Scholar] [CrossRef]
- Lin, B.; Yuan, L.; Chen, J. Selection pressure drives the co-evolution of several lipid metabolism genes in mammals. Chin. Sci. Bull. 2012, 57, 877. [Google Scholar] [CrossRef][Green Version]
- Hulbert, A.J.; Else, P.L. Evolution of mammalian endothermic metabolism: Mitochondrial activity and cell composition. Am. J. Physiol. 1989, 256, R63–R69. [Google Scholar] [CrossRef]
- O’brien, P.J. Catalytic promiscuity and the divergent evolution of DNA repair enzymes. Chem. Rev. 2006, 106, 720–752. [Google Scholar] [CrossRef]
- Allan, D.C.; Phillips, J.C. Why Ubiquitin Has Not Evolved. Int. J. Mol. Sci. 2017, 18, 1995. [Google Scholar] [CrossRef]
- Van dam, T.J.; Zwartkruis, F.J.; Bos, J.L.; Snel, B. Evolution of the TOR pathway. J. Mol. Evol. 2011, 73, 209–220. [Google Scholar] [CrossRef]
- Signor, S.A.; Nuzhdin, S.V. The Evolution of Gene Expression in cis and trans. Trends Genet. 2018, 34, 532–544. [Google Scholar] [CrossRef]
- Soyer, O.S.; Bonhoeffer, S. Evolution of complexity in signaling pathways. Proc. Natl. Acad. Sci. USA 2006, 103, 16337–16342. [Google Scholar] [CrossRef]
- Ko, J.Y.; Oh, S.; Yoo, K.H. Functional Enhancers as Master Regulators of Tissue-Specific Gene Regulation and Cancer Development. Mol. Cells. 2017, 40, 169–177. [Google Scholar]
- Zabidi, M.A.; Stark, A. Regulatory Enhancer-Core-Promoter Communication via Transcription Factors and Cofactors. Trends Genet. 2016, 32, 801–814. [Google Scholar] [CrossRef]
- Hancks, D.C.; Kazazian, H.H. Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef]
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013, 23, 345–356. [Google Scholar] [CrossRef]






{kind=link}
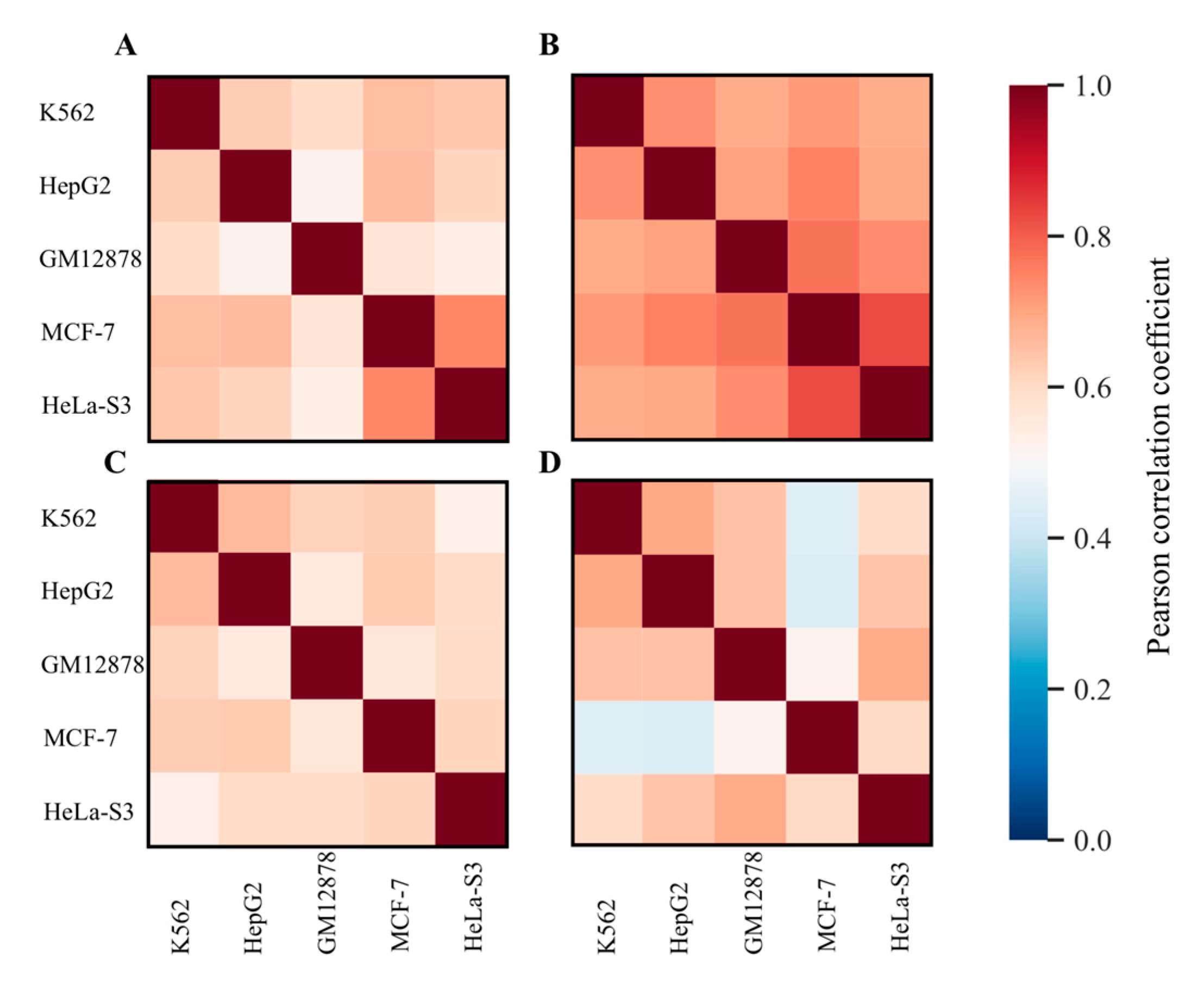{kind=link}
