The Role of Immune Checkpoint Receptors in Regulating Immune Reactivity in Lupus

 ,
, 
 ,
,
Abstract
1. Introduction
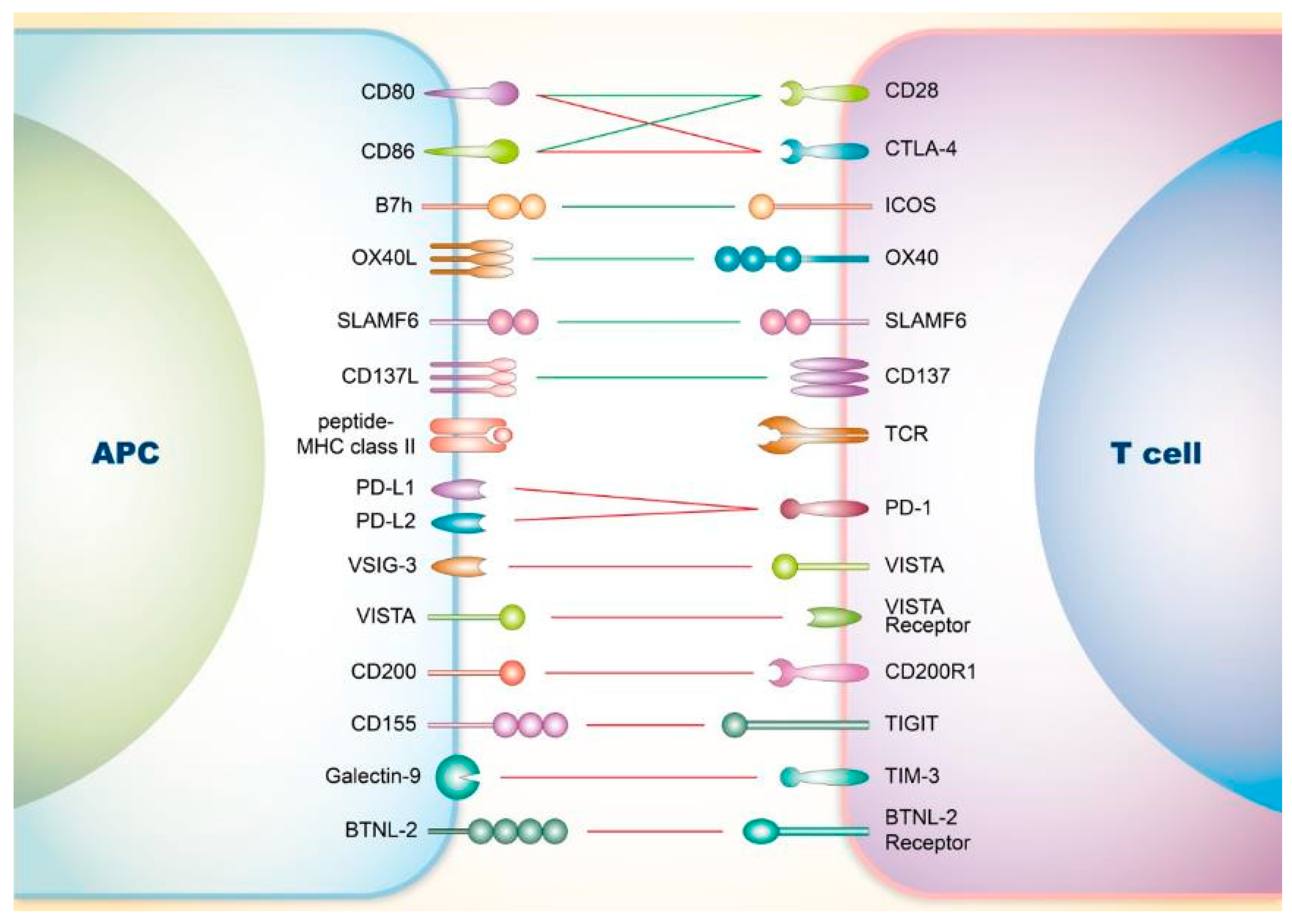
2. Co-Signaling Axes in T Cells Relating to SLE
2.1. Involvement of Co-Stimulatory Receptors on T Cells in SLE
2.1.1. CD28
2.1.2. Inducible Co-Stimulator (ICOS)
2.1.3. OX40
2.1.4. Signaling Lymphocyte Activation Molecule Family (SLAMF)
2.1.5. CD137
2.2. Involvement of Co-Inhibitory Receptors on T Cells in SLE
2.2.1. CTLA-4
2.2.2. PD-1
2.2.3. V-Domain Ig Suppressor of T Cell Activation (VISTA)
2.2.4. CD200
2.2.5. T-Cell Immunoreceptor with Ig and ITIM Domains (TIGIT)
2.2.6. T-Cell Immunoglobulin and Mucin-Domain Containing-3 (TIM-3)
2.2.7. Others
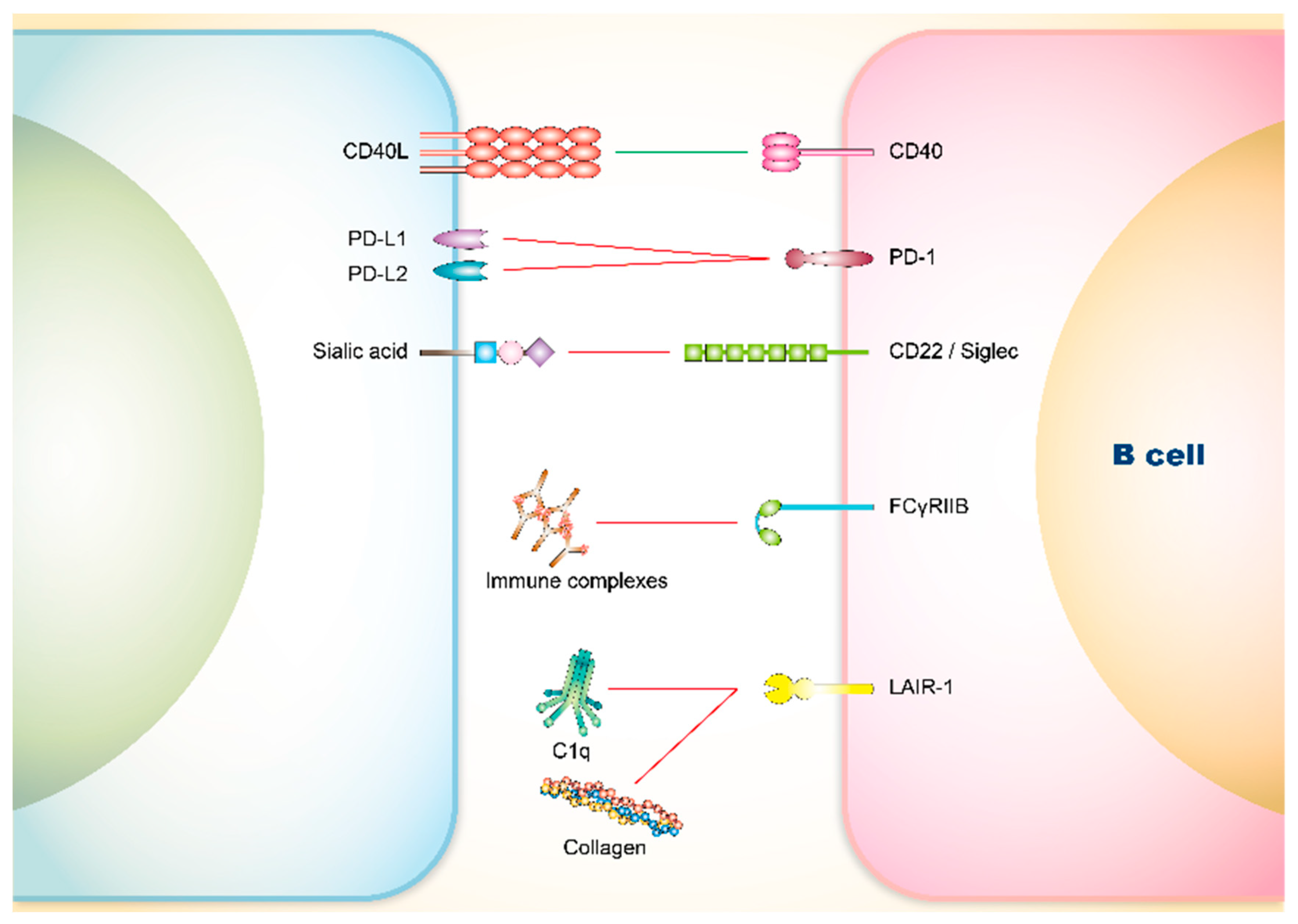
3. Co-Signaling Axes in B Cells Relating to SLE
3.1. Involvement of Co-Stimulatory Receptors on B Cells in SLE
CD40
3.2. Involvement of Co-Inhibitory Receptors on B Cells in SLE
3.2.1. PD-1
3.2.2. CD22 and Siglec
3.2.3. FCγRIIB
3.2.4. Leukocyte Associated Immunoglobulin-Like Receptor (LAIR)-1
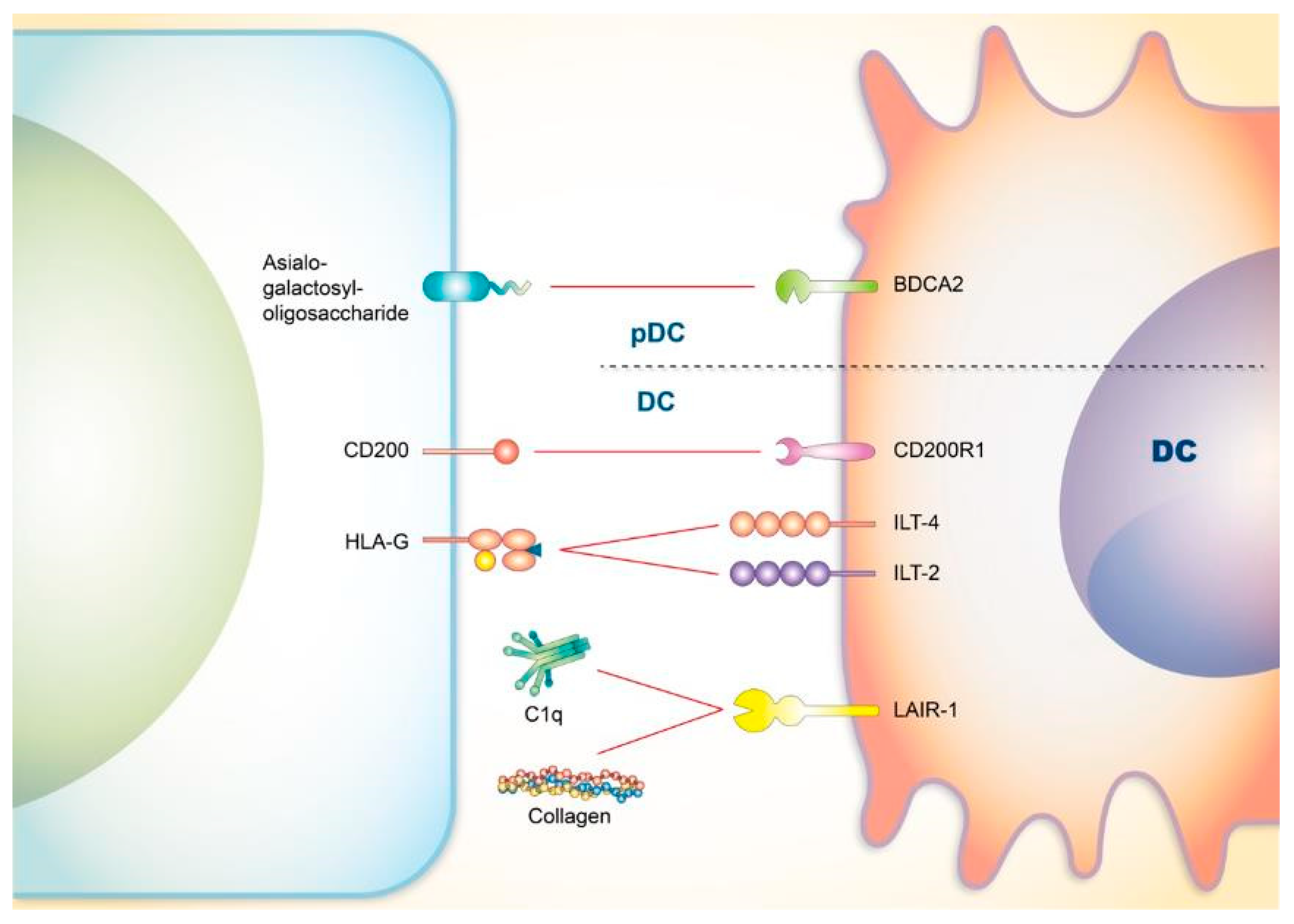
4. Co-Signaling Axes in Dendritic Cells (DCs) Relating to SLE
4.1. CD200
4.2. Blood-Derived Dendritic Cell Antigen 2 (BDCA2)
4.3. Immunoglobulin-Like Transcript 4 (ILT4) and ILT2
4.4. LAIR-1
5. Co-Signaling Axes in Neutrophils Relating to SLE
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Vignali, D.A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.K.; Kalra, B.S. Belimumab: Targeted therapy for lupus. Int. J. Rheum. Dis. 2013, 16, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Khamashta, M. The efficacy of novel B cell biologics as the future of SLE treatment: A review. Autoimmun. Rev. 2014, 13, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Del Prete, A.; Bosisio, D. Dendritic cell recruitment and activation in autoimmunity. J. Autoimmun. 2017, 85, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Vassilopoulos, D.; Boletis, J.; Boumpas, D.T. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): Implications for the pathogenesis and treatment. Autoimmun. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vlist, M.; Kuball, J.; Radstake, T.R.; Meyaard, L. Immune checkpoints and rheumatic diseases: What can cancer immunotherapy teach us? Nat. Rev. Rheumatol. 2016, 12, 593. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227. [Google Scholar] [CrossRef]
- June, C.H.; Ledbetter, J.A.; Gillespie, M.M.; Lindsten, T.; Thompson, C.B. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell. Biol. 1987, 7, 4472–4481. [Google Scholar] [CrossRef]
- Mueller, D.L.; Jenkins, M.K.; Schwartz, R.H. Clonal expansion versus functional clonal inactivation: A costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.D.; Le Gros, G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol. Cell Biol. 1999, 77, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Sulli, A.; Paolino, S.; Pizzorni, C. CTLA-4 blockade in the treatment of rheumatoid arthritis: An update. Expert Rev. Clin. Immunol. 2016, 12, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Goldzweig, O.; Hashkes, P.J. Abatacept in the treatment of polyarticular JIA: Development, clinical utility, and place in therapy. Drug Des. Dev. Ther. 2011, 5, 61. [Google Scholar]
- Taha Khalaf, A.; Song, J.-Q.; Gao, T.-T.; Yu, X.-P.; Lei, T.-C. CTLA-4 gene polymorphism and the risk of systemic lupus erythematosus in the Chinese population. Biomed Res. Int. 2011, 2011, 167395. [Google Scholar]
- Sage, P.T.; Paterson, A.M.; Lovitch, S.B.; Sharpe, A.H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014, 41, 1026–1039. [Google Scholar] [CrossRef]
- Finck, B.K.; Linsley, P.S.; Wofsy, D. Treatment of murine lupus with CTLA4Ig. Science 1994, 265, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Daikh, D.I.; Wofsy, D. Cutting edge: Reversal of murine lupus nephritis with CTLA4Ig and cyclophosphamide. J. Immunol. 2001, 166, 2913–2916. [Google Scholar] [CrossRef] [PubMed]
- Khatri, M.L. Rowell’s syndrome. Indian J. Derm. Venereol. Leprol. 2000, 66, 262–263. [Google Scholar]
- Zeitouni, N.C.; Funaro, D.; Cloutier, R.A.; Gagne, E.; Claveau, J. Redefining Rowell’s syndrome. Br. J. Derm. 2000, 142, 343–346. [Google Scholar] [CrossRef]
- Shteyngarts, A.R.; Warner, M.R.; Camisa, C. Lupus erythematosus associated with erythema multiforme: Does Rowell’s syndrome exist? J. Am. Acad. Derm. 1999, 40, 773–777. [Google Scholar] [CrossRef]
- Child, F.J.; Kapur, N.; Creamer, D.; Kobza Black, A. Rowell’s syndrome. Clin. Exp. Derm. 1999, 24, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Dogra, A.; Minocha, Y.C.; Gupta, M.; Capalash, P. Rowell’s Syndrome. Indian J. Derm. Venereol. Leprol. 2000, 66, 324–325. [Google Scholar]
- Fitzgerald, E.A.; Purcell, S.M.; Kantor, G.R.; Goldman, H.M. Rowell’s syndrome: Report of a case. J. Am. Acad. Derm. 1996, 35, 801–803. [Google Scholar] [CrossRef]
- Nurieva, R.I.; Liu, X.; Dong, C. Yin–Yang of costimulation: Crucial controls of immune tolerance and function. Immunol. Rev. 2009, 229, 88–100. [Google Scholar] [CrossRef]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263. [Google Scholar] [CrossRef] [PubMed]
- McAdam, A.J.; Chang, T.T.; Lumelsky, A.E.; Greenfield, E.A.; Boussiotis, V.A.; Duke-Cohan, J.S.; Chernova, T.; Malenkovich, N.; Jabs, C.; Kuchroo, V.K. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J. Immunol. 2000, 165, 5035–5040. [Google Scholar] [CrossRef] [PubMed]
- Swallow, M.M.; Wallin, J.J.; William, C.S. B7h, a novel costimulatory homolog of B7. 1 and B7. 2, is induced by TNFα. Immunity 1999, 11, 423–432. [Google Scholar] [CrossRef]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827. [Google Scholar] [CrossRef]
