MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer


Abstract
1. Introduction
2. Breast Cancer Epigenetics
2.1. DNA Methylation
2.2. Histone Modification
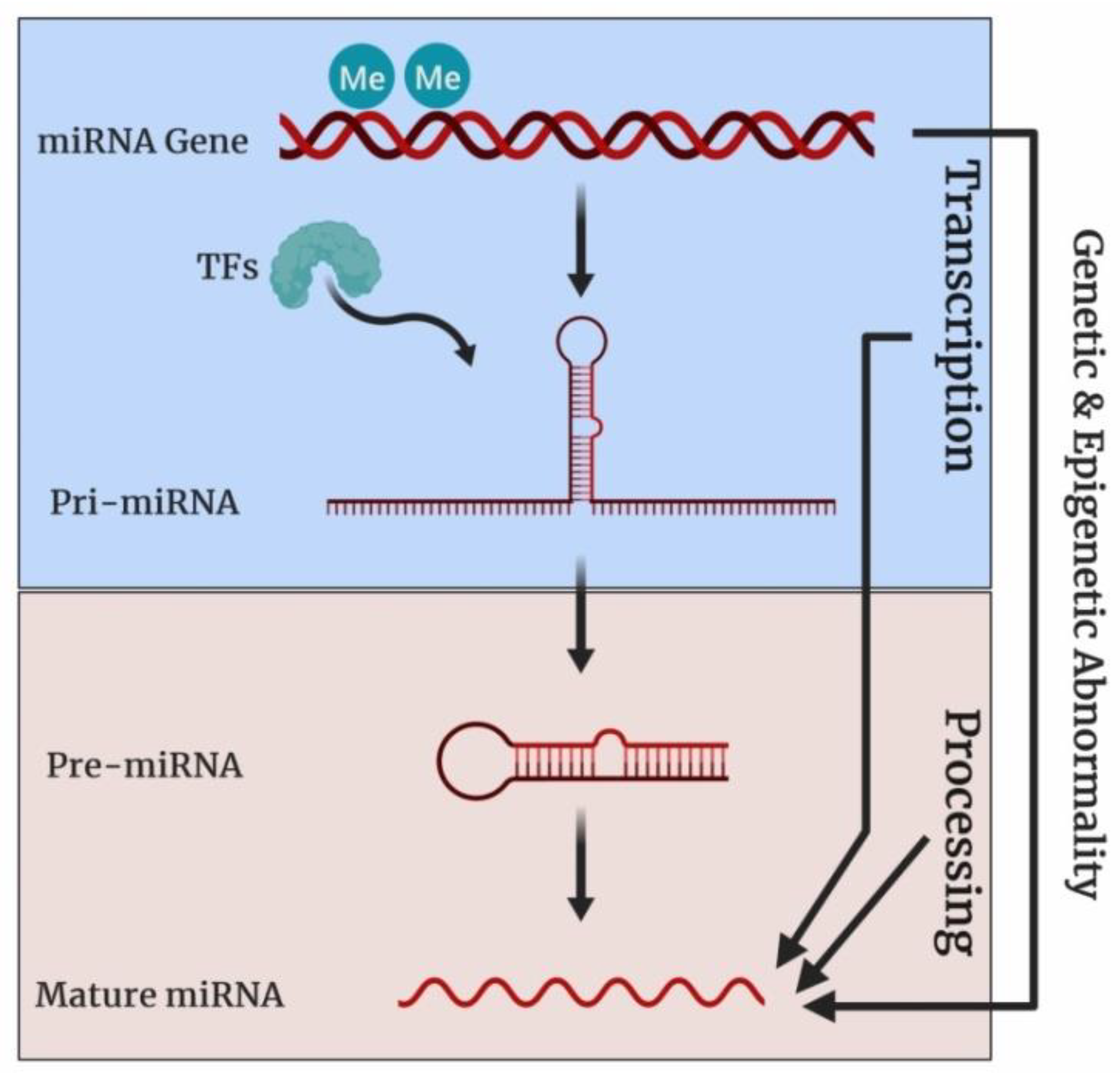
2.3. MicroRNAs (miRNAs)
2.3.1. MicroRNAs and Cancer
2.3.2. MicroRNAs and Breast Cancer
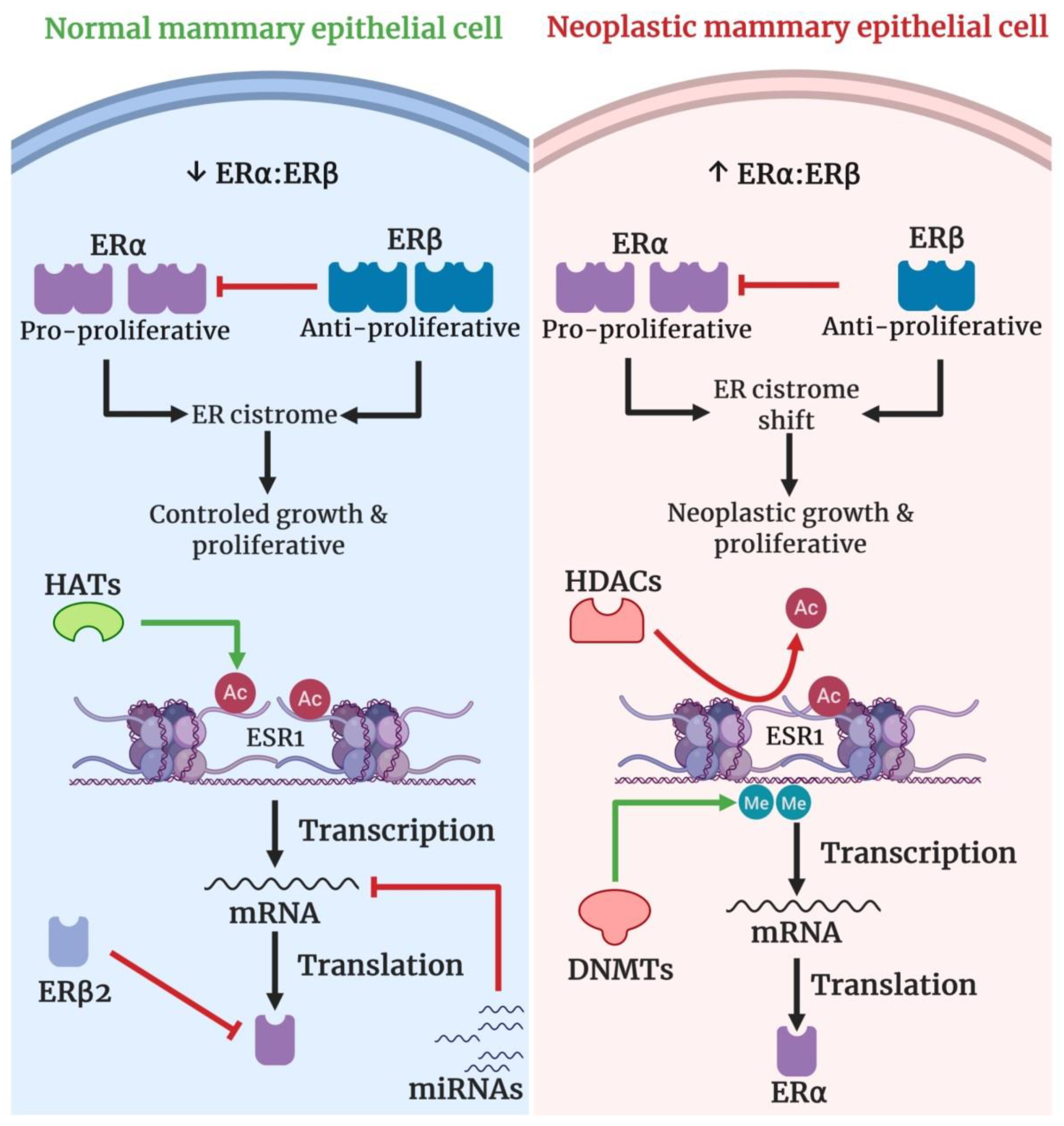
2.4. Estrogen Receptors
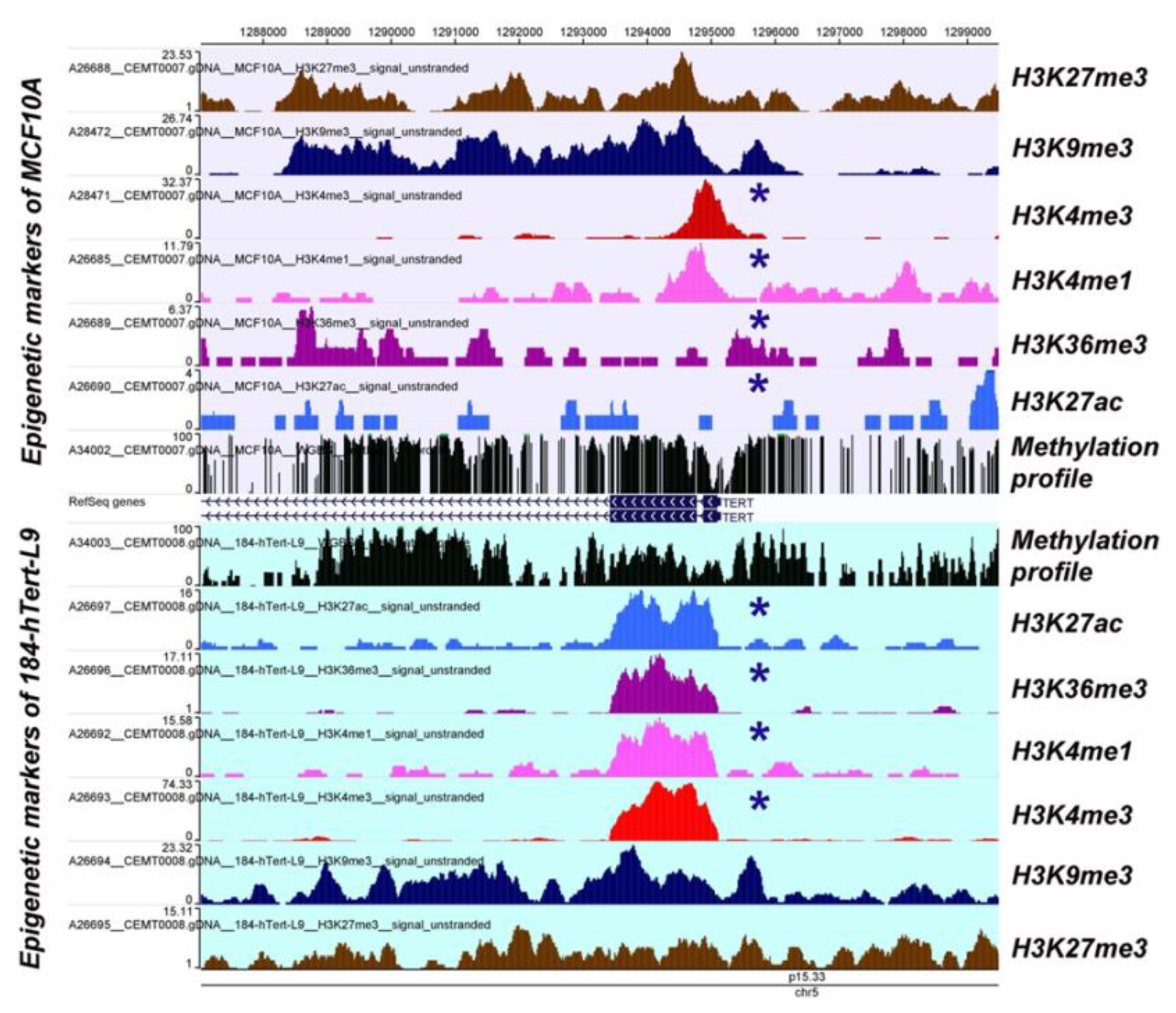
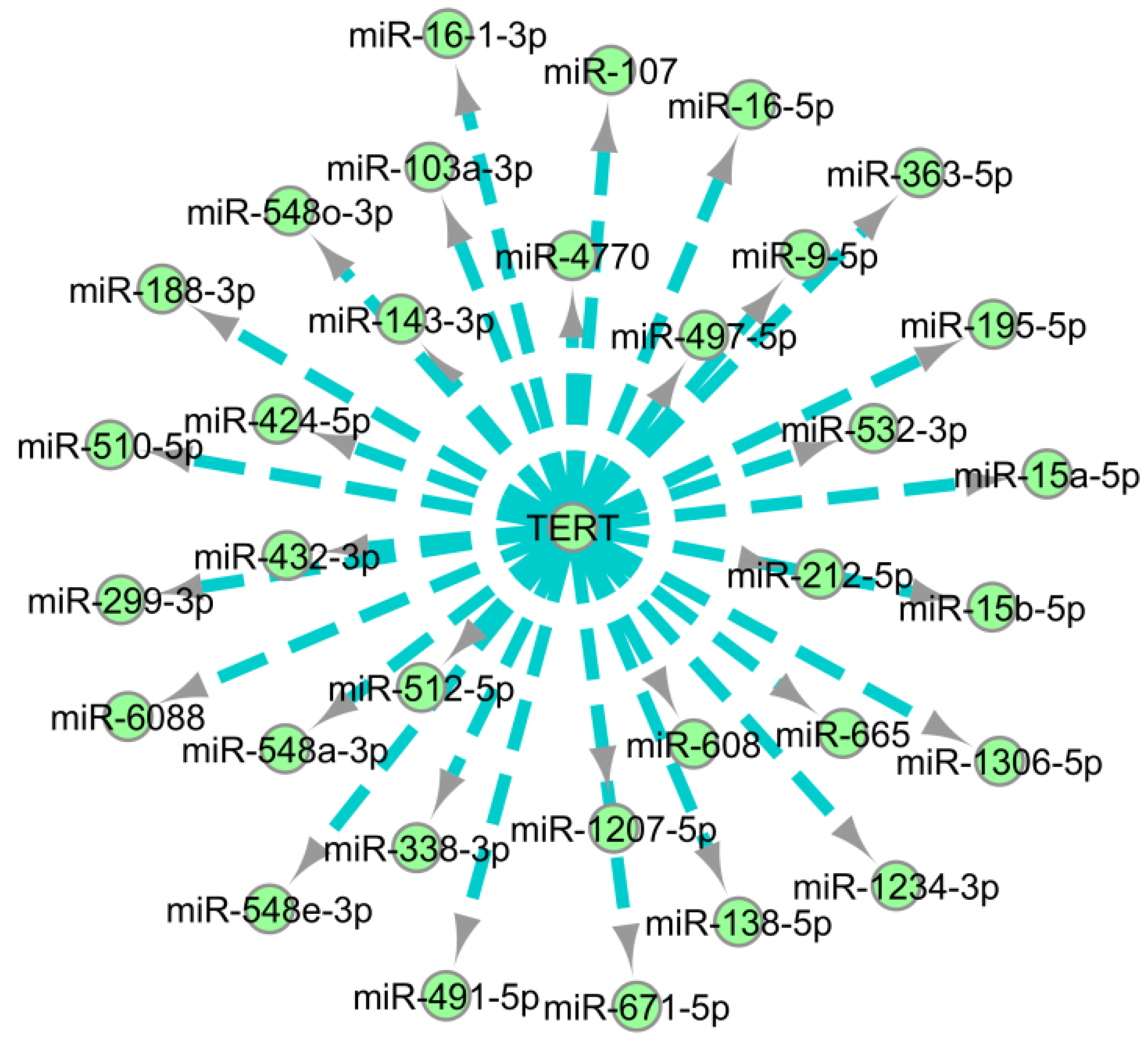
2.5. Human Telomerase Reverse Transcriptase (hTERT)
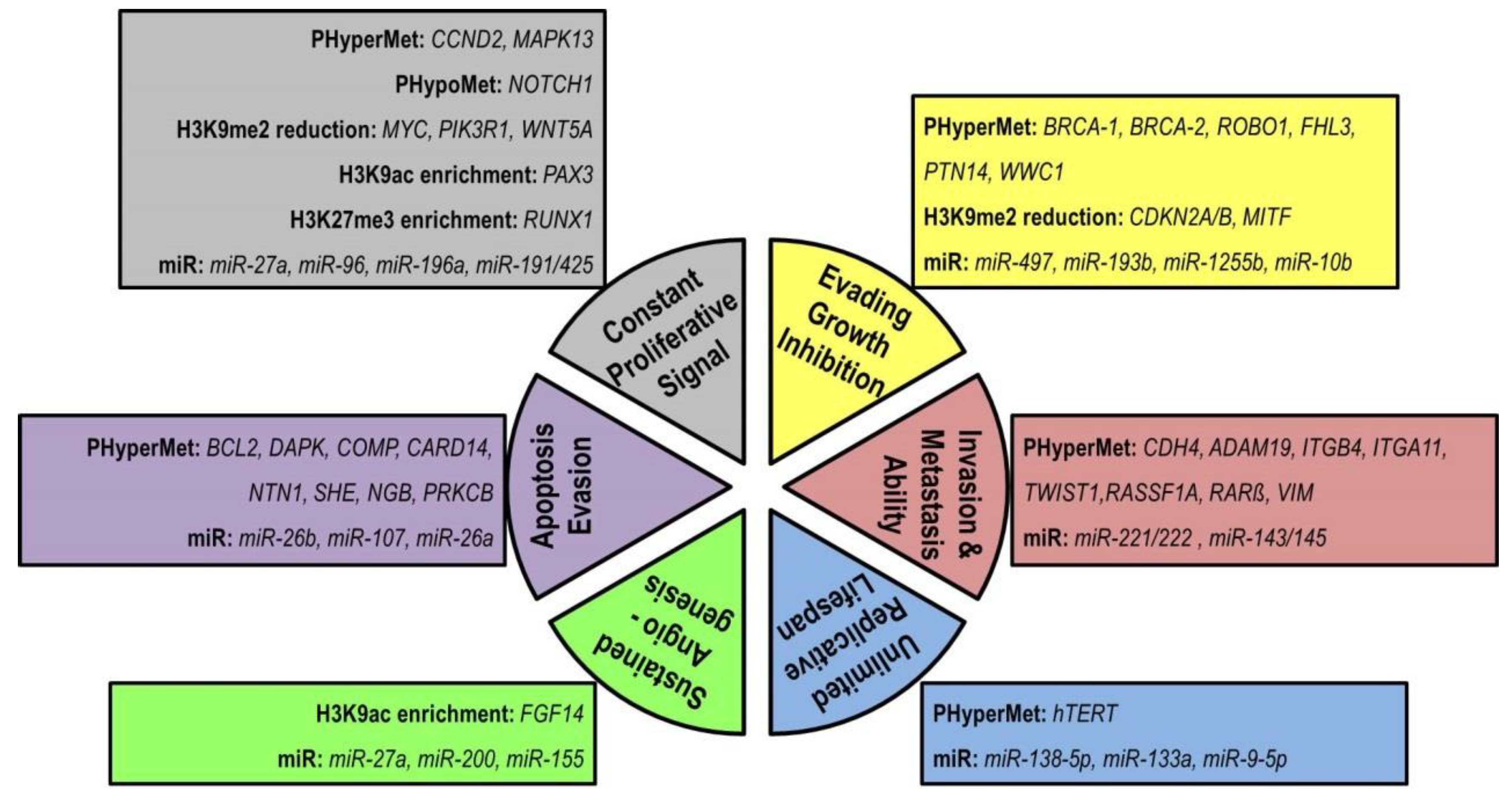
3. Epigenetic Version of Breast Cancer Hallmarks: An Avenue to Reversibility
4. Epigenetics Strategies of Breast Cancer Reversal