{kind=link}
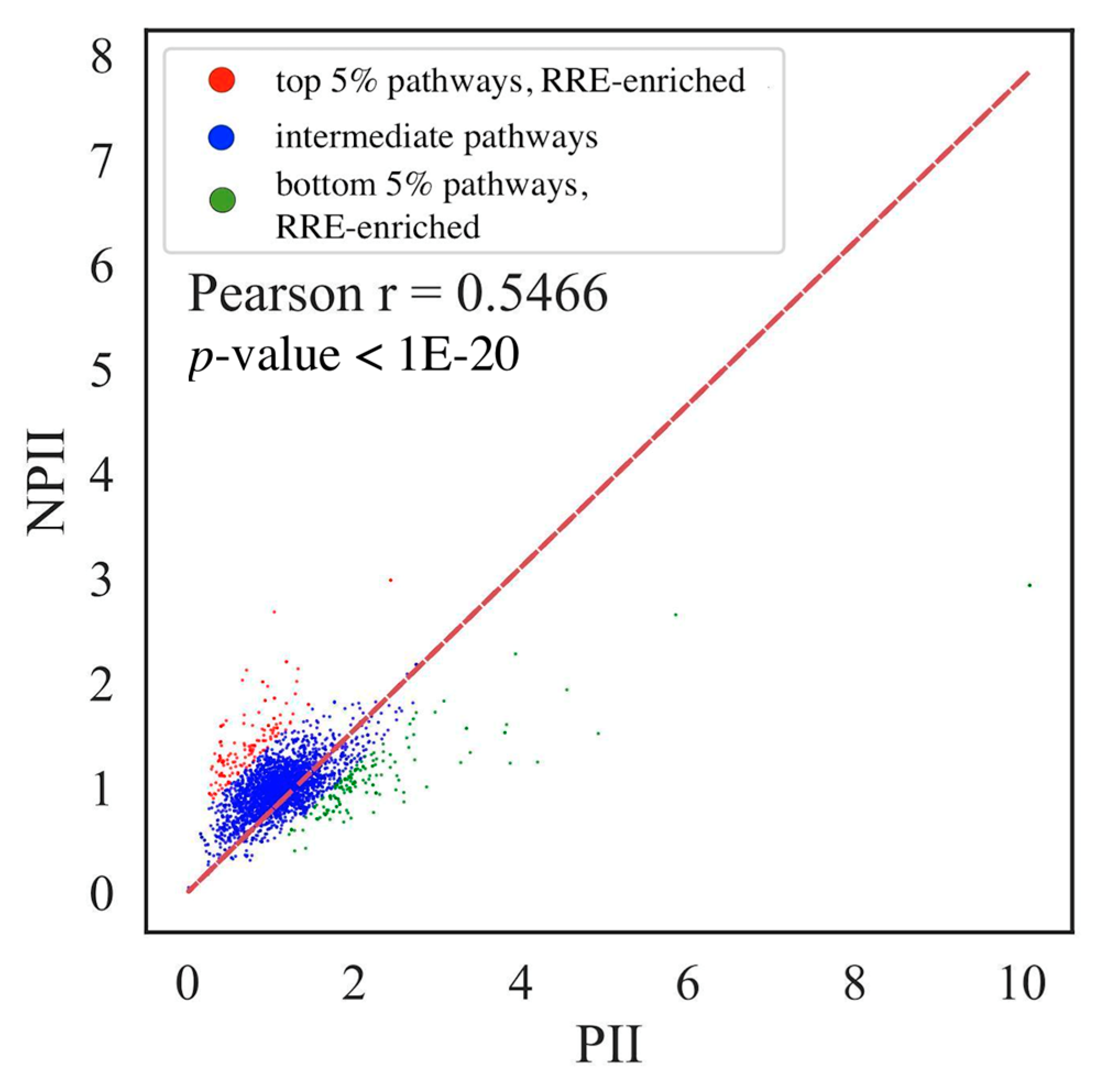{kind=link}
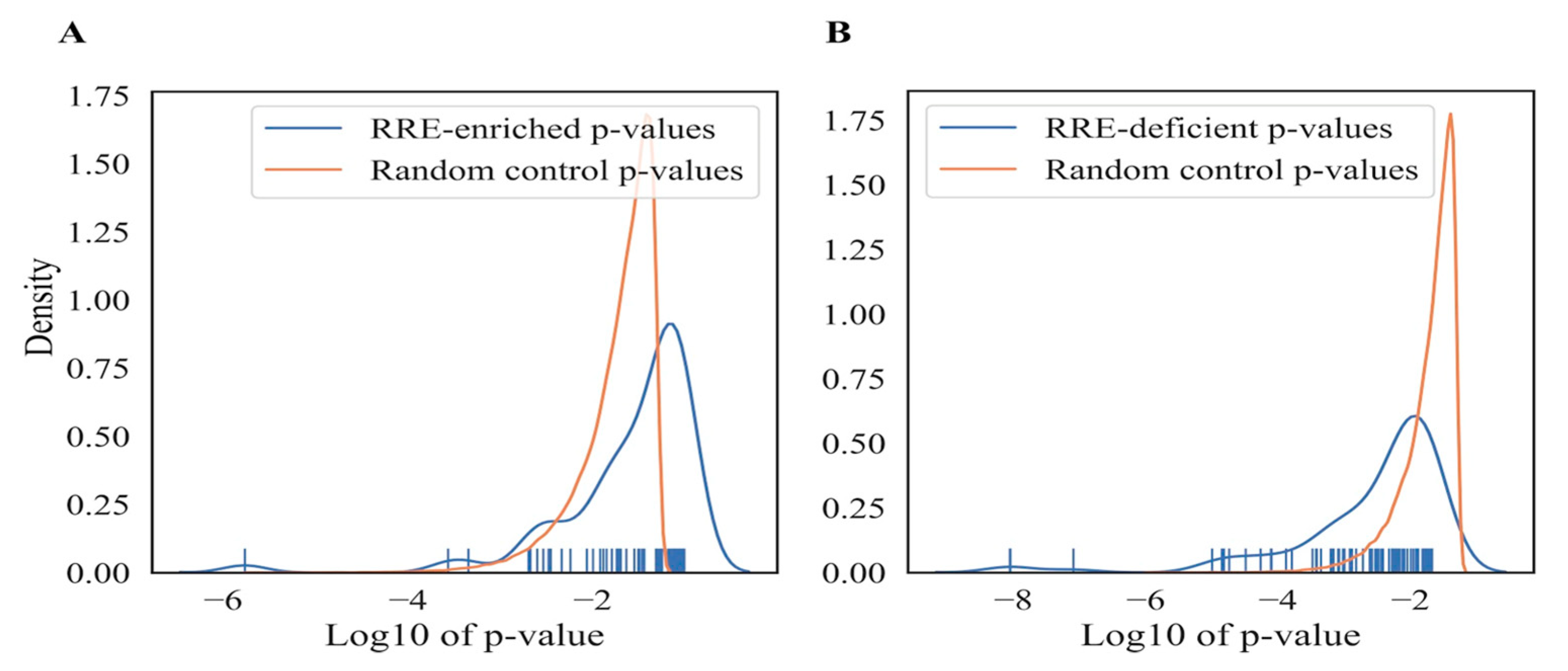{kind=link}
| Group | Non-Coding RNA Class | Number of Non-Coding RNA Genes in the Sample | Expected Number of Non-Coding RNA Genes in the Sample, Random Distribution Model | Hypergeometric p-Value for Hypothesis That Non-Coding RNA Genes are Overrepresented in the Respective Gene Set | Hypergeometric p-Value for Hypothesis That Non-Coding RNA Genes are Underrepresented in the Respective Gene Set | Conclusion |
|---|---|---|---|---|---|---|
| RRE-enriched | lncRNA | 145 | 74 | 2.22 × 10−8 | 0.99999998 | lncRNAs are overrepresented |
| RRE-deficient | lncRNA | 54 | 74 | 0.9951 | 0.0049 | lncRNAs are underrepresented |
| RRE-enriched | microRNA | 145 | 90 | 3.49 × 10−8 | 0.99999997 | microRNAs are overrepresented |
| RRE-deficient | microRNA | 94 | 90 | 0.33 | 0.67 | microRNAs are neither overrepresented nor underrepresented |
| Group of Processes | Pathway Analysis | GO Analysis | Overall Status | ||
|---|---|---|---|---|---|
| Enriched | Deficient | Enriched | Deficient | ||
| Posttranscriptional silencing by small RNAs | 1 | 0 | 1 | 0 | enriched |
| DNA Metabolism and Chromatin Structure | 7 | 6 | 4 | 3 | enriched |
| Sensory Perception and Neurotransmission | 3 | 0 | 2 | 0 | enriched |
| Lipids Metabolism | 12 | 8 | 3 | 0 | enriched |
| Endocytosis | 5 | 4 | 0 | 0 | enriched |
| Immune System | 11 | 21 | 2 | 8 | deficient |
| Protein Ubiquitination and Degradation | 0 | 5 | 0 | 2 | deficient |
| Cell Adhesion, Migration and Interaction | 13 | 15 | 0 | 12 | deficient |
| Metals Metabolism and Ion Transport | 0 | 3 | 3 | 4 | deficient |
| Cell Death | 7 | 13 | 0 | 11 | deficient |