- Odegard, J.M.; Marks, B.R.; DiPlacido, L.D.; Poholek, A.C.; Kono, D.H.; Dong, C.; Flavell, R.A.; Craft, J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med. 2008, 205, 2873–2886. [Google Scholar] [CrossRef]
- Hu, Y.-L.; Metz, D.P.; Chung, J.; Siu, G.; Zhang, M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J. Immunol. 2009, 182, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, L.L.; Cullen, J.L.; Kashgarian, M.; Dong, C.; Craft, J.; Shlomchik, M.J. Local triggering of the ICOS coreceptor by CD11c+ myeloid cells drives organ inflammation in lupus. Immunity 2015, 42, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Quintana, F.J.; Weiner, H.L. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4+ CD25− LAP+ regulatory T cell and is associated with down-regulation of IL-17+ CD4+ ICOS+ CXCR5+ follicular helper T cells. J. Immunol. 2008, 181, 6038–6050. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Mai, X.M.; Forbush, K.; Bevan, M.J.; Dong, C. B7h is required for T cell activation, differentiation, and effector function. Proc. Natl. Acad. Sci. USA 2003, 100, 14163–14168. [Google Scholar] [CrossRef] [PubMed]
- Melosky, B.; Burkes, R.; Rayson, D.; Alcindor, T.; Shear, N.; Lacouture, M. Management of skin rash during EGFR-targeted monoclonal antibody treatment for gastrointestinal malignancies: Canadian recommendations. Curr. Oncol. 2009, 16, 16–26. [Google Scholar] [CrossRef]
- Ling, V.; Wu, P.W.; Spaulding, V.; Kieleczawa, J.; Luxenberg, D.; Carreno, B.M.; Collins, M. Duplication of primate and rodent B7-H3 immunoglobulin V-and C-like domains: Divergent history of functional redundancy and exon loss. Genomics 2003, 82, 365–377. [Google Scholar] [CrossRef]
- Nguyen, T.; Liu, X.K.; Zhang, Y.; Dong, C. BTNL2, a butyrophilin-like molecule that functions to inhibit T cell activation. J. Immunol. 2006, 176, 7354–7360. [Google Scholar] [CrossRef]
- Prasad, D.V.; Richards, S.; Mai, X.M.; Dong, C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity 2003, 18, 863–873. [Google Scholar] [CrossRef]
- Orozco, G.; Eerligh, P.; Sánchez, E.; Zhernakova, S.; Roep, B.O.; González-Gay, M.A.; López-Nevot, M.A.; Callejas, J.L.; Hidalgo, C.; Pascual-Salcedo, D. Analysis of a functional BTNL2 polymorphism in type 1 diabetes, rheumatoid arthritis, and systemic lupus erythematosus. Hum. Immunol. 2005, 66, 1235–1241. [Google Scholar] [CrossRef]
- Rogers, P.R.; Song, J.; Gramaglia, I.; Killeen, N.; Croft, M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001, 15, 445–455. [Google Scholar] [CrossRef]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar] [PubMed]
- Brocker, T.; Gulbranson-Judge, A.; Flynn, S.; Riedinger, M.; Raykundalia, C.; Lane, P. CD4 T cell traffic control: In vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B follicles. Eur. J. Immunol. 1999, 29, 1610–1616. [Google Scholar] [CrossRef]
- Gramaglia, I.; Jember, A.; Pippig, S.D.; Weinberg, A.D.; Killeen, N.; Croft, M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000, 165, 3043–3050. [Google Scholar] [CrossRef] [PubMed]
- Takeda, I.; Ine, S.; Killeen, N.; Ndhlovu, L.C.; Murata, K.; Satomi, S.; Sugamura, K.; Ishii, N. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J. Immunol. 2004, 172, 3580–3589. [Google Scholar] [CrossRef]
- Valzasina, B.; Guiducci, C.; Dislich, H.; Killeen, N.; Weinberg, A.D.; Colombo, M.P. Triggering of OX40 (CD134) on CD4+ CD25+ T cells blocks their inhibitory activity: A novel regulatory role for OX40 and its comparison with GITR. Blood 2005, 105, 2845–2851. [Google Scholar] [CrossRef]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2009, 28, 57–78. [Google Scholar] [CrossRef]
- Jacquemin, C.; Schmitt, N.; Contin-Bordes, C.; Liu, Y.; Narayanan, P.; Seneschal, J.; Maurouard, T.; Dougall, D.; Davizon, E.S.; Dumortier, H. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity 2015, 42, 1159–1170. [Google Scholar] [CrossRef]
- Graham, D.S.C.; Graham, R.R.; Manku, H.; Wong, A.K.; Whittaker, J.C.; Gaffney, P.M.; Moser, K.L.; Rioux, J.D.; Altshuler, D.; Behrens, T.W. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat. Genet. 2008, 40, 83. [Google Scholar] [CrossRef]
- Aten, J.; Roos, A.; Claessen, N.; Schilder-Tol, E.J.; TEN BERGE, I.J.; Weening, J.J. Strong and selective glomerular localization of CD134 ligand and TNF receptor-1 in proliferative lupus nephritis. J. Am. Soc. Nephrol. 2000, 11, 1426–1438. [Google Scholar]
- Patschan, S.; Dolff, S.; Kribben, A.; Dürig, J.; Patschan, D.; Wilde, B.; Specker, C.; Philipp, T.; Witzke, O. CD134 expression on CD4+ T cells is associated with nephritis and disease activity in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2006, 145, 235–242. [Google Scholar] [CrossRef]
- Farres, M.N.; Al-Zifzaf, D.S.; Aly, A.A.; Abd Raboh, N.M. OX40/OX40L in systemic lupus erythematosus: Association with disease activity and lupus nephritis. Ann. Saudi Med. 2011, 31, 29–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Y.-B.; Ye, R.-G.; Li, Y.-J.; Xie, C.-M.; Wu, Y.-H. Effect of anti-CD134L mAb and CTLA4Ig on ConA-induced proliferation, Th cytokine secretion, and anti-dsDNA antibody production in spleen cells from lupus-prone BXSB mice. Autoimmunity 2008, 41, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-b.; Ye, R.-g.; Li, Y.-j.; Xie, C.-m. Targeting the CD134–CD134L interaction using anti-CD134 and/or rhCD134 fusion protein as a possible strategy to prevent lupus nephritis. Rheumatol. Int. 2009, 29, 417. [Google Scholar] [CrossRef] [PubMed]