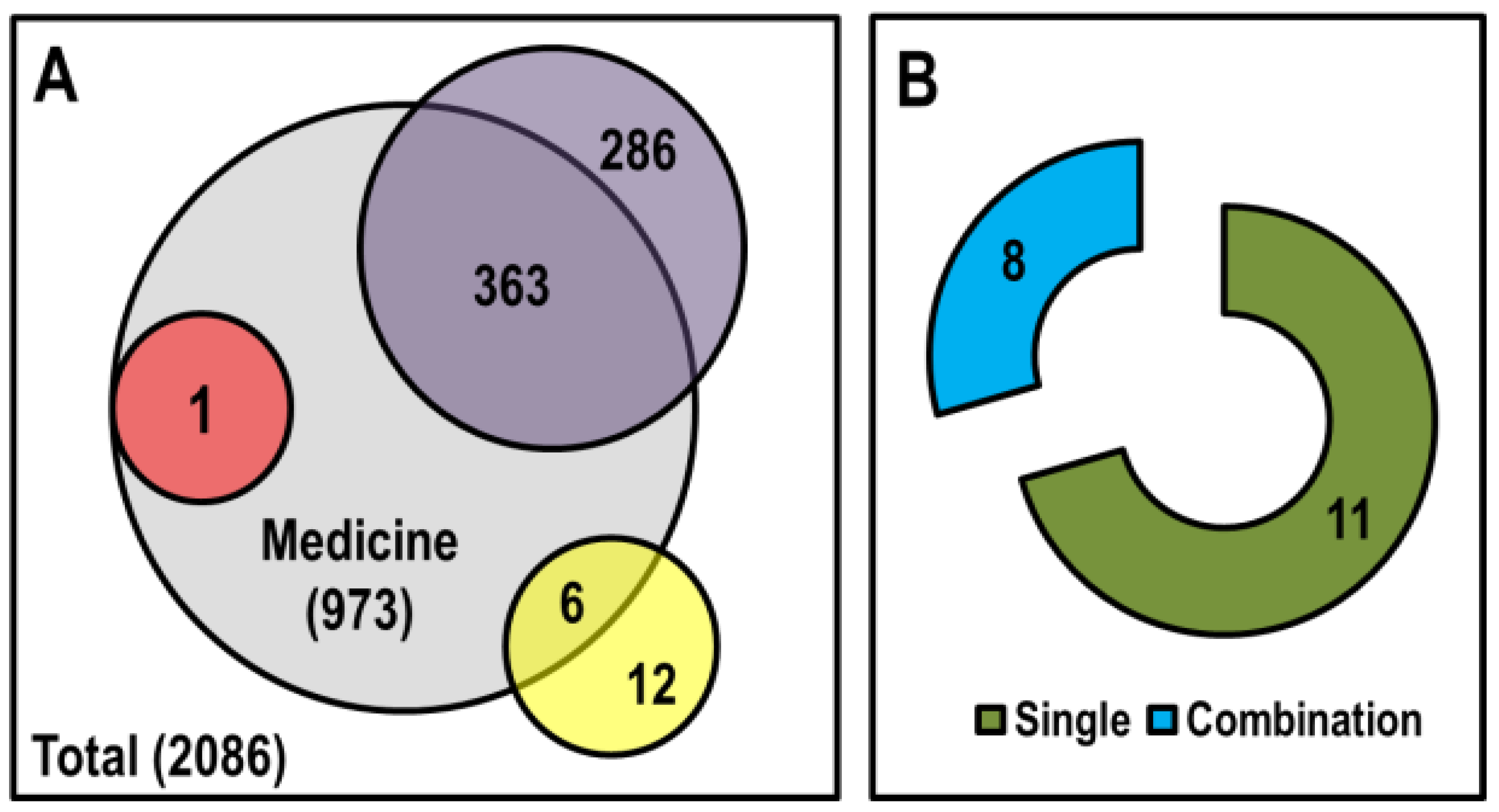
4.1. Epigenetic Drugs
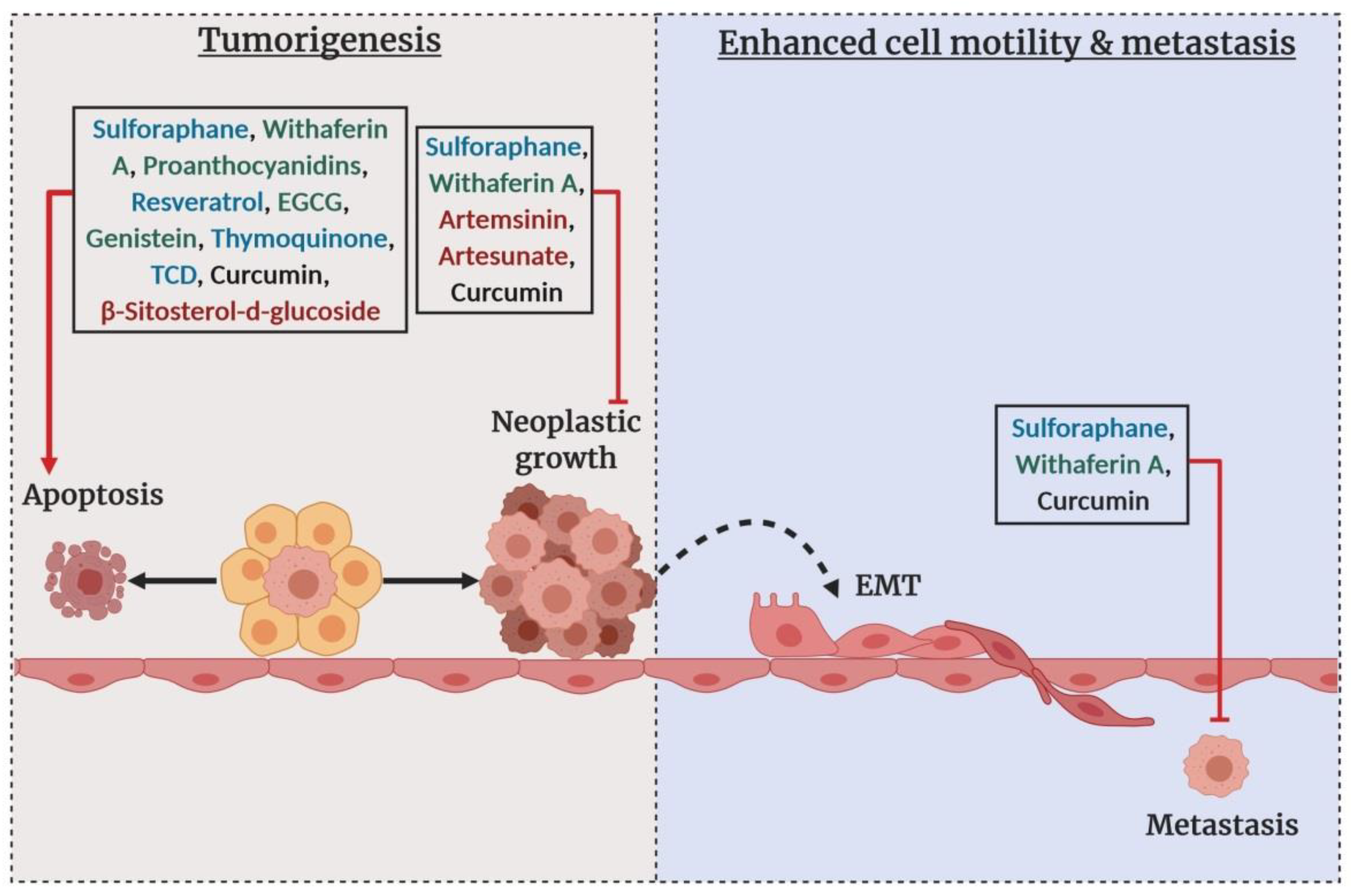
4.2. Epigenetic Diet
4.3. Epigenome Editing Tools
4.4. miRNA-Based Therapy
4.4.1. MiRNA Mimic
4.4.2. MiRNA Antagonists (antagomiRs)
Anti-miRNA-135a
Anti-miRNA-492
Anti-miRNA-203
Anti-miRNA-937
4.4.3. MiRNA Sponges
miRNA-933 Sponge
miRNA-23a Sponge
miRNA-10b Sponge
miRNA-203a-3p Sponge
miRNA-21 Sponge
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Fact Sheets by International Agency for Research on Cancer (IARC) and World Health Organization (WHO). Available online: http://gco.iarc.fr/today/data/factsheets/cancers/20-Breast-fact-sheet.pdf (accessed on 20 June 2019).