| General Signaling Pathways | 33 | 38 | 0 | 15 | deficient |
| Hormones Signaling Pathways | 0 | 3 | 0 | 4 | deficient |
| Stress Response | 0 | 0 | 0 | 2 | deficient |
| Response to Viruses | 0 | 0 | 0 | 2 | deficient |
| Amino Acids Metabolism | 2 | 4 | 0 | 0 | deficient |
| Detoxication of Xenobiotics | 0 | 8 | 0 | 0 | deficient |
| Protein Aggregation and Import | 0 | 0 | 0 | 7 | deficient |
| Morphogenesis | 16 | 9 | 0 | 45 | ambiguous |
| Cytoskeleton | 7 | 2 | 0 | 7 | ambiguous |
| RNA Synthesis and Degradation | 3 | 0 | 6 | 23 | ambiguous |
| Translation, Protein Export and Folding | 0 | 3 | 3 | 0 | ambiguous |
| Cell Cycle and Mitosis | 22 | 3 | 2 | 6 | ambiguous |
| Other Processes | 14 | 10 | 7 | 18 | ambiguous |
| Group of Processes | Current Study | TFBS Study | Comment | ||
|---|---|---|---|---|---|
| Overall Status | Type of Analysis | Status | Type of Analysis | ||
| (Pathways/GO/Consensus) | (Pathways/GO/Consensus) | ||||
| Consensus Match | |||||
| Sensory Perception and Neurotransmission | enriched | consensus | enriched | consensus | |
| Lipids Metabolism | enriched | consensus | enriched | consensus | |
| Protein Ubiquitination and Degradation | deficient | consensus | deficient | consensus | Corresponds to “Translation and Protein Quality Control” in the TFBS study |
| Posttranscriptional silencing by small RNAs | enriched | consensus | enriched | consensus | Identified by Gorilla software and validated using hypergeometric enrichment in both studies |
| DNA Metabolism and Chromatin Structure | enriched | consensus | enriched | consensus | Corresponds to “DNA repair” in the TFBS study |
| General Signaling Pathways | deficient | consensus | deficient | consensus | |
| Match by Overall Status | |||||