- Sitrin, J.; Suto, E.; Wuster, A.; Eastham-Anderson, J.; Kim, J.M.; Austin, C.D.; Lee, W.P.; Behrens, T.W. The Ox40/Ox40 ligand pathway promotes pathogenic Th cell responses, plasmablast accumulation, and lupus nephritis in NZB/W F1 mice. J. Immunol. 2017, 199, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Detre, C.; Keszei, M.; Romero, X.; Tsokos, G.C.; Terhorst, C. SLAM family receptors and the SLAM-associated protein (SAP) modulate T cell functions. Semin. Immunopathol. 2010, 32, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Rauen, T.; Kis-Toth, K.; Kyttaris, V.C.; Hedrich, C.M.; Terhorst, C.; Tsokos, G.C. Increased expression of SLAM receptors SLAMF3 and SLAMF6 in systemic lupus erythematosus T lymphocytes promotes Th17 differentiation. J. Immunol. 2012, 188, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, M.A.; Adam, K.; Strazza, M.; Tocheva, A.S.; Peled, M.; Mor, A. SLAMF6 clustering is required to augment T cell activation. PLoS ONE 2019, 14, e0218109. [Google Scholar] [CrossRef]
- Chatterjee, M.; Kis-Toth, K.; Thai, T.-H.; Terhorst, C.; Tsokos, G.C. SLAMF6-driven co-stimulation of human peripheral T cells is defective in SLE T cells. Autoimmunity 2011, 44, 211–218. [Google Scholar] [CrossRef]
- Brown, D.R.; Calpe, S.; Keszei, M.; Wang, N.; McArdel, S.; Terhorst, C.; Sharpe, A.H. Cutting edge: An NK cell-independent role for Slamf4 in controlling humoral autoimmunity. J. Immunol. 2011, 187, 21–25. [Google Scholar] [CrossRef]
- Koh, A.E.; Njoroge, S.W.; Feliu, M.; Cook, A.; Selig, M.K.; Latchman, Y.E.; Sharpe, A.H.; Colvin, R.B.; Paul, E. The SLAM family member CD48 (Slamf2) protects lupus-prone mice from autoimmune nephritis. J. Autoimmun. 2011, 37, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Karampetsou, M.P.; Comte, D.; Kis-Toth, K.; Kyttaris, V.C.; Tsokos, G.C. Expression patterns of signaling lymphocytic activation molecule family members in peripheral blood mononuclear cell subsets in patients with systemic lupus erythematosus. PLoS ONE 2017, 12, e0186073. [Google Scholar] [CrossRef] [PubMed]
- Stratigou, V.; Doyle, A.F.; Carlucci, F.; Stephens, L.; Foschi, V.; Castelli, M.; McKenna, N.; Cook, H.T.; Lightstone, L.; Cairns, T.D. Altered expression of signalling lymphocyte activation molecule receptors in T-cells from lupus nephritis patients—A potential biomarker of disease activity. Rheumatology 2017, 56, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. Therapeutic potential of anti-CD137 (4-1BB) monoclonal antibodies. Expert Opin. Ther. Targets 2016, 20, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Mittler, R.S.; Vella, A.T. Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J. Immunol. 1999, 162, 5037–5040. [Google Scholar] [PubMed]
- Hurtado, J.C.; Kim, Y.-J.; Kwon, B.S. Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J. Immunol. 1997, 158, 2600–2609. [Google Scholar]
- Mak, A.; Dharmadhikari, B.; Kow, N.Y.; Thamboo, T.; Tang, Q.; Wong, L.W.; Sreedharan, S.K.; Schwarz, H. Deletion of CD137 ligand exacerbates renal and cutaneous but alleviates cerebral manifestations in lupus. Front. Immunol. 2019, 10, 1411. [Google Scholar] [CrossRef]
- Vinay, D.S.; Choi, J.H.; Kim, J.D.; Choi, B.K.; Kwon, B.S. Role of endogenous 4-1BB in the development of systemic lupus erythematosus. Immunology 2007, 122, 394–400. [Google Scholar] [CrossRef]
- Vinay, D.S.; Kim, J.D.; Asai, T.; Choi, B.K.; Kwon, B.S. Absence of 4–1BB Gene Function Exacerbates Lacrimal Gland Inflammation in Autoimmune-Prone MRL-Fas lpr Mice. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4608–4615. [Google Scholar] [CrossRef]
- Foell, J.; Strahotin, S.; O’Neil, S.P.; McCausland, M.M.; Suwyn, C.; Haber, M.; Chander, P.N.; Bapat, A.S.; Yan, X.-J.; Chiorazzi, N. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB× NZW F 1 mice. J. Clin. Investig. 2003, 111, 1505–1518. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, H.M.; Subudhi, S.K.; Chen, J.; Koka, R.; Chen, L.; Fu, Y.-X. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat. Med. 2002, 8, 1405. [Google Scholar] [CrossRef]
- Mittler, R.S.; Bailey, T.S.; Klussman, K.; Trailsmith, M.D.; Hoffmann, M.K. Anti–4-1BB monoclonal antibodies abrogate T cell–dependent humoral immune responses in vivo through the induction of helper T cell anergy. J. Exp. Med. 1999, 190, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Wu, M.Y.; Ng, C.Y.; Lu, C.W.; Wu, J.; Kao, P.H.; Yang, C.K.; Peng, M.T.; Huang, C.Y.; Chang, W.C.; et al. Severe cutaneous adverse reactions induced by targeted anticancer therapies and immunotherapies. Cancer Manag. Res. 2018, 10, 1259–1273. [Google Scholar] [CrossRef]
- Klocke, K.; Sakaguchi, S.; Holmdahl, R.; Wing, K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc. Natl. Acad. Sci. USA 2016, 113, E2383–E2392. [Google Scholar] [CrossRef]
- Verma, N.; Burns, S.O.; Walker, L.S.; Sansom, D.M. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin. Exp. Immunol. 2017, 190, 1–7. [Google Scholar] [CrossRef]
- Kuehn, H.S.; Ouyang, W.; Lo, B.; Deenick, E.K.; Niemela, J.E.; Avery, D.T.; Schickel, J.-N.; Tran, D.Q.; Stoddard, J.; Zhang, Y. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014, 345, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.-E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Med. 2015, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Lidar, M.; Giat, E.; Garelick, D.; Horowitz, Y.; Amital, H.; Steinberg-Silman, Y.; Schachter, J.; Shapira-Frommer, R.; Markel, G. Rheumatic manifestations among cancer patients treated with immune checkpoint inhibitors. Autoimmun. Rev. 2018, 17, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Fadel, F.; Karoui, K.E.; Knebelmann, B. Anti-CTLA4 antibody–induced lupus nephritis. N. Engl. J. Med. 2009, 361, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261. [Google Scholar] [CrossRef]
- Wang, J.; Yoshida, T.; Nakaki, F.; Hiai, H.; Okazaki, T.; Honjo, T. Establishment of NOD-Pdcd1-/-mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 11823–11828. [Google Scholar] [CrossRef]