- American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018; pp. 12–15. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, Y.; Kiani, M.F.; Wang, B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin. Breast Cancer 2016, 16, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed]
- Rizzolo, P.; Silvestri, V.; Falchetti, M.; Ottini, L. Inherited and acquired alterations in development of breast cancer. Appl. Clin. Genet. 2011, 4, 145–158. [Google Scholar] [PubMed]
- Mandujano-Tinoco, E.A.; Garcia-Venzor, A.; Melendez-Zajgla, J.; Maldonado, V. New emerging roles of microRNAs in breast cancer. Breast Cancer Res. Treat. 2018, 171, 247–259. [Google Scholar] [CrossRef]
- Pasculli, B.; Barbano, R.; Parrella, P. Epigenetics of breast cancer: Biology and clinical implication in the era of precision medicine. Semin. Cancer Biol. 2018, 51, 22–35. [Google Scholar] [CrossRef]
- Mese, G.; Yalcin-Ozuysal, O. Epigenetics of Breast Cancer: DNA Methylome and Global Histone Modifications. In Epigenetic Advancements in Cancer; Mishra, M.K., Bishnupuri, K.S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 207–228. [Google Scholar]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef]
- Zhang, Z.; Atwell, L.L.; Farris, P.E.; Ho, E.; Shannon, J. Associations between cruciferous vegetable intake and selected biomarkers among women scheduled for breast biopsies. Public Health Nutr. 2016, 19, 1288–1295. [Google Scholar] [CrossRef]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase I study of panobinostat (LBH589) and letrozole in postmenopausal metastatic breast cancer patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef]
- Fedele, P.; Orlando, L.; Cinieri, S. Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert. Opin. Investig. Drugs 2017, 26, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Lustberg, M.B.; Ramaswamy, B. Epigenetic therapy in breast cancer. Curr. Breast Cancer Rep. 2011, 3, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenetics Chromatin 2017, 10, 23. [Google Scholar] [CrossRef]
- Casillas, M.A., Jr.; Lopatina, N.; Andrews, L.G.; Tollefsbol, T.O. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol. Cell. Biochem. 2003, 252, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in cancer and aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef]
- Han, L.; Zheng, S.; Sun, S.; Huang, T.H.M.; Zhao, Z. Genome-Wide DNA Methylation Profiling in 40 Breast Cancer Cell Lines. In International Conference on Intelligent Computing; Huang, D.-S., Zhao, Z., Bevilacqua, V., Figueroa, J.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 277–284. [Google Scholar]
- Wang, F.; Yang, Y.; Fu, Z.; Xu, N.; Chen, F.; Yin, H.; Lu, X.; Shen, R.; Lu, C. Differential DNA methylation status between breast carcinomatous and normal tissues. Biomed. Pharmacother. 2014, 68, 699–707. [Google Scholar] [CrossRef]
- Holm, K.; Staaf, J.; Lauss, M.; Aine, M.; Lindgren, D.; Bendahl, P.O.; Vallon-Christersson, J.; Barkardottir, R.B.; Hoglund, M.; Borg, A.; et al. An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells. Breast Cancer Res. 2016, 18, 27. [Google Scholar] [CrossRef]
- Tanas, A.S.; Sigin, V.O.; Kalinkin, A.I.; Litviakov, N.V.; Slonimskaya, E.M.; Ibragimova, M.K.; Ignatova, E.O.; Simonova, O.A.; Kuznetsova, E.B.; Kekeeva, T.V.; et al. Genome-wide methylotyping resolves breast cancer epigenetic heterogeneity and suggests novel therapeutic perspectives. Epigenomics 2019, 11, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, L.; Shu, X.O.; Cai, Q.; Shu, X.; Li, B.; Guo, X.; Ye, F.; Michailidou, K.; Bolla, M.K.; et al. Genetically predicted levels of DNA methylation biomarkers and breast cancer risk: Data from 228,951 women of European descent. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, B.P.; Apolonio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Z.; Cai, X. Identification of epigenetic modulators in human breast cancer by integrated analysis of DNA methylation and RNA-Seq data. Epigenetics 2018, 13, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.K.; Usmani, N.; Michiels, S.; Metzger-Filho, O.; Saini, K.S.; Kovalchuk, O.; Parliament, M. Towards understanding the breast cancer epigenome: A comparison of genome-wide DNA methylation and gene expression data. Oncotarget 2016, 7, 3002–3017. [Google Scholar] [CrossRef]
- Li, G.; Wang, D.; Ma, W.; An, K.; Liu, Z.; Wang, X.; Yang, C.; Du, F.; Han, X.; Chang, S.; et al. Transcriptomic and epigenetic analysis of breast cancer stem cells. Epigenomics 2018, 10, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef]
- Locke, W.J.; Zotenko, E.; Stirzaker, C.; Robinson, M.D.; Hinshelwood, R.A.; Stone, A.; Reddel, R.R.; Huschtscha, L.I.; Clark, S.J. Coordinated epigenetic remodelling of transcriptional networks occurs during early breast carcinogenesis. Clin. Epigenetics 2015, 7, 52. [Google Scholar] [CrossRef]
- Nickel, A.; Stadler, S.C. Role of epigenetic mechanisms in epithelial-to-mesenchymal transition of breast cancer cells. Transl. Res. 2015, 165, 126–142. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Lameirinhas, A.; Henrique, R.; Jeronimo, C. Metabolism and epigenetic interplay in cancer: Regulation and putative therapeutic targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef]
- Mathot, P.; Grandin, M.; Devailly, G.; Souaze, F.; Cahais, V.; Moran, S.; Campone, M.; Herceg, Z.; Esteller, M.; Juin, P.; et al. DNA methylation signal has a major role in the response of human breast cancer cells to the microenvironment. Oncogenesis 2017, 6, e390. [Google Scholar] [CrossRef]
- Zheng, C.; Hayes, J.J. Structures and interactions of the core histone tail domains. Biopolymers 2003, 68, 539–546. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Bian, J.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Correction: Role of novel histone modifications in cancer. Oncotarget 2018, 9, 19460. [Google Scholar] [CrossRef] [PubMed]
- Espino, P.S.; Drobic, B.; Dunn, K.L.; Davie, J.R. Histone modifications as a platform for cancer therapy. J. Cell. Biochem. 2005, 94, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Chervona, Y.; Costa, M. Histone modifications and cancer: Biomarkers of prognosis? Am. J. Cancer Res. 2012, 2, 589–597. [Google Scholar]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Xue-Gang, L.; Shu, G.; Yu, G.; Chun-Ling, Z. Histone modification and breast cancer. Breast Cancer 2011. [Google Scholar] [CrossRef]
- Magnani, L.; Louloupi, A.; Zwart, W. Chapter 23 - Histone Posttranslational Modifications in Breast Cancer and Their Use in Clinical Diagnosis and Prognosis. In Epigenetic Biomarkers and Diagnostics; García-Giménez, J.L., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 467–477. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Guo, S.W. Epigenetics of endometriosis. Mol. Hum. Reprod. 2009, 15, 587–607. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J. A structural perspective on readout of epigenetic histone and DNA methylation marks. Cold Spring Harb. Perspect. Biol. 2016, 8, a018754. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Healey, M.A.; Hu, R.; Beck, A.H.; Collins, L.C.; Schnitt, S.J.; Tamimi, R.M.; Hazra, A. Association of H3K9me3 and H3K27me3 repressive histone marks with breast cancer subtypes in the Nurses’ Health Study. Breast Cancer Res. Treat. 2014, 147, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, H.; He, L.; Yu, X.; Liu, X.; Zhong, R.; Shu, M. A novel subtype classification and risk of breast cancer by histone modification profiling. Breast Cancer Res. Treat. 2016, 157, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Noberini, R.; Uggetti, A.; Pruneri, G.; Minucci, S.; Bonaldi, T. Pathology tissue-quantitative mass spectrometry analysis to profile histone post-translational modification patterns in patient samples. Mol. Cell. Proteom. 2016, 15, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Noberini, R.; Osti, D.; Miccolo, C.; Richichi, C.; Lupia, M.; Corleone, G.; Hong, S.P.; Colombo, P.; Pollo, B.; Fornasari, L.; et al. Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 2018, 46, 3817–3832. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenetics 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Lebert, A.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. The epigenetic landscape of promoter genome-wide analysis in breast cancer. Sci Rep. 2017, 7, 6597. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef]
- Olena, A.F.; Patton, J.G. Genomic organization of microRNAs. J. Cell. Physiol. 2010, 222, 540–545. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol. Lett. 2018, 15, 2735–2742. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef]
- Lewis, K.A.; Jordan, H.R.; Tollefsbol, T.O. Effects of SAHA and EGCG on growth potentiation of triple-negative breast cancer cells. Cancers 2018, 11. [Google Scholar] [CrossRef]
- Martin, S.L.; Kala, R.; Tollefsbol, T.O. Mechanisms for the inhibition of colon cancer cells by sulforaphane through epigenetic modulation of microrna-21 and human telomerase reverse transcriptase (hTERT) down-regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef]
- Ramchandran, R.; Chaluvally-Raghavan, P. miRNA-mediated RNA activation in mammalian cells. Adv. Exp. Med. Biol. 2017, 983, 81–89. [Google Scholar]
- Vaschetto, L.M. miRNA activation is an endogenous gene expression pathway. RNA Biol. 2018, 15, 826–828. [Google Scholar] [CrossRef]
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Huang, S.; Zhang, Z.; Qian, X.; Sun, P.; Zhou, X. Pan-cancer analysis on microRNA-associated gene activation. EBioMedicine 2019, 43, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, Y.; Liang, Z.; Wang, X.; Meng, S.; Xu, X.; Xu, X.; Wu, J.; Ji, A.; Hu, Z.; et al. Up-regulation of p16 by miR-877-3p inhibits proliferation of bladder cancer. Oncotarget 2016, 7, 51773–51783. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.R.; Park, K.H.; Yang, J.O.; Lee, C.W.; Oh, S.J.; Yun, J.; Lee, M.Y.; Han, S.B.; Kang, J.S. miR-6734 Up-regulates p21 gene expression and induces cell cycle arrest and apoptosis in colon cancer cells. PLoS ONE 2016, 11, e0160961. [Google Scholar] [CrossRef] [PubMed]
- Chaluvally-Raghavan, P.; Jeong, K.J.; Pradeep, S.; Silva, A.M.; Yu, S.; Liu, W.; Moss, T.; Rodriguez-Aguayo, C.; Zhang, D.; Ram, P.; et al. Direct upregulation of STAT3 by microRNA-551b-3p deregulates growth and metastasis of ovarian cancer. Cell. Rep. 2016, 15, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tong, D.; Han, C.; Zhao, Z.; Wang, X.; Jiang, T.; Li, Q.; Liu, S.; Chen, L.; Chen, Y.; et al. Blockade of miR-3614 maturation by IGF2BP3 increases TRIM25 expression and promotes breast cancer cell proliferation. EBioMedicine 2019, 41, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Chen, H.; Song, J.; Chen, X.; Lin, C.; Zhang, X.; Hou, N.; Pan, J.; Zhou, Z.; Wang, L.; et al. MiR-454-3p-mediated wnt/beta-catenin signaling antagonists suppression promotes breast cancer metastasis. Theranostics 2019, 9, 449–465. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, S.; Zhao, Y.; Zhang, Z.; Qin, C.; Yang, X. MicroRNA-216a suppresses the proliferation and migration of human breast cancer cells via the Wnt/beta-catenin signaling pathway. Oncol. Rep. 2019, 41, 2647–2656. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Zhang, G. miR-4458 regulates cell proliferation and apoptosis through targeting SOCS1 in triple-negative breast cancer. J. Cell. Biochem. 2019, 120, 12943–12948. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, B.; Wang, Y.; Chen, G.; Lian, Q.; Wang, H. miR-140-3p inhibits breast cancer proliferation and migration by directly regulating the expression of tripartite motif 28. Oncol. Lett. 2019, 17, 3835–3841. [Google Scholar] [CrossRef]