| Stress Response | deficient | GO | deficient | GO | |
| Cell Adhesion, Migration and Interaction | deficient | consensus | deficient | GO | |
| Cell Death | deficient | consensus | deficient | GO | |
| Protein Aggregation and Import | deficient | GO | deficient | GO | Corresponds to “Protein Localization and Modification” in the TFBS study |
| Response to Viruses | deficient | GO | deficient | consensus | |
| Does not Match | |||||
| Immune System | deficient | consensus | ambiguous | - | |
| Metals Metabolism and Ion Transport | deficient | consensus | enriched | GO | |
| Hormones Signaling Pathways | deficient | consensus | enriched | pathways | Corresponds to “Hormones” in the TFBS study |
| Amino Acids Metabolism | deficient | pathways | enriched | consensus | |
| Detoxication of Xenobiotics | deficient | pathways | enriched | consensus | |
| RNA Synthesis and Degradation | ambiguous | - | deficient | GO | |
| Translation, Protein Export and Folding | ambiguous | - | deficient | consensus | Corresponds to “Translation and Protein Quality Control” in the TFBS study |
| Cell Cycle and Mitosis | ambiguous | - | deficient | GO | |
| Group Appears Only in one of the Studies | |||||
| Endocytosis | enriched | pathways | - | ||
| Morphogenesis | ambiguous | - | - | - | |
| Cytoskeleton | ambiguous | - | - | - | |
| Cellular immune response (T cells and NK cells) | - | - | enriched | consensus | |
| Fertilization | - | - | enriched | consensus | |
| Vitamin metabolism | - | - | enriched | pathways | |
| Molecular transport | - | - | enriched | pathways | |
| Sulfur metabolism and linked redox reactions | - | - | enriched | pathways | |
| Response to phorbol acetate | - | - | deficient | GO | |