- Ansari, M.J.I.; Salama, A.D.; Chitnis, T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.D.; Chitnis, T.; Imitola, J.; Ansari, M.J.I.; Akiba, H.; Tushima, F.; Azuma, M.; Yagita, H.; Sayegh, M.H.; Khoury, S.J. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2003, 198, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365. [Google Scholar] [CrossRef] [PubMed]
- Kasagi, S.; Kawano, S.; Okazaki, T.; Honjo, T.; Morinobu, A.; Hatachi, S.; Shimatani, K.; Tanaka, Y.; Minato, N.; Kumagai, S. Anti-programmed cell death 1 antibody reduces CD4+ PD-1+ T cells and relieves the lupus-like nephritis of NZB/W F1 mice. J. Immunol. 2010, 184, 2337–2347. [Google Scholar] [CrossRef]
- Wong, M.; La Cava, A.; Singh, R.P.; Hahn, B.H. Blockade of programmed death-1 in young (New Zealand black× New Zealand white) F1 mice promotes the activity of suppressive CD8+ T cells that protect from lupus-like disease. J. Immunol. 2010, 185, 6563–6571. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; La Cava, A.; Hahn, B.H. Blockade of programmed death-1 in young (New Zealand Black× New Zealand White) F1 mice promotes the suppressive capacity of CD4+ regulatory T cells protecting from lupus-like disease. J. Immunol. 2013, 190, 5402–5410. [Google Scholar] [CrossRef] [PubMed]
- Sage, P.T.; Francisco, L.M.; Carman, C.V.; Sharpe, A.H. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat. Immunol. 2013, 14, 152. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.-F.; Gondek, D.; Wang, Y.; Fava, R.A. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 14846–14851. [Google Scholar] [CrossRef]
- Ceeraz, S.; Sergent, P.A.; Plummer, S.F.; Schned, A.R.; Pechenick, D.; Burns, C.M.; Noelle, R.J. VISTA deficiency accelerates the development of fatal murine lupus nephritis. Arthritis Rheumatol. 2017, 69, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Sergent, P.; Plummer, S.; Pettus, J.; Mabaera, R.; DeLong, J.; Pechenick, D.; Burns, C.; Noelle, R.; Ceeraz, S. Blocking the VISTA pathway enhances disease progression in (NZB× NZW) F1 female mice. Lupus 2018, 27, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Ceeraz, S.; Sergent, P.; Schned, A.; Burns, C.; Noelle, R. Therapeutic role of the novel checkpoint regulator VISTA in murine autoimmune disease models.(P5174). Am. Assoc. Immnol. 2013, 190, 194. [Google Scholar]
- Caserta, S.; Nausch, N.; Sawtell, A.; Drummond, R.; Barr, T.; MacDonald, A.S.; Mutapi, F.; Zamoyska, R. Chronic infection drives expression of the inhibitory receptor CD200R, and its ligand CD200, by mouse and human CD4 T cells. PLoS ONE 2012, 7, e35466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bando, Y.; Vargas-Lowy, D.; Elyaman, W.; Khoury, S.J.; Huang, T.; Reif, K.; Chitnis, T. CD200R1 agonist attenuates mechanisms of chronic disease in a murine model of multiple sclerosis. J. Neurosci. 2010, 30, 2025–2038. [Google Scholar] [CrossRef]
- Šimelyte, E.; Criado, G.; Essex, D.; Uger, R.A.; Feldmann, M.; Williams, R.O. CD200-FC, a novel antiarthritic biologic agent that targets proinflammatory cytokine expression in the joints of mice with collagen-induced arthritis. Arthritis Rheum. 2008, 58, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhao, L.; Zhang, F.; Zhang, X. Impact of CD200-Fc on dendritic cells in lupus-prone NZB/WF1 mice. Sci. Rep. 2016, 6, 31874. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, L.-d.; Tong, L.-s.; Qian, S.-n.; Ren, Y.; Zhang, L.; Ding, X.; Chen, Y.; Wang, Y.-x.; Zhang, W. Aberrant CD200/CD200R1 expression and function in systemic lupus erythematosus contributes to abnormal T-cell responsiveness and dendritic cell activity. Arthritis Res. Ther. 2012, 14, R123. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, H.; Xiang, W.; He, X.; Liao, W.; Yi, Z. CD200R1 agonist attenuates LPS-induced inflammatory response in human renal proximal tubular epithelial cells by regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem. Biophys. Res. Commun. 2015, 460, 287–294. [Google Scholar] [CrossRef]
- Lee, L.; Liu, J.; Manuel, J.; Gorczynski, R. A role for the immunomodulatory molecules CD200 and CD200R in regulating bone formation. Immunol. Lett. 2006, 105, 150–158. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, B.; Yin, Y.; Leng, X.; Jiang, Y.; Zhang, L.; Li, Y.; Li, X.; Zhang, F.; He, W. Aberrant CD200/CD200R1 expression and its potential role in Th17 cell differentiation, chemotaxis and osteoclastogenesis in rheumatoid arthritis. Rheumatology 2014, 54, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Pontikoglou, C.; Labat, E.; Deschaseaux, F.; Sensebé, L. CD200R/CD200 inhibits osteoclastogenesis: New mechanism of osteoclast control by mesenchymal stem cells in human. PLoS ONE 2013, 8, e72831. [Google Scholar] [CrossRef] [PubMed]
- Sakisaka, T.; Takai, Y. Biology and pathology of nectins and nectin-like molecules. Curr. Opin. Cell Biol. 2004, 16, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Colonna, M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin. Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Hou, H.; Wu, S.; Zhou, Y.; Wang, J.; Yu, J.; Wu, X.; Lu, Y.; Mao, L.; Bosco, M.J. TIGIT signalling pathway negatively regulates CD 4+ T-cell responses in systemic lupus erythematosus. Immunology 2017, 151, 280–290. [Google Scholar] [CrossRef]
- Luo, Q.; Ye, J.; Zeng, L.; Li, X.; Fang, L.; Ju, B.; Huang, Z.; Li, J. Elevated expression of TIGIT on CD3+ CD4+ T cells correlates with disease activity in systemic lupus erythematosus. Allergy Asthma Clin. Immunol. 2017, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef]