- Cui, K.; Zhang, H.; Wang, G.Z. MiR-483 suppresses cell proliferation and promotes cell apoptosis by targeting SOX3 in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Z.; Li, S.; Dong, L.; Li, Y.; Mao, Y.; Liang, Y.; Tao, Y.; Ma, J. Inhibition of miR214 attenuates the migration and invasion of triplenegative breast cancer cells. Mol. Med. Rep. 2019, 19, 4035–4042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, P.; Zou, J.Y.; Zou, G.; Wang, W.Z.; Liu, Y.L.; Zhao, H.W.; Fang, A.P. MiR-216a-5p act as a tumor suppressor, regulating the cell proliferation and metastasis by targeting PAK2 in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2469–2475. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Miao, J.; Ding, Y.; Zhang, Y.; Huang, X.; Zhou, X.; Tang, R. miR-4458 inhibits breast cancer cell growth, migration, and invasion by targeting CPSF4. Biochem. Cell. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Tang, J.; Zhu, X.; Jiang, H. Silencing of hsa_circ_0004771 inhibits proliferation and induces apoptosis in breast cancer through activation of miR-653 by targeting ZEB2 signaling pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Liu, M.; Han, H. Overexpression of microRNA-190 inhibits migration, invasion, epithelial-mesenchymal transition, and angiogenesis through suppression of protein kinase B-extracellular signal-regulated kinase signaling pathway via binding to stanniocalicin 2 in breast cancer. J. Cell. Physiol. 2019, 234, 17824–17838. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, X. microRNA-370 promotes cell growth by targeting WNK2 in breast cancer. DNA Cell. Biol. 2019, 38, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yang, S.; Wang, M.; Liu, D.; Liu, Y.; Zhang, Y.; Zhang, Q. Epigenetically altered miR193a3p promotes HER2 positive breast cancer aggressiveness by targeting GRB7. Int. J. Mol. Med. 2019, 43, 2352–2360. [Google Scholar] [CrossRef]
- Huang, X.; Tang, F.; Weng, Z.; Zhou, M.; Zhang, Q. MiR-591 functions as tumor suppressor in breast cancer by targeting TCF4 and inhibits Hippo-YAP/TAZ signaling pathway. Cancer Cell. Int. 2019, 19, 108. [Google Scholar] [CrossRef]
- Zuo, Z.; Ye, F.; Liu, Z.; Huang, J.; Gong, Y. MicroRNA-153 inhibits cell proliferation, migration, invasion and epithelial-mesenchymal transition in breast cancer via direct targeting of RUNX2. Exp. Ther. Med. 2019, 17, 4693–4702. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, H.; Wang, J.; Qian, X.; Li, N. miR-4513 promotes breast cancer progression through targeting TRIM3. Am. J. Transl. Res. 2019, 11, 2431–2438. [Google Scholar] [PubMed]
- Cui, Y.; Wang, J.; Liu, S.; Qu, D.; Jin, H.; Zhu, L.; Yang, J.; Zhang, J.; Li, Q.; Zhang, Y.; et al. miR-216a promotes breast cancer cell apoptosis by targeting PKCalpha. Fundam. Clin. Pharmacol. 2019, 33, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Tian, W.; Chen, Y.; Wang, L.; Jiang, Y.; Gao, B.; Luo, D. MicroRNA-374c-5p inhibits the development of breast cancer through TATA-box binding protein associated factor 7-mediated transcriptional regulation of DEP domain containing 1. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, C.; Niu, R.; Hu, G.; Gu, Z.; Zhuang, Z. MiR-890 inhibits proliferation and invasion and induces apoptosis in triple-negative breast cancer cells by targeting CD147. BMC Cancer 2019, 19, 577. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Gjerstorff, M.F.; Shirjang, S.; Asadzadeh, Z.; Khaze, V.; Holmskov, U.; Kazemi, T.; Duijf, P.H.G.; Baradaran, B. miR-142-3p is a tumor suppressor that inhibits estrogen receptor expression in ER-positive breast cancer. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yang, X.; He, X.; Ma, W.; Wang, J.; Zhou, Q.; Li, M.; Yu, S. MicroRNA-449b-5p suppresses the growth and invasion of breast cancer cells via inhibiting CREPT-mediated Wnt/beta-catenin signaling. Chem. Biol. Interact. 2019, 302, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhou, B.; Xiong, Y.; Cai, H. miR-135 regulated breast cancer proliferation and epithelial-mesenchymal transition acts by the Wnt/beta-catenin signaling pathway. Int. J. Mol. Med. 2019, 43, 1623–1634. [Google Scholar] [CrossRef]
- Schwarzenbacher, D.; Klec, C.; Pasculli, B.; Cerk, S.; Rinner, B.; Karbiener, M.; Ivan, C.; Barbano, R.; Ling, H.; Wulf-Goldenberg, A.; et al. MiR-1287-5p inhibits triple negative breast cancer growth by interaction with phosphoinositide 3-kinase CB, thereby sensitizing cells for PI3Kinase inhibitors. Breast Cancer Res. 2019, 21, 20. [Google Scholar] [CrossRef]
- Xiao, B.; Shi, X.; Bai, J. miR-30a regulates the proliferation and invasion of breast cancer cells by targeting Snail. Oncol. Lett. 2019, 17, 406–413. [Google Scholar] [CrossRef]
- Lv, Z.D.; Xin, H.N.; Yang, Z.C.; Wang, W.J.; Dong, J.J.; Jin, L.Y.; Li, F.N. miR-135b promotes proliferation and metastasis by targeting APC in triple-negative breast cancer. J. Cell. Physiol. 2019, 234, 10819–10826. [Google Scholar] [CrossRef]
- Ji, H.; Sang, M.; Liu, F.; Ai, N.; Geng, C. miR-124 regulates EMT based on ZEB2 target to inhibit invasion and metastasis in triple-negative breast cancer. Pathol. Res. Pract. 2019, 215, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jiang, G.Q. MiR-4282 inhibits proliferation, invasion and metastasis of human breast cancer by targeting Myc. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8763–8771. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.C.; Han, S.H.; Xing, Y.F. Overexpression of miR-3196 suppresses cell proliferation and induces cell apoptosis through targeting ERBB3 in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8383–8390. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Xie, X.X.; Bai, J.; Wang, C.; Zhao, L.; Jiang, D.Q. Increased expression of miR-1179 inhibits breast cancer cell metastasis by modulating Notch signaling pathway and correlates with favorable prognosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8374–8382. [Google Scholar] [CrossRef] [PubMed]
- Abdolvahabi, Z.; Nourbakhsh, M.; Hosseinkhani, S.; Hesari, Z.; Alipour, M.; Jafarzadeh, M.; Ghorbanhosseini, S.S.; Seiri, P.; Yousefi, Z.; Yarahmadi, S.; et al. MicroRNA-590-3P suppresses cell survival and triggers breast cancer cell apoptosis via targeting sirtuin-1 and deacetylation of p53. J. Cell. Biochem. 2019, 120, 9356–9368. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Lei, M.; Han, Y.; Zhao, D.; Zhang, X.; Yang, Y.; Liu, R. MicroRNA-645 targets urokinase plasminogen activator and decreases the invasive growth of MDA-MB-231 triple-negative breast cancer cells. Onco. Targets Ther. 2018, 11, 7733–7743. [Google Scholar] [CrossRef]
- Qin, H.; Liu, W. MicroRNA-99a-5p suppresses breast cancer progression and cell-cycle pathway through downregulating CDC25A. J. Cell. Physiol. 2019, 234, 3526–3537. [Google Scholar] [CrossRef]
- Zhu, X.; Rao, X.; Yao, W.; Zou, X. Downregulation of MiR-196b-5p impedes cell proliferation and metastasis in breast cancer through regulating COL1A1. Am. J. Transl. Res. 2018, 10, 3122–3132. [Google Scholar]
- Wang, Y.; Liu, Z.; Shen, J. MicroRNA-421-targeted PDCD4 regulates breast cancer cell proliferation. Int. J. Mol. Med. 2019, 43, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.J.; Zeng, H.X.; Huang, P.; Wang, S.; Xie, C.H.; Li, S.J. MiR-508-3p inhibits cell invasion and epithelial-mesenchymal transition by targeting ZEB1 in triple-negative breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6379–6385. [Google Scholar] [CrossRef]
- Kong, P.; Chen, L.; Yu, M.; Tao, J.; Liu, J.; Wang, Y.; Pan, H.; Zhou, W.; Wang, S. miR-3178 inhibits cell proliferation and metastasis by targeting Notch1 in triple-negative breast cancer. Cell. Death. Dis. 2018, 9, 1059. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Chen, X.; Liu, H.; Yu, K.; Bao, Y.; Chai, J.; Gao, H.; Zou, L. LGR5 acts as a target of miR-340-5p in the suppression of cell progression and drug resistance in breast cancer via Wnt/beta-catenin pathway. Gene 2019, 683, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Pang, W.; Yang, N.; Hao, L.; Wang, L. MicroRNA-511 inhibits malignant behaviors of breast cancer by directly targeting SOX9 and regulating the PI3K/Akt pathway. Int. J. Oncol. 2018, 53, 2715–2726. [Google Scholar] [CrossRef]
- Xie, D.; Song, H.; Wu, T.; Li, D.; Hua, K.; Xu, H.; Zhao, B.; Wu, C.; Hu, J.; Ji, C.; et al. MicroRNA-424 serves an antioncogenic role by targeting cyclindependent kinase 1 in breast cancer cells. Oncol. Rep. 2018, 40, 3416–3426. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Li, D.; Wu, T.; Xie, D.; Hua, K.; Hu, J.; Deng, X.; Ji, C.; Deng, Y.; Fang, L. MicroRNA-301b promotes cell proliferation and apoptosis resistance in triple-negative breast cancer by targeting CYLD. BMB Rep. 2018, 51, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xiao, Z.; Zhang, S. Knockdown of miR-194-5p inhibits cell proliferation, migration and invasion in breast cancer by regulating the Wnt/beta-catenin signaling pathway. Int. J. Mol. Med. 2018, 42, 3355–3363. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Chen, Y.; Liu, Y.; Lai, Y.; Liu, D. miR-199b-5p inhibits triple negative breast cancer cell proliferation, migration and invasion by targeting DDR1. Oncol. Lett. 2018, 16, 4889–4896. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Li, Y.; Feng, Y.; Lu, M.; Yuan, H.; Yi, Z.; Wu, Y.; Xiang, T.; Li, H.; Ren, G. Downregulated miR-1247-5p associates with poor prognosis and facilitates tumor cell growth via DVL1/Wnt/beta-catenin signaling in breast cancer. Biochem. Biophys. Res. Commun. 2018, 505, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Gao, B.; Yan, X.; Lei, Z.; Chen, K.; Li, Y.; Zeng, Q.; Chen, Z.; Li, H. MicroRNA 628 suppresses migration and invasion of breast cancer stem cells through targeting SOS1. Onco. Targets Ther. 2018, 11, 5419–5428. [Google Scholar] [CrossRef]
- Gao, J.; Yu, S.R.; Yuan, Y.; Zhang, L.L.; Lu, J.W.; Feng, J.F.; Hu, S.N. MicroRNA-590-5p functions as a tumor suppressor in breast cancer conferring inhibitory effects on cell migration, invasion, and epithelial-mesenchymal transition by downregulating the Wnt-beta-catenin signaling pathway. J. Cell. Physiol. 2019, 234, 1827–1841. [Google Scholar] [CrossRef]