| Electron transfer reactions | - | - | deficient | GO | |
| Mitochondria | - | - | deficient | GO | |
| Nucleic Base, Nucleosides and Nucleotides metabolism | - | - | deficient | consensus | |
| Carbohydrates metabolism | - | - | ambiguous | - | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikitin, D.; Kolosov, N.; Murzina, A.; Pats, K.; Zamyatin, A.; Tkachev, V.; Sorokin, M.; Kopylov, P.; Buzdin, A. Retroelement-Linked H3K4me1 Histone Tags Uncover Regulatory Evolution Trends of Gene Enhancers and Feature Quickly Evolving Molecular Processes in Human Physiology. Cells 2019, 8, 1219. https://doi.org/10.3390/cells8101219
Nikitin D, Kolosov N, Murzina A, Pats K, Zamyatin A, Tkachev V, Sorokin M, Kopylov P, Buzdin A. Retroelement-Linked H3K4me1 Histone Tags Uncover Regulatory Evolution Trends of Gene Enhancers and Feature Quickly Evolving Molecular Processes in Human Physiology. Cells. 2019; 8(10):1219. https://doi.org/10.3390/cells8101219
Chicago/Turabian StyleNikitin, Daniil, Nikita Kolosov, Anastasiia Murzina, Karina Pats, Anton Zamyatin, Victor Tkachev, Maxim Sorokin, Philippe Kopylov, and Anton Buzdin. 2019. "Retroelement-Linked H3K4me1 Histone Tags Uncover Regulatory Evolution Trends of Gene Enhancers and Feature Quickly Evolving Molecular Processes in Human Physiology" Cells 8, no. 10: 1219. https://doi.org/10.3390/cells8101219
APA StyleNikitin, D., Kolosov, N., Murzina, A., Pats, K., Zamyatin, A., Tkachev, V., Sorokin, M., Kopylov, P., & Buzdin, A. (2019). Retroelement-Linked H3K4me1 Histone Tags Uncover Regulatory Evolution Trends of Gene Enhancers and Feature Quickly Evolving Molecular Processes in Human Physiology. Cells, 8(10), 1219. https://doi.org/10.3390/cells8101219