- Liu, S.; Sun, L.; Wang, C.; Cui, Y.; Ling, Y.; Li, T.; Lin, F.; Fu, W.; Ding, M.; Zhang, S. Treatment of murine lupus with TIGIT-Ig. Clin. Immunol. 2019, 203, 72–80. [Google Scholar] [CrossRef]
- Luo, Q.; Li, X.; Fu, B.; Zhang, L.; Deng, Z.; Qing, C.; Su, R.; Xu, J.; Guo, Y.; Huang, Z. Decreased expression of TIGIT in NK cells correlates negatively with disease activity in systemic lupus erythematosus. Int. J. Clin. Exp. Pathol. 2018, 11, 2408–2418. [Google Scholar]
- Wang, X.; Shu, Q.; Dong, L.; Xingfu, L. The expression and significance of T cell immunoglobulin domain and mucin domain-3 and its ligand Galectin-9 in the peripheral blood of initial systemic lupus erythematosus patients. Chin. J. Rheumatol. 2011, 15, 220–223. [Google Scholar]
- Zheng, H.; Guo, X.; Tian, Q.; Li, H.; Zhu, Y. Distinct role of Tim-3 in systemic lupus erythematosus and clear cell renal cell carcinoma. Int. J. Clin. Exp. Med. 2015, 8, 7029. [Google Scholar] [PubMed]
- Jin, L.; Bai, R.; Zhou, J.; Shi, W.; Xu, L.; Sheng, J.; Peng, H.; Jin, Y.; Yuan, H. Association of Serum T cell Immunoglobulin Domain and Mucin-3 and Interleukin-17 with Systemic Lupus Erythematosus. Med. Sci. Monit. Basic Res. 2018, 24, 168. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yang, X.; Xia, Q.; Zhen, J.; Zhuang, X.; Peng, T. Expression of human T cell immunoglobulin domain and mucin-3 (TIM-3) on kidney tissue from systemic lupus erythematosus (SLE) patients. Clin. Exp. Med. 2014, 14, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Q.; Qian, Q.; Zhao, Z.; Fang, F.; Hu, X.; An, J.; Wu, J.; Liu, C. Expression of human T cell immunoglobulin domain and mucin-3 (TIM-3) and TIM-3 ligands in peripheral blood from patients with systemic lupus erythematosus. Arch. Dermatol. Res. 2016, 308, 553–561. [Google Scholar] [CrossRef]
- Zhao, D.; Guo, M.; Liu, B.; Lin, Q.; Xie, T.; Zhang, Q.; Jia, X.; Shu, Q.; Liang, X.; Gao, L. Frontline Science: Tim-3-mediated dysfunctional engulfment of apoptotic cells in SLE. J. Leukoc. Biol. 2017, 102, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Meng, J.; Wang, X.; Liu, S.; Shu, Q.; Gao, L.; Ju, Y.; Zhang, L.; Sun, W.; Ma, C. Expression of human TIM-1 and TIM-3 on lymphocytes from systemic lupus erythematosus patients. Scand. J. Immunol. 2008, 67, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Moritoki, M.; Kadowaki, T.; Niki, T.; Nakano, D.; Soma, G.; Mori, H.; Kobara, H.; Masaki, T.; Kohno, M.; Hirashima, M. Galectin-9 ameliorates clinical severity of MRL/lpr lupus-prone mice by inducing plasma cell apoptosis independently of Tim-3. PLoS ONE 2013, 8, e60807. [Google Scholar] [CrossRef]
- Oya, Y.; Watanabe, N.; Kobayashi, Y.; Owada, T.; Oki, M.; Ikeda, K.; Suto, A.; Kagami, S.-i.; Hirose, K.; Kishimoto, T. Lack of B and T lymphocyte attenuator exacerbates autoimmune disorders and induces Fas-independent liver injury in MRL-lpr/lpr mice. Int. Immunol. 2011, 23, 335–344. [Google Scholar] [CrossRef]
- Wang, L.; Kang, N.; Zhou, J.; Guo, Y.; Zhang, X.; Cui, L.; Ba, D.; He, W. Downregulation of CD94/NKG2A inhibitory receptor on decreased γδ T cells in patients with systemic lupus erythematosus. Scand. J. Immunol. 2012, 76, 62–69. [Google Scholar] [CrossRef]
- Hagberg, N.; Theorell, J.; Hjorton, K.; Spee, P.; Eloranta, M.L.; Bryceson, Y.T.; Rönnblom, L. Functional anti-CD94/NKG2A and anti-CD94/NKG2C autoantibodies in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2015, 67, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Furie, R.A.; Tanaka, Y.; Kalunian, K.C.; Mosca, M.; Petri, M.A.; Dörner, T.; Cardiel, M.H.; Bruce, I.N.; Gomez, E. Baricitinib for systemic lupus erythematosus: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 222–231. [Google Scholar] [CrossRef]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472. [Google Scholar] [CrossRef]
- Palkowitsch, L.; Marienfeld, U.; Brunner, C.; Eitelhuber, A.; Krappmann, D.; Marienfeld, R.B. The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-κB activation. J. Biol. Chem. 2011, 286, 7522–7534. [Google Scholar] [CrossRef] [PubMed]
- Hayden-Martinez, K.; Kane, L.P.; Hedrick, S.M. Effects of a constitutively active form of calcineurin on T cell activation and thymic selection. J. Immunol. 2000, 165, 3713–3721. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Barr, V.A.; Akpan, I.; Mittelstadt, P.R.; Singha, L.I.; Samelson, L.E.; Ashwell, J.D. Recruitment of calcineurin to the TCR positively regulates T cell activation. Nat. Immunol. 2017, 18, 196. [Google Scholar] [CrossRef] [PubMed]
- Clipstone, N.A.; Crabtree, G.R. Calcineurin Is a Key Signaling Enzyme in T Lymphocyte Activation and the Target of the Immunosuppressive Drugs Cyclosporin A and FK506 a. Ann. N. Y. Acad. Sci. 1993, 696, 20–30. [Google Scholar] [CrossRef]
- Barbarino, J.M.; Staatz, C.E.; Venkataramanan, R.; Klein, T.E.; Altman, R.B. PharmGKB summary: Cyclosporine and tacrolimus pathways. Pharm. Genom. 2013, 23, 563. [Google Scholar] [CrossRef]
- Dooley, M.A.; Pendergraft, I.; Ginzler, E.M.; Olsen, N.J.; Tumlin, J.; Rovin, B.H.; Houssiau, F.; Wofsy, D.; Isenberg, D.; Solomons, N. Speed of remission with the use of voclosporin, MMF and low dose steroids: Results of a global lupus nephritis study. Arthritis Rheumatol. 2016, 68, 5. [Google Scholar]
- Cortés-Hernández, J.; Torres-Salido, M.T.; Medrano, A.S.; Tarrés, M.V.; Ordi-Ros, J. Long-term outcomes—Mycophenolate mofetil treatment for lupus nephritis with addition of tacrolimus for resistant cases. Nephrol. Dial. Transplant. 2010, 25, 3939–3948. [Google Scholar] [CrossRef]