- Huang, X.; Lyu, J. Tumor suppressor function of miR-483-3p on breast cancer via targeting of the cyclin E1 gene. Exp. Ther. Med. 2018, 16, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Fan, H.; Zhang, Z.; Li, N. miR-125b-5p inhibits breast cancer cell proliferation, migration and invasion by targeting KIAA1522. Biochem. Biophys. Res. Commun. 2018, 504, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, P.; Wang, J.; Wang, L.; Ren, M.; Zhang, R.; He, J. MicroRNA-1254 exerts oncogenic effects by directly targeting RASSF9 in human breast cancer. Int. J. Oncol. 2018, 53, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Rohini, M.; Gokulnath, M.; Miranda, P.J.; Selvamurugan, N. miR-590-3p inhibits proliferation and promotes apoptosis by targeting activating transcription factor 3 in human breast cancer cells. Biochimie 2018, 154, 10–18. [Google Scholar] [CrossRef]
- Yan, L.; Yu, M.C.; Gao, G.L.; Liang, H.W.; Zhou, X.Y.; Zhu, Z.T.; Zhang, C.Y.; Wang, Y.B.; Chen, X. MiR-125a-5p functions as a tumour suppressor in breast cancer by downregulating BAP1. J. Cell. Biochem. 2018, 119, 8773–8783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, F.; Luo, Q.; Yan, D.; Sun, S. miR-1284 inhibits the growth and invasion of breast cancer cells by targeting ZIC2. Oncol. Res. 2019, 27, 253–260. [Google Scholar] [CrossRef]
- Liu, F.; Sang, M.; Meng, L.; Gu, L.; Liu, S.; Li, J.; Geng, C. miR92b promotes autophagy and suppresses viability and invasion in breast cancer by targeting EZH2. Int. J. Oncol. 2018, 53, 1505–1515. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, J.; Lu, Y.; Bo, S.; Li, L.; Wang, L.; Zhang, Q.; Mao, J. miR-140-5p inhibits the proliferation and enhances the efficacy of doxorubicin to breast cancer stem cells by targeting Wnt1. Cancer Gene Ther. 2019, 26, 74–82. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, G.; Sun, R.; Pan, X.; Wang, X.; Li, H.; Sun, Y. miR1855p inhibits Factin polymerization and reverses epithelial mesenchymal transition of human breast cancer cells by modulating RAGE. Mol. Med. Rep. 2018, 18, 2621–2630. [Google Scholar] [CrossRef]
- Guan, J.; Zhou, Y.; Mao, F.; Lin, Y.; Shen, S.; Zhang, Y.; Sun, Q. MicroRNA320a suppresses tumor cell growth and invasion of human breast cancer by targeting insulinlike growth factor 1 receptor. Oncol. Rep. 2018, 40, 849–858. [Google Scholar] [CrossRef]
- Chai, C.; Wu, H.; Wang, B.; Eisenstat, D.D.; Leng, R.P. MicroRNA-498 promotes proliferation and migration by targeting the tumor suppressor PTEN in breast cancer cells. Carcinogenesis 2018, 39, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Cao, C.; Dai, Q.; Chen, J.; Tu, J. miR-202 acts as a potential tumor suppressor in breast cancer. Oncol. Lett. 2018, 16, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Yan, B.; Shen, Y. MiR-1301-3p inhibits human breast cancer cell proliferation by regulating cell cycle progression and apoptosis through directly targeting ICT1. Breast Cancer 2018, 25, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Fang, J.; Yu, Y.; Hou, L.K.; Chi, J.R.; Chen, A.X.; Zhao, Y.; Cao, X.C. miR-129-5p suppresses breast cancer proliferation by targeting CBX4. Neoplasma 2018, 65, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Yu, J.; Dai, Y.; Li, J.; Guo, M.; Song, J.; Zhou, X. Overexpression of miR-361-5p in triple-negative breast cancer (TNBC) inhibits migration and invasion by targeting RQCD1 and inhibiting the EGFR/PI3K/Akt pathway. Bosn. J. Basic Med. Sci. 2019, 19, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jiang, K.; Zhu, X.; Zhao, G.; Wu, H.; Deng, G.; Qiu, C. miR-433 inhibits breast cancer cell growth via the MAPK signaling pathway by targeting Rap1a. Int. J. Biol. Sci. 2018, 14, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Han, G.; Liu, Y.; Jiang, H.; He, Q. MiRNA-20a-5p promotes the growth of triple-negative breast cancer cells through targeting RUNX3. Biomed. Pharmacother. 2018, 103, 1482–1489. [Google Scholar] [CrossRef]
- Du, H.Y.; Liu, B. MiR-1271 as a tumor suppressor in breast cancer proliferation and progression via targeting SPIN1. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhang, J.; Li, J.; Shao, J.; Fang, L. MiR-130a-3p inhibits migration and invasion by regulating RAB5B in human breast cancer stem cell-like cells. Biochem. Biophys. Res. Commun. 2018, 501, 486–493. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Wang, J. MicroRNA-384 inhibits the progression of breast cancer by targeting ACVR1. Oncol. Rep. 2018, 39, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiu, H.; Tang, R.; Song, H.; Pan, H.; Feng, Z.; Chen, L. miR30a inhibits epithelialmesenchymal transition and metastasis in triplenegative breast cancer by targeting ROR1. Oncol. Rep. 2018, 39, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Zhang, X.; Wang, S.; Shi, B. MiR-449a suppresses cell migration and invasion by targeting PLAGL2 in breast cancer. Pathol. Res. Pract. 2018, 214, 790–795. [Google Scholar] [CrossRef]
- Luo, T.; Yan, Y.; He, Q.; Ma, X.; Wang, W. miR-328-5p inhibits MDA-MB-231 breast cancer cell proliferation by targeting RAGE. Oncol. Rep. 2018, 39, 2906–2914. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Guan, W.; Han, S.; Hong, D.K.; Kim, L.S.; Kim, H. MicroRNA-708-3p mediates metastasis and chemoresistance through inhibition of epithelial-to-mesenchymal transition in breast cancer. Cancer Sci. 2018, 109, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Cai, J.; Meng, F.; Sui, C.; Jiang, Y. MiR-144 suppresses proliferation, invasion, and migration of breast cancer cells through inhibiting CEP55. Cancer Biol. Ther. 2018, 19, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, J.; He, W.; Zhao, P.; Ye, C. MicroRNA-433 targets AKT3 and inhibits cell proliferation and viability in breast cancer. Oncol. Lett. 2018, 15, 3998–4004. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bi, L.; Wang, Q.; Wen, M.; Li, C.; Ren, Y.; Jiao, Q.; Mao, J.H.; Wang, C.; Wei, G.; et al. miR-1204 targets VDR to promotes epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene 2018, 37, 3426–3439. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, S.; Zhou, J.; Qian, Q. miR-424-5p regulates cell proliferation, migration and invasion by targeting doublecortin-like kinase 1 in basal-like breast cancer. Biomed. Pharmacother. 2018, 102, 147–152. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, H.; Liu, Y.; Zheng, S. Promotional effect of microRNA-194 on breast cancer cells via targeting F-box/WD repeat-containing protein 7. Oncol. Lett. 2018, 15, 4439–4444. [Google Scholar] [CrossRef]
- Yin, C.; Mou, Q.; Pan, X.; Zhang, G.; Li, H.; Sun, Y. MiR-577 suppresses epithelial-mesenchymal transition and metastasis of breast cancer by targeting Rab25. Thorac. Cancer 2018, 9, 472–479. [Google Scholar] [CrossRef]
- Yu, Y.; Luo, W.; Yang, Z.J.; Chi, J.R.; Li, Y.R.; Ding, Y.; Ge, J.; Wang, X.; Cao, X.C. miR-190 suppresses breast cancer metastasis by regulation of TGF-beta-induced epithelial-mesenchymal transition. Mol. Cancer 2018, 17, 70. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, Y.; Li, J.; Ma, D. MicroRNA-664 targets insulin receptor substrate 1 to suppress cell proliferation and invasion in breast cancer. Oncol. Res. 2019, 27, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Yang, R.; Zhao, S.; Chen, Y.; Hong, S.; Wang, K.; Wang, T.; Cheng, J.; Zhang, T.; Chen, D. Decreased miR-320 expression is associated with breast cancer progression, cell migration, and invasiveness via targeting Aquaporin 1. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Liu, X.; Bai, X.; Zhao, T.; Wang, M.; Xu, R.; Li, M.; Hu, Y.; Li, W.; Yang, L.; et al. MiR-519d suppresses breast cancer tumorigenesis and metastasis via targeting MMP3. Int. J. Biol. Sci. 2018, 14, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, H.; Gao, C.; Lv, Y.; Chen, X.; Xu, R.; Sun, M.; Liu, X.; Lu, X.; Pei, X.; et al. Tumor-promoting properties of miR-8084 in breast cancer through enhancing proliferation, suppressing apoptosis and inducing epithelial-mesenchymal transition. J. Transl. Med. 2018, 16, 38. [Google Scholar] [CrossRef]
- Cheng, X.; Chen, J.; Huang, Z. miR-372 promotes breast cancer cell proliferation by directly targeting LATS2. Exp. Ther. Med. 2018, 15, 2812–2817. [Google Scholar] [CrossRef]
- Yin, R.; Guo, L.; Gu, J.; Li, C.; Zhang, W. Over expressing miR-19b-1 suppress breast cancer growth by inhibiting tumor microenvironment induced angiogenesis. Int. J. Biochem. Cell. Biol. 2018, 97, 43–51. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Liu, B.; Han, J. MicroRNA-124-3p directly targets PDCD6 to inhibit metastasis in breast cancer. Oncol. Lett. 2018, 15, 984–990. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, M.; Huang, J.; Li, Y.; Wang, S.; Harrington, C.A.; Qian, D.Z.; Sun, X.X.; Dai, M.S. microRNA-130a suppresses breast cancer cell migration and invasion by targeting FOSL1 and upregulating ZO-1. J. Cell. Biochem. 2018, 119, 4945–4956. [Google Scholar] [CrossRef]
- Li, Y.; Liang, Y.; Sang, Y.; Song, X.; Zhang, H.; Liu, Y.; Jiang, L.; Yang, Q. MiR-770 suppresses the chemo-resistance and metastasis of triple negative breast cancer via direct targeting of STMN1. Cell Death Dis. 2018, 9, 14. [Google Scholar] [CrossRef]
- Chen, H.; Pan, H.; Qian, Y.; Zhou, W.; Liu, X. MiR-25-3p promotes the proliferation of triple negative breast cancer by targeting BTG2. Mol. Cancer 2018, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, J. miR-3188 regulates cell proliferation, apoptosis, and migration in breast cancer by targeting TUSC5 and regulating the p38 MAPK signaling pathway. Oncol. Res. 2018, 26, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome. Biol. 2007, 8, R214. [Google Scholar] [CrossRef]
- Hamam, R.; Hamam, D.; Alsaleh, K.A.; Kassem, M.; Zaher, W.; Alfayez, M.; Aldahmash, A.; Alajez, N.M. Circulating microRNAs in breast cancer: Novel diagnostic and prognostic biomarkers. Cell. Death. Dis. 2017, 8, e3045. [Google Scholar] [CrossRef] [PubMed]
- Fkih M’hamed, I.; Privat, M.; Trimeche, M.; Penault-Llorca, F.; Bignon, Y.J.; Kenani, A. miR-10b, miR-26a, miR-146a and miR-153 expression in triple negative vs. non triple negative breast cancer: Potential biomarkers. Pathol. Oncol. Res. 2017, 23, 815–827. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Helland, A.; Gromov, P.; Wielenga, V.T.; Talman, M.M.; Brunner, N.; Sandhu, V.; Borresen-Dale, A.L.; Gromova, I.; Haakensen, V.D. Profiling of microRNAs in tumor interstitial fluid of breast tumors - a novel resource to identify biomarkers for prognostic classification and detection of cancer. Mol. Oncol. 2017, 11, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gutierrez, A.D.; Catalan, O.M.; Vazquez-Romo, R.; Porras Reyes, F.I.; Alvarado-Miranda, A.; Lara Medina, F.; Bargallo-Rocha, J.E.; Orozco Moreno, L.T.; Cantu De Leon, D.; Herrera, L.A.; et al. miRNA profile obtained by nextgeneration sequencing in metastatic breast cancer patients is able to predict the response to systemic treatments. Int. J. Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Makar, S.; Swetha, R.; Gutti, G.; Singh, S.K. Estrogen signaling: An emanating therapeutic target for breast cancer treatment. Eur. J. Med. Chem. 2019, 177, 116–143. [Google Scholar] [CrossRef] [PubMed]
- Jameera Begam, A.; Jubie, S.; Nanjan, M.J. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorg. Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Chambliss, K.L.; Wu, Q.; Oltmann, S.; Konaniah, E.S.; Umetani, M.; Korach, K.S.; Thomas, G.D.; Mineo, C.; Yuhanna, I.S.; Kim, S.H.; et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J. Clin. Invest. 2010, 120, 2319–2330. [Google Scholar] [CrossRef]