- Mok, C.; To, C.; Yu, K.; Ho, L. Combined low-dose mycophenolate mofetil and tacrolimus for lupus nephritis with suboptimal response to standard therapy: A 12-month prospective study. Lupus 2013, 22, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Crampton, S.P.; Morawski, P.A.; Bolland, S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis. Models Mech. 2014, 7, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Lederman, S.; Yellin, M.; Inghirami, G.; Lee, J.; Knowles, D.; Chess, L. Molecular interactions mediating TB lymphocyte collaboration in human lymphoid follicles. Roles of T cell-B-cell-activating molecule (5c8 antigen) and CD40 in contact-dependent help. J. Immunol. 1992, 149, 3817–3826. [Google Scholar] [PubMed]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Lederman, S.; Yellin, M.J.; Cleary, A.M.; Pernis, A.; Inghirami, G.; Cohn, L.E.; Covey, L.R.; Lee, J.J.; Rothman, P.; Chess, L. T-BAM/CD40-L on helper T lymphocytes augments lymphokine-induced B cell Ig isotype switch recombination and rescues B cells from programmed cell death. J. Immunol. 1994, 152, 2163–2171. [Google Scholar] [PubMed]
- Qian, J.; Burkly, L.C.; Smith, E.P.; Ferrant, J.L.; Hoyer, L.W.; Scott, D.W.; Haudenschild, C.C. Role of CD154 in the secondary immune response: The reduction of pre-existing splenic germinal centers and anti-factor VIII inhibitor titer. Eur. J. Immunol. 2000, 30, 2548–2554. [Google Scholar] [CrossRef]
- Masoud, S.; McAdoo, S.P.; Bedi, R.; Cairns, T.D.; Lightstone, L. Ofatumumab for B cell depletion in patients with systemic lupus erythematosus who are allergic to rituximab. Rheumatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Arnold, D.M. The effect of rituximab on humoral and cell mediated immunity and infection in the treatment of autoimmune diseases. Br. J. Haematol. 2010, 149, 3–13. [Google Scholar] [CrossRef]
- Koshy, M.; Berger, D.; Crow, M.K. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J. Clin. Investig. 1996, 98, 826–837. [Google Scholar] [CrossRef]
- Blossom, S.; Chu, E.B.; Weigle, W.O.; Gilbert, K.M. CD40 ligand expressed on B cells in the BXSB mouse model of systemic lupus erythematosus. J. Immunol. 1997, 159, 4580–4586. [Google Scholar] [PubMed]
- Wang, X.; Huang, W.; Schiffer, L.E.; Mihara, M.; Akkerman, A.; Hiromatsu, K.; Davidson, A. Effects of anti-CD154 treatment on B cells in murine systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Folzenlogen, D.; Hofer, M.F.; Leung, D.Y.; Freed, J.H.; Newell, M.K. Analysis of CD80 and CD86 expression on peripheral blood B lymphocytes reveals increased expression of CD86 in lupus patients. Clin. Immunol. Immunopathol. 1997, 83, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, H.; Shimoyama, Y.; Kashiwakura, J.; Takeno, M.; Sakane, T.; Suzuki, N. Preferential expression of B7.2 (CD86), but not B7.1 (CD80), on B cells induced by CD40/CD40L interaction is essential for anti-DNA autoantibody production in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 2003, 21, 71–77. [Google Scholar] [PubMed]
- Kalunian, K.C.; Davis, J.C., Jr.; Merrill, J.T.; Totoritis, M.C.; Wofsy, D.; Group, I.L.S. Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 3251–3258. [Google Scholar] [CrossRef] [PubMed]
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A.; Group, B.L.N.T. A short course of BG9588 (anti–CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Daikh, D.I.; Finck, B.K.; Linsley, P.S.; Hollenbaugh, D.; Wofsy, D. Long-term inhibition of murine lupus by brief simultaneous blockade of the B7/CD28 and CD40/gp39 costimulation pathways. J. Immunol. 1997, 159, 3104–3108. [Google Scholar]
- Wang, X.; Huang, W.; Mihara, M.; Sinha, J.; Davidson, A. Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J. Immunol. 2002, 168, 2046–2053. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Khan, A.R.; Hams, E.; Floudas, A.; Sparwasser, T.; Weaver, C.T.; Fallon, P.G. PD-L1hi B cells are critical regulators of humoral immunity. Nat. Commun. 2015, 6, 5997. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.Y.; Zhu, Q.Q.; Wang, Y.Y.; Lu, Y.; Li, Z.J.; Li, B.Q.; Tang, J.; Wang, H.T.; Song, C.W.; Xie, C.H.; et al. The role and clinical significance of programmed cell death- ligand 1 expressed on CD19(+)B-cells and subsets in systemic lupus erythematosus. Clin. Immunol. 2019, 198, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Valle, J.; Perez-Fernandez, V.A.; Correa-Rocha, R.; Pion, M. Generation of Human Breg-Like Phenotype with Regulatory Function In Vitro with Bacteria-Derived Oligodeoxynucleotides. Int. J. Mol. Sci. 2018, 19, 1737. [Google Scholar] [CrossRef] [PubMed]
- Stefanski, A.L.; Wiedemann, A.; Reiter, K.; Hiepe, F.; Lino, A.C.; Dorner, T. Enhanced Programmed Death 1 and Diminished Programmed Death Ligand 1 Up-Regulation Capacity of Post-Activated Lupus B Cells. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hou, S.; Fang, Q.; Liu, X.; Liu, X.; Qi, H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity 2018, 49, 264–274.e264. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Daridon, C.; Blassfeld, D.; Reiter, K.; Mei, H.E.; Giesecke, C.; Goldenberg, D.M.; Hansen, A.; Hostmann, A.; Frölich, D.; Dörner, T. Epratuzumab targeting of CD22 affects adhesion molecule expression and migration of B-cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R204. [Google Scholar] [CrossRef]
- Clark, E.A. CD22, a B cell-specific receptor, mediates adhesion and signal transduction. J. Immunol. 1993, 150, 4715–4718. [Google Scholar]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef]
- Jellusova, J.; Wellmann, U.; Amann, K.; Winkler, T.H.; Nitschke, L. CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J. Immunol. 2010, 184, 3618–3627. [Google Scholar] [CrossRef]
- Meyer, S.J.; Linder, A.T.; Brandl, C.; Nitschke, L. B Cell Siglecs-News on Signaling and Its Interplay with Ligand Binding. Front. Immunol. 2018, 9, 2820. [Google Scholar] [CrossRef] [PubMed]
- Carnahan, J.; Wang, P.; Kendall, R.; Chen, C.; Hu, S.; Boone, T.; Juan, T.; Talvenheimo, J.; Montestruque, S.; Sun, J.; et al. Epratuzumab, a Humanized Monoclonal Antibody Targeting CD22. Clin. Cancer Res. 2003, 9, 3982s. [Google Scholar] [PubMed]
- Dörner, T.; Shock, A.; Goldenberg, D.M.; Lipsky, P.E. The mechanistic impact of CD22 engagement with epratuzumab on B cell function: Implications for the treatment of systemic lupus erythematosus. Autoimmun. Rev. 2015, 14, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Clowse, M.E.B.; Wallace, D.J.; Furie, R.A.; Petri, M.A.; Pike, M.C.; Leszczyński, P.; Neuwelt, C.M.; Hobbs, K.; Keiserman, M.; Duca, L.; et al. Efficacy and Safety of Epratuzumab in Moderately to Severely Active Systemic Lupus Erythematosus: Results From Two Phase III Randomized, Double-Blind, Placebo-Controlled Trials. Arthritis Rheumatol. 2017, 69, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Geh, D.; Gordon, C. Epratuzumab for the treatment of systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2018, 14, 245–258. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Dorner, T.; Bootsma, H.; Devauchelle-Pensec, V.; Bowman, S.J.; Mariette, X.; Bartz, H.; Oortgiesen, M.; Shock, A.; Koetse, W.; et al. Efficacy of Epratuzumab, an Anti-CD22 Monoclonal IgG Antibody, in Systemic Lupus Erythematosus Patients With Associated Sjogren’s Syndrome: Post Hoc Analyses From the EMBODY Trials. Arthritis Rheumatol. 2018, 70, 763–773. [Google Scholar] [CrossRef]
- Bolland, S.; Ravetch, J.V. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000, 13, 277–285. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Lehmann, B.; Schwab, I.; Bohm, S.; Lux, A.; Biburger, M.; Nimmerjahn, F. FcgammaRIIB: A modulator of cell activation and humoral tolerance. Expert Rev. Clin. Immunol. 2012, 8, 243–254. [Google Scholar] [CrossRef]
- Tiller, T.; Kofer, J.; Kreschel, C.; Busse, C.E.; Riebel, S.; Wickert, S.; Oden, F.; Mertes, M.M.; Ehlers, M.; Wardemann, H. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J. Exp. Med. 2010, 207, 2767–2778. [Google Scholar] [CrossRef]
- Siriboonrit, U.; Tsuchiya, N.; Sirikong, M.; Kyogoku, C.; Bejrachandra, S.; Suthipinittharm, P.; Luangtrakool, K.; Srinak, D.; Thongpradit, R.; Fujiwara, K.; et al. Association of Fcgamma receptor IIb and IIIb polymorphisms with susceptibility to systemic lupus erythematosus in Thais. Tissue Antigens 2003, 61, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Wang, C.M.; Ma, C.C.; Luo, S.F.; Edberg, J.C.; Kimberly, R.P.; Wu, J. Association of a transmembrane polymorphism of Fcgamma receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum. 2006, 54, 3908–3917. [Google Scholar] [CrossRef] [PubMed]
- Mackay, M.; Stanevsky, A.; Wang, T.; Aranow, C.; Li, M.; Koenig, S.; Ravetch, J.V.; Diamond, B. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J. Exp. Med. 2006, 203, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Yang, H.; Li, X.; Li, X.; Gibson, A.W.; Cafardi, J.M.; Zhou, T.; Edberg, J.C.; Kimberly, R.P. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J. Immunol. 2007, 178, 3272–3280. [Google Scholar] [CrossRef] [PubMed]
- Meyaard, L. The inhibitory collagen receptor LAIR-1 (CD305). J. Leukoc. Biol. 2008, 83, 799–803. [Google Scholar] [CrossRef]
- Poggi, A.; Tomasello, E.; Ferrero, E.; Zocchi, M.R.; Moretta, L. p40/LAIR-1 regulates the differentiation of peripheral blood precursors to dendritic cells induced by granulocyte-monocyte colony-stimulating factor. Eur. J. Immunol. 1998, 28, 2086–2091. [Google Scholar] [CrossRef]
- Jansen, C.A.; Cruijsen, C.W.; de Ruiter, T.; Nanlohy, N.; Willems, N.; Janssens-Korpela, P.L.; Meyaard, L. Regulated expression of the inhibitory receptor LAIR-1 on human peripheral T cells during T cell activation and differentiation. Eur. J. Immunol. 2007, 37, 914–924. [Google Scholar] [CrossRef]
- Merlo, A.; Tenca, C.; Fais, F.; Battini, L.; Ciccone, E.; Grossi, C.E.; Saverino, D. Inhibitory receptors CD85j, LAIR-1, and CD152 down-regulate immunoglobulin and cytokine production by human B lymphocytes. Clin. Diagn. Lab. Immunol. 2005, 12, 705–712. [Google Scholar] [CrossRef]
- Colombo, B.M.; Canevali, P.; Magnani, O.; Rossi, E.; Puppo, F.; Zocchi, M.R.; Poggi, A. Defective expression and function of the leukocyte associated Ig-like receptor 1 in B lymphocytes from systemic lupus erythematosus patients. PLoS ONE 2012, 7, e31903. [Google Scholar] [CrossRef]
- Worbs, T.; Hammerschmidt, S.I.; Forster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef]
- Gill, M.A.; Blanco, P.; Arce, E.; Pascual, V.; Banchereau, J.; Palucka, A.K. Blood dendritic cells and DC-poietins in systemic lupus erythematosus. Hum. Immunol. 2002, 63, 1172–1180. [Google Scholar] [CrossRef]
- Khan, S.A.; Nowatzky, J.; Jimenez-Branda, S.; Greenberg, J.D.; Clancy, R.; Buyon, J.; Bhardwaj, N. Active systemic lupus erythematosus is associated with decreased blood conventional dendritic cells. Exp. Mol. Pathol. 2013, 95, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Tripodo, C.; Gong, M.; Sangaletti, S.; Colombo, M.P.; Coffman, R.L.; Barrat, F.J. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med. 2010, 207, 2931–2942. [Google Scholar] [CrossRef]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, N.; Ronnblom, L. Systemic Lupus Erythematosus--A Disease with A Dysregulated Type I Interferon System. Scand. J. Immunol. 2015, 82, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Caraux, A.; De Vos, J.; Fiol, G.; Larroque, M.; Cognot, C.; Bret, C.; Duperray, C.; Hose, D.; Klein, B. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 2009, 114, 5173–5181. [Google Scholar] [CrossRef]