- Saha Roy, S.; Vadlamudi, R.K. Role of estrogen receptor signaling in breast cancer metastasis. Int. J. Breast Cancer 2012, 2012, 654698. [Google Scholar] [CrossRef]
- Kulkoyluoglu-Cotul, E.; Arca, A.; Madak-Erdogan, Z. Crosstalk between estrogen signaling and breast cancer metabolism. Trends Endocrinol. Metab. 2019, 30, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.; Mattar, A.; Logullo, A.F.; Soares, F.A.; Gebrim, L.H. Estrogen receptor alpha/beta ratio and estrogen receptor beta as predictors of endocrine therapy responsiveness-a randomized neoadjuvant trial comparison between anastrozole and tamoxifen for the treatment of postmenopausal breast cancer. BMC Cancer 2013, 13, 425. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.P.; Chu, Y.W.; Wang, J. Analysis of hormone receptor status in primary and recurrent breast cancer via data mining pathology reports. Open Med. (Wars) 2019, 14, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef]
- Chi, D.; Singhal, H.; Li, L.; Xiao, T.; Liu, W.; Pun, M.; Jeselsohn, R.; He, H.; Lim, E.; Vadhi, R.; et al. Estrogen receptor signaling is reprogrammed during breast tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 11437–11443. [Google Scholar] [CrossRef]
- Lopez-Tarruella, S.; Schiff, R. The dynamics of estrogen receptor status in breast cancer: Re-shaping the paradigm. Clin. Cancer Res. 2007, 13, 6921–6925. [Google Scholar] [CrossRef]
- Jimenez-Garduno, A.M.; Mendoza-Rodriguez, M.G.; Urrutia-Cabrera, D.; Dominguez-Robles, M.C.; Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induced methylation of the estrogen receptor ERalpha gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef]
- Tsuboi, K.; Nagatomo, T.; Gohno, T.; Higuchi, T.; Sasaki, S.; Fujiki, N.; Kurosumi, M.; Takei, H.; Yamaguchi, Y.; Niwa, T.; et al. Single CpG site methylation controls estrogen receptor gene transcription and correlates with hormone therapy resistance. J. Steroid. Biochem. Mol. Biol. 2017, 171, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, C.; Jiang, H.; Liang, L.; Shi, W.; Zhang, Q.; Sun, P.; Xiang, R.; Wang, Y.; Yang, S. ZEB1 induces ER-alpha promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell. Death Dis. 2017, 8, e2732. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, Y.Y.; Meeran, S.M.; Tollefsbol, T.O. Synergistic epigenetic reactivation of estrogen receptor-alpha (ERalpha) by combined green tea polyphenol and histone deacetylase inhibitor in ERalpha-negative breast cancer cells. Mol. Cancer 2010, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic reactivation of estrogen receptor-alpha (ERalpha) by genistein enhances hormonal therapy sensitivity in ERalpha-negative breast cancer. Mol. Cancer 2013, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Park, S.J.; Lee, Y.S.; Kong, H.K.; Park, J.H. miRNAs involved in LY6K and estrogen receptor alpha contribute to tamoxifen-susceptibility in breast cancer. Oncotarget 2016, 7, 42261–42273. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.C.; Conger, A.K.; Yan, T.J.; Hoang, V.T.; Miller, D.F.; Buechlein, A.; Rusch, D.B.; Nephew, K.P.; Collins-Burow, B.M.; Burow, M.E. MicroRNA-335-5p and -3p synergize to inhibit estrogen receptor alpha expression and promote tamoxifen resistance. FEBS Lett. 2017, 591, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, N.; Liu, L.; Dong, H.; Wu, C. Correlation between microRNA-21, microRNA-206 and estrogen receptor, progesterone receptor, human epidermal growth factor receptor 2 in breast cancer. Clin. Biochem. 2019. [Google Scholar] [CrossRef]
- Xu, Y.; Chao, L.; Wang, J.; Sun, Y. miRNA-148a regulates the expression of the estrogen receptor through DNMT1-mediated DNA methylation in breast cancer cells. Oncol. Lett. 2017, 14, 4736–4740. [Google Scholar] [CrossRef]
- Ljepoja, B.; Garcia-Roman, J.; Sommer, A.K.; Wagner, E.; Roidl, A. MiRNA-27a sensitizes breast cancer cells to treatment with Selective Estrogen Receptor Modulators. Breast 2019, 43, 31–38. [Google Scholar] [CrossRef]
- Luengo-Gil, G.; Garcia-Martinez, E.; Chaves-Benito, A.; Conesa-Zamora, P.; Navarro-Manzano, E.; Gonzalez-Billalabeitia, E.; Garcia-Garre, E.; Martinez-Carrasco, A.; Vicente, V.; Ayala de la Pena, F. Clinical and biological impact of miR-18a expression in breast cancer after neoadjuvant chemotherapy. Cell. Oncol. (Dordr) 2019. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1-integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Pestana, A.; Vinagre, J.; Sobrinho-Simoes, M.; Soares, P. TERT biology and function in cancer: Beyond immortalisation. J. Mol. Endocrinol. 2017, 58, R129–R146. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.A.; Tollefsbol, T.O. Regulation of the telomerase reverse transcriptase subunit through epigenetic mechanisms. Front. Genet. 2016, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Leao, R.; Apolonio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Gollahon, L.; Kataoka, T.; Kuroi, K.; Yokoyama, T.; Gazdar, A.F.; Hiyama, K.; Piatyszek, M.A.; Shay, J.W. Telomerase activity in human breast tumors. J. Natl. Cancer Inst. 1996, 88, 116–122. [Google Scholar] [CrossRef]
- Kirkpatrick, K.L.; Clark, G.; Ghilchick, M.; Newbold, R.F.; Mokbel, K. hTERT mRNA expression correlates with telomerase activity in human breast cancer. Eur. J. Surg. Oncol. 2003, 29, 321–326. [Google Scholar] [CrossRef]
- Hannen, R.; Bartsch, J.W. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef]
- Bodvarsdottir, S.K.; Steinarsdottir, M.; Hilmarsdottir, H.; Jonasson, J.G.; Eyfjord, J.E. MYC amplification and TERT expression in breast tumor progression. Cancer Genet. Cytogenet. 2007, 176, 93–99. [Google Scholar] [CrossRef]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033. [Google Scholar] [CrossRef]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akincilar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Invest. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Xiong, W.; Liu, H.; Jia, C.; Zhang, H.; Cui, Z.; Zhang, Y.; Cui, J. Inhibition of telomerase activity by dominant-negative hTERT retards the growth of breast cancer cells. Breast Cancer 2016, 23, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Rubis, B.; Holysz, H.; Gladych, M.; Toton, E.; Paszel, A.; Lisiak, N.; Kaczmarek, M.; Hofmann, J.; Rybczynska, M. Telomerase downregulation induces proapoptotic genes expression and initializes breast cancer cells apoptosis followed by DNA fragmentation in a cell type dependent manner. Mol. Biol. Rep. 2013, 40, 4995–5004. [Google Scholar] [CrossRef]
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999, 59, 551–557. [Google Scholar] [PubMed]
- Shimoi, T.; Yoshida, M.; Kitamura, Y.; Yoshino, T.; Kawachi, A.; Shimomura, A.; Noguchi, E.; Yunokawa, M.; Yonemori, K.; Shimizu, C.; et al. TERT promoter hotspot mutations in breast cancer. Breast Cancer 2018, 25, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Canadian Epigenomes - CEEHRC. Available online: http://www.epigenomes.ca/data-release/ (accessed on 21 June 2019).
- Gay-Bellile, M.; Veronese, L.; Combes, P.; Eymard-Pierre, E.; Kwiatkowski, F.; Dauplat, M.M.; Cayre, A.; Privat, M.; Abrial, C.; Bignon, Y.J.; et al. TERT promoter status and gene copy number gains: Effect on TERT expression and association with prognosis in breast cancer. Oncotarget 2017, 8, 77540–77551. [Google Scholar] [CrossRef] [PubMed]
- Zinn, R.L.; Pruitt, K.; Eguchi, S.; Baylin, S.B.; Herman, J.G. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007, 67, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef]
- Qing, H.; Aono, J.; Findeisen, H.M.; Jones, K.L.; Heywood, E.B.; Bruemmer, D. differential regulation of telomerase reverse transcriptase promoter activation and protein degradation by histone deacetylase inhibition. J. Cell. Physiol. 2016, 231, 1276–1282. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Liu, Y.B.; Zhang, Y.; Shaw, J.; Valeriote, F.A.; Gautam, S.C. Inhibition of hTERT in pancreatic cancer cells by pristimerin involves suppression of epigenetic regulators of gene transcription. Oncol. Rep. 2017, 37, 1914–1920. [Google Scholar] [CrossRef]
- Zhang, D.; Xiao, Y.F.; Zhang, J.W.; Xie, R.; Hu, C.J.; Tang, B.; Wang, S.M.; Wu, Y.Y.; Hao, N.B.; Yang, S.M. miR-1182 attenuates gastric cancer proliferation and metastasis by targeting the open reading frame of hTERT. Cancer Lett. 2015, 360, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Wang, H.; Wang, A.H.; Zhang, L.Y.; Bai, J. MicroRNA-532 and microRNA-3064 inhibit cell proliferation and invasion by acting as direct regulators of human telomerase reverse transcriptase in ovarian cancer. PLoS ONE 2017, 12, e0173912. [Google Scholar] [CrossRef] [PubMed]
- Ohira, T.; Naohiro, S.; Nakayama, Y.; Osaki, M.; Okada, F.; Oshimura, M.; Kugoh, H. miR-19b regulates hTERT mRNA expression through targeting PITX1 mRNA in melanoma cells. Sci. Rep. 2015, 5, 8201. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Xiao, Y.F.; Tang, B.; Wu, Y.Y.; Hu, C.J.; Xie, R.; Yang, X.; Yu, S.T.; Dong, H.; Zhao, X.Y.; et al. hTERT mediates gastric cancer metastasis partially through the indirect targeting of ITGB1 by microRNA-29a. Sci. Rep. 2016, 6, 21955. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Y.H.; Diao, H.Y.; Ma, J.; Yao, Y.L. MiR-661 inhibits glioma cell proliferation, migration and invasion by targeting hTERT. Biochem. Biophys. Res. Commun. 2015, 468, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.J.; Chu, R.Q.; Ma, J.; Wang, Z.X.; Zhang, G.J.; Yang, X.F.; Song, Z.; Ma, Y.Y. MicroRNA138 regulates keratin 17 protein expression to affect HaCaT cell proliferation and apoptosis by targeting hTERT in psoriasis vulgaris. Biomed. Pharmacother. 2017, 85, 169–176. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, J.J.; Tao, H.; Jin, W.S. MicroRNA-21 controls hTERT via PTEN in human colorectal cancer cell proliferation. J. Physiol. Biochem. 2015, 71, 59–68. [Google Scholar] [CrossRef]
- Yan, T.; Ooi, W.F.; Qamra, A.; Cheung, A.; Ma, D.; Sundaram, G.M.; Xu, C.; Xing, M.; Poon, L.; Wang, J.; et al. HoxC5 and miR-615-3p target newly evolved genomic regions to repress hTERT and inhibit tumorigenesis. Nat. Commun. 2018, 9, 100. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic instability in cancer: Teetering on the limit of tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Kaboli, P.J.; Rahmat, A.; Ismail, P.; Ling, K.H. MicroRNA-based therapy and breast cancer: A comprehensive review of novel therapeutic strategies from diagnosis to treatment. Pharmacol. Res. 2015, 97, 104–121. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Home - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 8 July 2019).
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-mesenchymal transition: Epigenetic reprogramming driving cellular plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Qi, Y.; Wang, D.; Wang, D.; Jin, T.; Yang, L.; Wu, H.; Li, Y.; Zhao, J.; Du, F.; Song, M.; et al. HEDD: The human epigenetic drug database. Database (Oxford) 2016, 2016. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://www.drugbank.ca/ (accessed on 1 July 2019).
- Liu, X.R.; Zhou, L.H.; Hu, J.X.; Liu, L.M.; Wan, H.P.; Zhang, X.Q. UNC0638, a G9a inhibitor, suppresses epithelialmesenchymal transitionmediated cellular migration and invasion in triple negative breast cancer. Mol. Med. Rep. 2018, 17, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.; Lenz, D.; Fickel, J. Environmental change-dependent inherited epigenetic response. Genes 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, C.; Issa, J.P. Diet, nutrition, and cancer epigenetics. Annu. Rev. Nutr. 2016, 36, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.M.; Tollefsbol, T.O. Epigenetic diet: Impact on the epigenome and cancer. Epigenomics 2011, 3, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Ahmed, A.; Tollefsbol, T.O. Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin. Epigenetics 2010, 1, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.; Srinivasan, A. Epigenetic gene regulation by dietary compounds in cancer prevention. Adv. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Bullo, M.M.H.; Ahmed, Z.; Imtiaz, A.; Yaqoob, E.; Jadoon, M.; Ahmed, H.; Afreen, A.; Yaqoob, S. Nutrigenomics: Epigenetics and cancer prevention: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Buckhaults, P.; Li, S.; Tollefsbol, T. Temporal efficacy of a sulforaphane-based broccoli sprout diet in prevention of breast cancer through modulation of epigenetic mechanisms. Cancer Prev. Res. (Phila) 2018, 11, 451–464. [Google Scholar] [CrossRef]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T.O. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell. Res. 2018, 368, 67–74. [Google Scholar] [CrossRef]
- Boyanapalli, S.S.; Kong, A.T. “Curcumin, the king of spices”: Epigenetic regulatory mechanisms in the prevention of cancer, neurological, and inflammatory diseases. Curr. Pharmacol. Rep. 2015, 1, 129–139. [Google Scholar] [CrossRef]
- Royston, K.J.; Udayakumar, N.; Lewis, K.; Tollefsbol, T.O. A novel combination of withaferin a and sulforaphane inhibits epigenetic machinery, cellular viability and induces apoptosis of breast cancer cells. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Szarc Vel Szic, K.; Declerck, K.; Crans, R.A.J.; Diddens, J.; Scherf, D.B.; Gerhauser, C.; Vanden Berghe, W. Epigenetic silencing of triple negative breast cancer hallmarks by Withaferin, A. Oncotarget 2017, 8, 40434–40453. [Google Scholar] [CrossRef] [PubMed]
- Hargraves, K.G.; He, L.; Firestone, G.L. Phytochemical regulation of the tumor suppressive microRNA, miR-34a, by p53-dependent and independent responses in human breast cancer cells. Mol. Carcinog. 2016, 55, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Parbin, S.; Shilpi, A.; Kar, S.; Pradhan, N.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Insights into the molecular interactions of thymoquinone with histone deacetylase: Evaluation of the therapeutic intervention potential against breast cancer. Mol. Biosyst. 2016, 12, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Chiu, C.F.; Chu, P.C.; Lin, W.Y.; Chiu, S.J.; Weng, J.R. A triterpenoid from wild bitter gourd inhibits breast cancer cells. Sci. Rep. 2016, 6, 22419. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.; Han, B.; Li, Z.; Wang, B.; Jiang, P.; Zhang, J.; Ma, W.; Zhou, D.; Li, X.; et al. Anti-breast-cancer activity exerted by beta-sitosterol-d-glucoside from sweet potato via upregulation of microRNA-10a and via the PI3K-Akt signaling pathway. J. Agric. Food Chem. 2018, 66, 9704–9718. [Google Scholar] [CrossRef]
- Gao, Y.; Tollefsbol, T.O. Combinational proanthocyanidins and resveratrol synergistically inhibit human breast cancer cells and impact epigenetic (-) mediating machinery. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Deregowska, A.; Wnuk, M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics 2017, 7, 3461–3477. [Google Scholar] [CrossRef]
- Ross, S.L.; Morris, T.; Campone, M.; Cortes, J.; Duhoux, F.P.; Howell, S.J. Abstract OT1-03-03: STEM: SFX-01 in the treatment and evaluation of metastatic breast cancer. Cancer Res. 2019, 79 (Suppl. 4), OT1-03-03. [Google Scholar] [CrossRef]
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cancer induction and suppression with transcriptional control and epigenome editing technologies. J. Hum. Genet. 2018, 63, 187–194. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Brocken, D.J.W.; Tark-Dame, M.; Dame, R.T. dCas9: A versatile tool for epigenome editing. Curr. Issues Mol. Biol. 2018, 26, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, I.; Ramachandran, S.; Srivastava, S.K. CRISPR-Cas9: A multifaceted therapeutic strategy for cancer treatment. Semin. Cell. Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Epigenome editing made easy. Nat. Biotechnol. 2015, 33, 606–607. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S.; Yuan, X.; Rots, M.G.; Strahl, B.D.; Blancafort, P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012, 7, 350–360. [Google Scholar] [CrossRef]
- Stolzenburg, S.; Rots, M.G.; Beltran, A.S.; Rivenbark, A.G.; Yuan, X.; Qian, H.; Strahl, B.D.; Blancafort, P. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res. 2012, 40, 6725–6740. [Google Scholar] [CrossRef]
- Falahi, F.; Huisman, C.; Kazemier, H.G.; van der Vlies, P.; Kok, K.; Hospers, G.A.; Rots, M.G. Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol. Cancer Res. 2013, 11, 1029–1039. [Google Scholar] [CrossRef]
- Garcia-Bloj, B.; Moses, C.; Sgro, A.; Plani-Lam, J.; Arooj, M.; Duffy, C.; Thiruvengadam, S.; Sorolla, A.; Rashwan, R.; Mancera, R.L.; et al. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget 2016, 7, 60535–60554. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Stepper, P.; Gomm, J.J.; Hoa, L.; Morgan, A.; Allen, M.D.; Jones, J.L.; Gribben, J.G.; Jurkowski, T.P.; Ficz, G. Hit-and-run epigenetic editing prevents senescence entry in primary breast cells from healthy donors. Nat. Commun. 2017, 8, 1450. [Google Scholar] [CrossRef]
- Mabe, N.W.; Fox, D.B.; Lupo, R.; Decker, A.E.; Phelps, S.N.; Thompson, J.W.; Alvarez, J.V. Epigenetic silencing of tumor suppressor Par-4 promotes chemoresistance in recurrent breast cancer. J. Clin Invest. 2018, 128, 4413–4428. [Google Scholar] [CrossRef] [PubMed]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN tumor suppressor expression with the CRISPR/dCas9 system. Mol. Ther. Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Asiaf, A.; Ahmad, S.T.; Arjumand, W.; Zargar, M.A. MicroRNAs in breast cancer: Diagnostic and therapeutic potential. Methods Mol. Biol. 2018, 1699, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Sarkar, B.K.; Lee, S.S. The novel strategies for next-generation cancer treatment: miRNA combined with chemotherapeutic agents for the treatment of cancer. Oncotarget 2018, 9, 10164–10174. [Google Scholar] [CrossRef] [PubMed]
- Haghi, M.; Taha, M.F.; Javeri, A. Suppressive effect of exogenous miR-16 and miR-34a on tumorigenesis of breast cancer cells. J. Cell. Biochem. 2019, 120, 13342–13353. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Xu, L.; Wang, R.; Lu, X.; Sang, J. miR-381 overcomes cisplatin resistance in breast cancer by targeting MDR1. Cell. Biol. Int. 2019, 43, 12–21. [Google Scholar] [CrossRef]
- Ahmad, A.; Zhang, W.; Wu, M.; Tan, S.; Zhu, T. Tumor-suppressive miRNA-135a inhibits breast cancer cell proliferation by targeting ELK1 and ELK3 oncogenes. Genes Genomics 2018, 40, 243–251. [Google Scholar] [CrossRef] [PubMed]
- .Wang, M.J.; Zhang, H.; Li, J.; Zhao, H.D. microRNA-98 inhibits the proliferation, invasion, migration and promotes apoptosis of breast cancer cells by binding to HMGA2. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Wang, H.; Zhao, J.; Xu, C.; Du, Y.; Luo, X.; Zheng, F.; Liu, R.; Zhang, H.; et al. miRNA-135a promotes breast cancer cell migration and invasion by targeting HOXA10. BMC Cancer 2012, 12, 111. [Google Scholar] [CrossRef]
- Shen, F.; Cai, W.S.; Feng, Z.; Li, J.L.; Chen, J.W.; Cao, J.; Xu, B. MiR-492 contributes to cell proliferation and cell cycle of human breast cancer cells by suppressing SOX7 expression. Tumour Biol. 2015, 36, 1913–1921. [Google Scholar] [CrossRef]
- He, S.; Zhang, G.; Dong, H.; Ma, M.; Sun, Q. miR-203 facilitates tumor growth and metastasis by targeting fibroblast growth factor 2 in breast cancer. Onco. Targets Ther. 2016, 9, 6203–6210. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Jiang, W.; Jing, Z.; Mu, X.; Xiong, Z. miR-937 regulates the proliferation and apoptosis via targeting APAF1 in breast cancer. Onco. Targets Ther. 2019, 12, 5687–5699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Guo, H.; Tang, J. Long non-coding RNA TFAP2A-AS1 inhibits cell proliferation and invasion in breast cancer via miR-933/SMAD2. Med. Sci. Monit. 2019, 25, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wang, Y.; Wang, X.; Zhou, D.; Wang, X.; Zhou, M.; He, Z. Effect of the lncRNA GAS5-MiR-23a-ATG3 axis in regulating autophagy in patients with breast cancer. Cell. Physiol. Biochem. 2018, 48, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.L.; Zhang, T.T.; Zhou, N.; Wu, C.Y.; Lin, M.H.; Liu, Y.J. MiRNA-10b sponge: An anti-breast cancer study in vitro. Oncol. Rep. 2016, 35, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Shao, C.C.; Wang, X.J.; Zhao, X.; Chen, J.Q.; Ouyang, Y.X.; Feng, J.; Zhang, F.; Huang, W.H.; Ying, Q.; et al. circTADA2As suppress breast cancer progression and metastasis via targeting miR-203a-3p/SOCS3 axis. Cell. Death Dis. 2019, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Tian, H.; Guo, Y.; Li, Y.; Guo, Z.; Zhu, X.; Chen, X. miRNA oligonucleotide and sponge for miRNA-21 inhibition mediated by PEI-PLL in breast cancer therapy. Acta Biomater. 2015, 25, 184–193. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA Name | Target Gene/Signaling Pathway | Effect on Breast Cancer Process | References | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Proliferation and/or Growth | Apoptosis | Migration | Invasion | EMT | Angiogenesis | Metastasis | |||
| miR-3614 | TRIM25 | *↓ | [72] | ||||||
| miR-454-3p | Wnt/β-catenin signaling | ↑ | [73] | ||||||
| miR-216a | Wnt/β-catenin signaling | ↓ | ↓ | [74] | |||||
| miR-4458 | SOCS1 | ↓ | ↑ | [75] | |||||
| miR-140-3p | TRIM28 | ↓ | ↓ | [76] | |||||
| miR-483 | SOX3 | ↓ | [77] | ||||||
| miR-214 | α1-AT; PI3K/Akt/mTOR signaling | ↑ | ↑ | [78] | |||||
| miR-216a-5p | PAK2 | ↓ | ↓ | [79] | |||||
| miR-4458 | CPSF4 | ↓ | ↓ | [80] | |||||
| miR-653 | ZEB2 signaling | ↓ | ↑ | [81] | |||||
| miR-190 | AKT-ERK signaling | ↓ | ↓ | [82] | |||||
| miR-370 | WNK2 | ↑ | [83] | ||||||
| miR-193a-3p | GRB7 | ↓ | ↓ | ↓ | [84] | ||||
| miR-591 | TCF4; Hippo-YAP/TAZ signaling | ↑ | ↑ | [85] | |||||
| miR-153 | RUNX2 | ↓ | ↓ | ↓ | ↓ | [86] | |||
| miR-4513 | TRIM3 | ↑ | ↑ | ↑ | [87] | ||||
| miR-216a | PKCα | ↑ | [88] | ||||||
| miR-374c-5p | DEPDC1 | ↓ | ↑ | ↓ | ↓ | [89] | |||
| miR-890 | CD147 | ↓ | ↑ | ↓ | [90] | ||||
| miR-142-3p | ESR1 | ↓ | ↑ | [91] | |||||
| miR-449b-5p | Wnt/β-catenin signaling | ↓ | ↓ | [92] | |||||
| miR-135 | Wnt/β-catenin signaling | ↓ | ↓ | ↓ | ↓ | [93] | |||
| miR-1287-5p | PIK3CB | ↓ | [94] | ||||||
| miR-30a | Snail | ↓ | ↓ | [95] | |||||
| miR-135b | APC | ↑ | ↑ | [96] | |||||
| miR-124 | ZEB2 signaling | ↓ | ↓ | [97] | |||||
| miR-4282 | Myc | ↓ | ↓ | ↓ | [98] | ||||
| miR-3196 | ERBB3 | ↑ | [99] | ||||||
| miR-1179 | Notch signaling | ↓ | [100] | ||||||
| miR-590-3P | sirtuin-1 | ↑ | [101] | ||||||
| miR-645 | uPA | ↓ | [102] | ||||||
| miR-99a-5p | CDC25A | ↓ | ↑ | ↓ | [103] | ||||
| miR-196b-5p | COL1A1 | ↑ | ↑ | [104] | |||||
| miR-421 | PDCD4 | ↓ | ↑ | ↓ | ↓ | [105] | |||
| miR-508-3p | ZEB1 | ↓ | ↓ | [106] | |||||
| miR-3178 | Notch1 | ↓ | ↓ | [107] | |||||
| miR-340-5p | LGR5; Wnt/β-catenin signaling | ↓ | [108] | ||||||
| miR-511 | SOX9; PI3K/Akt pathway | ↓ | ↑ | ↓ | [109] | ||||
| miR-424 | CDK1 | ↓ | [110] | ||||||
| miR-301b | CYLD | ↑ | ↑ | [111] | |||||
| miR-194-5p | Wnt/β-catenin signaling | ↓ | ↓ | ↓ | [112] | ||||
| miR-199b-5p | DDR1 | ↓ | ↓ | ↓ | [113] | ||||
| miR-1247-5p | DVL1/Wnt/β-catenin signaling | ↓ | [114] | ||||||
| miR-628 | SOS1 | ↓ | ↓ | [115] | |||||
| miR-590-5p | Wnt-β-catenin signaling | ↓ | ↓ | ↓ | [116] | ||||
| miR-483-3p | cyclin E1 | ↓ | [117] | ||||||
| miR-125b-5p | KIAA1522 | ↓ | ↓ | ↓ | [118] | ||||
| miR-1254 | RASSF9 | ↑ | ↓ | [119] | |||||
| miR-590-3p | ATF3 | ↓ | ↑ | [120] | |||||
| miR-125a-5p | BAP1 | ↓ | ↑ | [121] | |||||
| miR-1284 | ZIC2 | ↓ | ↓ | [122] | |||||
| miR-92b | EZH2 | ↓ | ↓ | [123] | |||||
| miR-140-5p | Wnt1 | ↓ | [124] | ||||||
| miR-185-5p | RAGE | ↓ | [125] | ||||||
| miR-320a | IGF-1R | ↓ | ↓ | [126] | |||||
| miR-498 | PTEN | ↑ | ↑ | [127] | |||||
| miR-202 | KRAS | ↓ | ↓ | ↓ | [128] | ||||
| miR-1301-3p | ICT1 | ↓ | [129] | ||||||
| miR-129-5p | CBX4 | ↓ | [130] | ||||||
| miR-361-5p | RQCD1; EGFR/PI3K/Akt pathway | ↓ | ↓ | [131] | |||||
| miR-433 | Rap1a; MAPK signaling | ↓ | [132] | ||||||
| miR-20a-5p | RUNX3 | ↑ | [133] | ||||||
| miR-1271 | SPIN1 | ↓ | [134] | ||||||
| miR-130a-3p | RAB5B | ↓ | ↓ | [135] | |||||
| miR-384 | ACVR1 | ↓ | ↓ | [136] | |||||
| miR-30a | ROR1 | ↓ | ↓ | [137] | |||||
| miR-449a | PLAGL2 | ↓ | ↓ | [138] | |||||
| miR-328-5p | RAGE | ↓ | [139] | ||||||
| miR-708-3p | ZEB1, CDH2 and vimentin | ↓ | [140] | ||||||
| miR-144 | CEP55 | ↓ | ↓ | ↓ | [141] | ||||
| miR-433 | AKT3 | ↓ | [142] | ||||||
| miR-1204 | VDR | ↓ | ↑ | ↑ | [143] | ||||
| miR-424-5p | DCLK1 | ↓ | ↓ | ↓ | [144] | ||||
| miR-194 | Fbxw-7 | ↑ | [145] | ||||||
| miR-577 | Rab25 | ↓ | ↓ | [146] | |||||
| miR-190 | SMAD2 | ↓ | [147] | ||||||
| miR-664 | IRS1 | ↓ | ↓ | [148] | |||||
| miR-320 | AQP1 | ↓ | ↓ | ↓ | [149] | ||||
| miR-519d | MMP3 | ↓ | ↓ | ↓ | [150] | ||||
| miR-8084 | ING2 | ↑ | ↓ | ↑ | [151] | ||||
| miR-372 | LATS2 | ↑ | [152] | ||||||
| miR-19b-1 | VEGF | ↓ | ↓ | [153] | |||||
| miR-124-3p | PDCD6 | ↓ | [154] | ||||||
| miR-130a | FOSL1 | ↓ | ↓ | [155] | |||||
| miR-770 | STMN1 | ↓ | ↓ | [156] | |||||
| miR-25-3p | BTG2 | ↑ | [157] | ||||||
| miR-3188 | TUSC5; p38-MAPK signaling | ↑ | ↓ | ↑ | [158] | ||||
| Breast Tumor Subtypes | MiRNA Expression Profile | |
|---|---|---|
| Upregulated miRNAs | Downregulated miRNAs | |
| Luminal A | miR-126, miR-136, miR-100, miR-99a, miR-145, miR-10a, miR-199a, miR-199b, miR-130a, miR-30a-3p, miR-30a-5p, miR-224, miR-214, let-7a*, let-7b*, let7c, let-7f, miR-342* | miR-150, miR-142-3p, miR-142-5p, miR-106a, miR-106b, miR-18a, miR-93, miR-25, miR-187, miR-135b |
| Luminal B | miR-106b, miR-93, miR-25, miR-10a, miR-30a-3p, miR-30a-5p, miR-224, let-7f | miR-150*, miR-142-3p, miR-142-5p*, miR-148a, miR-18a, miR-155*, miR-187, miR-135b, miR-126, miR-136, miR-100, miR-99a, miR-145, miR-130a |
| Basal-like | miR-150, miR-142-3p, miR-142-5p, miR-148a, miR-106a*, miR-106b, miR-18a*, miR-93, miR-155*, miR-25, miR-187, miR-135b* | miR-126, miR-136, miR-100, miR-99a, miR-145, miR-10a, miR-199a, miR-199b, miR-130a, miR-30a-3p, miR-30a-5p, miR-224, miR-214, let-7a, let-7b, let7c, let-7f, miR-342 |
| HER2+ | miR-150, miR-142-3p, miR-142-5p, miR-148a, miR-106b, miR-25, miR-187* | miR-106a, miR-18a, miR-93, miR-155, miR-135b, miR-126, miR-136, miR-100, miR-99a, miR-145, miR-10a, miR-199b, miR-130a*, miR-30a-3p*, miR-30a-5p*, miR-224*, let-7a, let-7b, let7c, let-7f, miR-342 |
| Normal-like | miR-135b, miR-126*, miR-136, miR-100, miR-99a, miR-145, miR-10a, miR-199a, miR-199b, miR-130a*, miR-30a-3p, miR-214, let7c | miR-142-3p, miR-148a, miR-106a, miR-106b*, miR-93*, miR-25, let-7f |
| MicroRNA Name | Mature Sequence of miRNA | Target Gene | Score Class | References | |
|---|---|---|---|---|---|
| ESR1 | ESR2 | ||||
| hsa-let-7a-5p | 6-UGAGGUAGUAGGUUGUAUAGUU-27 | Y | Y | High | [180,181] |
| hsa-let-7b-5p | 6-UGAGGUAGUAGGUUGUGUGGUU-27 | Y | Y | High | |
| hsa-let-7c-5p | 11-UGAGGUAGUAGGUUGUAUGGUU-32 | Y | Y | High | |
| hsa-let-7d-5p | 8-AGAGGUAGUAGGUUGCAUAGUU-29 | Y | Y | High | |
| hsa-let-7e-5p | 8-UGAGGUAGGAGGUUGUAUAGUU-29 | Y | Y | High | |
| hsa-let-7f-5p | 63-CUAUACAAUCUAUUGCCUUCCC-84 | Y | Y | High | |
| hsa-let-7g-5p | 5-UGAGGUAGUAGUUUGUACAGUU-26 | Y | Y | High | |
| hsa-let-7i-5p | 6-UGAGGUAGUAGUUUGUGCUGUU-27 | Y | Y | High | |
| hsa-miR-106a-5p | 13-AAAAGUGCUUACAGUGCAGGUAG-35 | Y | Y | High | |
| hsa-miR-122-5p | 15-UGGAGUGUGACAAUGGUGUUUG-36 | Y | Y | High | |
| hsa-miR-124-3p | 14-CGUGUUCACAGCGGACCUUGAU-35 | Y | Y | High | |
| hsa-miR-129-5p | 5-CUUUUUGCGGUCUGGGCUUGC-25 | Y | Y | High | |
| hsa-miR-140-5p | 23-CAGUGGUUUUACCCUAUGGUAG-44 | Y | Y | High | |
| hsa-miR-145-5p | 16-GUCCAGUUUUCCCAGGAAUCCCU-38 | Y | Y | High | |
| hsa-miR-15a-5p | 14-UAGCAGCACAUAAUGGUUUGUG-35 | Y | Y | High | |
| hsa-miR-15b-5p | 20-UAGCAGCACAUCAUGGUUUACA-41 | Y | Y | High | |
| hsa-miR-16-5p | 14-UAGCAGCACGUAAAUAUUGGCG-35 | Y | Y | High | |
| hsa-miR-17-5p | 14-CAAAGUGCUUACAGUGCAGGUAG-36 | Y | Y | High | |
| hsa-miR-195-5p | 15-UAGCAGCACAGAAAUAUUGGC-35 | Y | Y | High | |
| hsa-miR-196a-5p | 7-UAGGUAGUUUCAUGUUGUUGGG-28 | Y | Y | High | |
| hsa-miR-196b-5p | 15-UAGGUAGUUUCCUGUUGUUGGG-36 | Y | Y | High | |
| hsa-miR-204-5p | 33-UUCCCUUUGUCAUCCUAUGCCU-54 | Y | Y | High | |
| hsa-miR-205-5p | 34-UCCUUCAUUCCACCGGAGUCUG-55 | Y | Y | High | |
| hsa-miR-20a-5p | 8-UAAAGUGCUUAUAGUGCAGGUAG-30 | Y | Y | High | |
| hsa-miR-20b-5p | 6-CAAAGUGCUCAUAGUGCAGGUAG-28 | Y | Y | High | |
| hsa-miR-21-5p | 8-UAGCUUAUCAGACUGAUGUUGA-29 | Y | Y | High | |
| hsa-miR-211-5p | 26-UUCCCUUUGUCAUCCUUCGCCU-47 | Y | Y | High | |
| hsa-miR-214-3p | 30-UGCCUGUCUACACUUGCUGUGC-51 | Y | Y | High | |
| hsa-miR-24-3p | 7-UGCCUACUGAGCUGAUAUCAGU-28 | Y | Y | High | |
| hsa-miR-25-3p | 14-AGGCGGAGACUUGGGCAAUUG-34 | Y | Y | High | |
| hsa-miR-32-5p | 6-UAUUGCACAUUACUAAGUUGCA-27 | Y | Y | High | |
| hsa-miR-330-3p | 8-UCUCUGGGCCUGUGUCUUAGGC-39 | Y | Y | High | |
| hsa-miR-338-3p | 6-AACAAUAUCCUGGUGCUGAGUG-27 | Y | Y | High | |
| hsa-miR-3619-5p | 16-UCAGCAGGCAGGCUGGUGCAGC-37 | Y | Y | High | |
| hsa-miR-363-3p | 7-CGGGUGGAUCACGAUGCAAUUU-28 | Y | Y | High | |
| hsa-miR-367-3p | 6-ACUGUUGCUAAUAUGCAACUCU-27 | Y | Y | High | |
| hsa-miR-424-5p | 11-CAGCAGCAAUUCAUGUUUUGAA-32 | Y | Y | High | |
| hsa-miR-497-5p | 24-CAGCAGCACACUGUGGUUUGU-44 | Y | Y | High | |
| hsa-miR-507 | 56-UUUUGCACCUUUUGGAGUGAA-76 | Y | Y | High | |
| hsa-miR-548l | 15-AAAAGUAUUUGCGGGUUUUGUC-36 | Y | Y | High | |
| hsa-miR-573 | 16-CUGAAGUGAUGUGUAACUGAUCAG-39 | Y | Y | High | |
| hsa-miR-583 | 16-CAAAGAGGAAGGUCCCAUUAC-36 | Y | Y | High | |
| hsa-miR-590-5p | 16-GAGCUUAUUCAUAAAAGUGCAG-37 | Y | Y | High | |
| hsa-miR-7-5p | 24-UGGAAGACUAGUGAUUUUGUUGUU-47 | Y | Y | High | |
| hsa-miR-761 | 7-GCAGCAGGGUGAAACUGACACA-28 | Y | Y | High | |
| hsa-miR-766-3p | 29-AGGAGGAAUUGGUGCUGGUCUU-50 | Y | Y | High | |
| hsa-miR-92a-3p | 11- AGGUUGGGAUCGGUUGCAAUGCU-33 | Y | Y | High | |
| hsa-miR-93-5p | 11-CAAAGUGCUGUUCGUGCAGGUAG-33 | Y | Y | High | |
| hsa-miR-942-5p | 13-UCUUCUCUGUUUUGGCCAUGUG-34 | Y | Y | High | |
| hsa-miR-98-5p | 22-UGAGGUAGUAAGUUGUAUUGUU-43 | Y | Y | High | |
| Drug Category | Drug Name | References |
|---|---|---|
| DNA methyltransferase inhibitors | 5-Aza-2′-deoxycytidine, 5-Azacytidine, 5-Fluoro-2-Deoxycytidine | [228,229] |
| Histidine methyltransferase inhibitors | Curcumin | [228] |
| Histone deacetylase inhibitors | Belinostat, Entinostat, Panobinostat, Suberoylanilide hydroxamic acid (SAHA), Sodium butyrate, Vorinostat, Valproic acid, CUDC-101 | [228,229] |
| Histone methyltransferase inhibitors | EPZ004777, UNC0638 | [228,230] |
| Name | Effect on Breast Cancer Hallmarks | Target Gene/Signaling Pathway | References |
|---|---|---|---|
| miRNA-216a mimics | ↓Cell proliferation ↓Cell migration ↑Apoptosis | Wnt/β-catenin signaling | [74] |
| miRNA-449b-5p mimics | ↓ Growth ↓ Invasion | CREPT; Wnt/β-catenin/TCF-4 signaling | [92] |
| miRNA-301b mimics | ↓ Cell proliferation ↑ Apoptosis | CYLD | [111] |
| miRNA-1271 mimics | ↓ Cell proliferation ↓ Invasion ↓ Migration abilities | SPIN1 | [134] |
| miRNA-16 mimics | ↑ Apoptosis ↑ Cell-cycle arrest ↓ Invasion and migration | TGFBR2, SMAD2, SMAD3; TGF-β signaling | [264] |
| miRNA-34a mimics | ↑ Apoptosis ↑ Cell-cycle arrest ↓ Invasion and migration | TGFBR2, SMAD2, SMAD3; TGF-β signaling | [264] |
| miRNA-381 mimics | ↓ Cisplatin resistance | MDR1 | [265] |
| miRNA-135a mimics | ↓ Proliferation ↓ Clongenicity | ELK1, ELK3 | [266] |
| miRNA-27a mimics | ↓ Tamoxifen resistance | ERα | [185] |
| miRNA-98 mimics | ↓ Proliferation ↓ Invasion ↓ Migration ↑ Apoptosis | HMGA2 | [267] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019, 8, 1214. https://doi.org/10.3390/cells8101214
Rahman MM, Brane AC, Tollefsbol TO. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells. 2019; 8(10):1214. https://doi.org/10.3390/cells8101214
Chicago/Turabian StyleRahman, Mohammad Mijanur, Andrew C. Brane, and Trygve O. Tollefsbol. 2019. "MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer" Cells 8, no. 10: 1214. https://doi.org/10.3390/cells8101214
APA StyleRahman, M. M., Brane, A. C., & Tollefsbol, T. O. (2019). MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells, 8(10), 1214. https://doi.org/10.3390/cells8101214