- Mathian, A.; Gallegos, M.; Pascual, V.; Banchereau, J.; Koutouzov, S. Interferon-alpha induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZBxNZW)F1 mice but not in BALB/c mice. Eur. J. Immunol. 2011, 41, 863–872. [Google Scholar] [CrossRef]
- Ytterberg, S.R.; Schnitzer, T.J. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 401–406. [Google Scholar] [CrossRef]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Niewold, T.B.; Clark, D.N.; Salloum, R.; Poole, B.D. Interferon Alpha in Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2010, 2010, 948364. [Google Scholar] [CrossRef]
- Kuznik, A.; Bencina, M.; Svajger, U.; Jeras, M.; Rozman, B.; Jerala, R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011, 186, 4794–4804. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Woodward, J.J.; Sasaki, T.; Minie, M.; Elkon, K.B. Cutting edge: Antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 2015, 194, 4089–4093. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Gong, M.; Xu, Z.; Gill, M.; Chaussabel, D.; Meeker, T.; Chan, J.H.; Wright, T.; Punaro, M.; Bolland, S.; et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 2010, 465, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Tang, J.; Mok, M.Y.; Chan, A.W.; Wu, A.; Lau, C.S. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 2888–2897. [Google Scholar] [CrossRef]
- Fransen, J.H.; van der Vlag, J.; Ruben, J.; Adema, G.J.; Berden, J.H.; Hilbrands, L.B. The role of dendritic cells in the pathogenesis of systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, 207. [Google Scholar] [CrossRef]
- Jin, O.; Sun, L.Y.; Zhou, K.X.; Zhang, X.S.; Feng, X.B.; Mok, M.Y.; Lau, C.S. Lymphocyte apoptosis and macrophage function: Correlation with disease activity in systemic lupus erythematosus. Clin. Rheumatol. 2005, 24, 107–110. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three Markers for Distinct Subsets of Dendritic Cells in Human Peripheral Blood. J. Immunol. 2000, 165, 6037. [Google Scholar] [CrossRef]
- Jahn, P.S.; Zanker, K.S.; Schmitz, J.; Dzionek, A. BDCA-2 signaling inhibits TLR-9-agonist-induced plasmacytoid dendritic cell activation and antigen presentation. Cell. Immunol. 2010, 265, 15–22. [Google Scholar] [CrossRef]
- Riboldi, E.; Daniele, R.; Parola, C.; Inforzato, A.; Arnold, P.L.; Bosisio, D.; Fremont, D.H.; Bastone, A.; Colonna, M.; Sozzani, S. Human C-type lectin domain family 4, member C (CLEC4C/BDCA-2/CD303) is a receptor for asialo-galactosyl-oligosaccharides. J. Biol. Chem. 2011, 286, 35329–35333. [Google Scholar] [CrossRef]
- Furie, R.; Werth, V.P.; Merola, J.F.; Stevenson, L.; Reynolds, T.L.; Naik, H.; Wang, W.; Christmann, R.; Gardet, A.; Pellerin, A.; et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Investig. 2019, 129, 1359–1371. [Google Scholar] [CrossRef]
- Chaichian, Y.; Wallace, D.J.; Weisman, M.H. A promising approach to targeting type 1 IFN in systemic lupus erythematosus. J. Clin. Investig. 2019, 129, 958–961. [Google Scholar] [CrossRef] [PubMed]
- LeMaoult, J.; Zafaranloo, K.; Le Danff, C.; Carosella, E.D. HLA-G up-regulates ILT2, ILT3, ILT4, and KIR2DL4 in antigen presenting cells, NK cells, and T cells. FASEB J. 2005, 19, 662–664. [Google Scholar] [CrossRef]
- Rizzo, R.; Bortolotti, D.; Bolzani, S.; Fainardi, E. HLA-G Molecules in Autoimmune Diseases and Infections. Front. Immunol. 2014, 5, 592. [Google Scholar] [CrossRef]
- Guerra-de Blas, P.D.C.; Villaseñor-Talavera, Y.S.; Cruz-González, D.d.J.; Baranda, L.; Doníz-Padilla, L.; Abud-Mendoza, C.; González-Amaro, R.; Monsiváis-Urenda, A.E. Analysis of the Expression and Function of Immunoglobulin-Like Transcript 4 (ILT4, LILRB2) in Dendritic Cells from Patients with Systemic Lupus Erythematosus. J. Immunol. Res. 2016, 2016, 4163094. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, A.; Chimenti, M.S.; Baffari, E.; Guarino, M.D.; Gigliucci, G.; Perricone, C.; Perricone, R. Downregulation of immunoglobulin-like transcript-4 (ILT4) in patients with psoriatic arthritis. PLoS ONE 2014, 9, e92018. [Google Scholar] [CrossRef] [PubMed]
- Monsiváis-Urenda, A.; Gómez-Martin, D.; Santana-de-Anda, K.; Cruz-Martínez, J.; Alcocer-Varela, J.; González-Amaro, R. Defective expression and function of the ILT2/CD85j regulatory receptor in dendritic cells from patients with systemic lupus erythematosus. Hum. Immunol. 2013, 74, 1088–1096. [Google Scholar] [CrossRef]
- Monsiváis-Urenda, A.E.; Baranda, L.; Alvarez-Quiroga, C.; Abud-Mendoza, C.; González-Amaro, R. Expression and functional role of HLA-G in immune cells from patients with systemic lupus erythematosus. J. Clin. Immunol. 2011, 31, 369–378. [Google Scholar] [CrossRef]
- Son, M.; Santiago-Schwarz, F.; Al-Abed, Y.; Diamond, B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc. Natl. Acad. Sci. USA 2012, 109, E3160–E3167. [Google Scholar] [CrossRef]
- Fremeaux-Bacchi, V.; Weiss, L.; Demouchy, C.; Blouin, J.; Kazatchkine, M.D. Autoantibodies to the collagen-like region of C1q are strongly associated with classical pathway-mediated hypocomplementemia in systemic lupus erythematosus. Lupus 1996, 5, 216–220. [Google Scholar] [CrossRef]
- Trendelenburg, M.; Lopez-Trascasa, M.; Potlukova, E.; Moll, S.; Regenass, S.; Fremeaux-Bacchi, V.; Martinez-Ara, J.; Jancova, E.; Picazo, M.L.; Honsova, E.; et al. High prevalence of anti-C1q antibodies in biopsy-proven active lupus nephritis. Nephrol. Dial. Transpl. 2006, 21, 3115–3121. [Google Scholar] [CrossRef]
- Son, M.; Diamond, B.; Volpe, B.; Aranow, C.; Mackay, M.; Santiago-Schwarz, F. Evidence for C1q-mediated crosslinking of CD33/LAIR-1 inhibitory immunoreceptors and biological control of CD33/LAIR-1 expression. Sci. Rep. 2017, 7, 270. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi, I.; Cantoni, C.; Carrega, P.; Oliveri, D.; Lui, G.; Conte, R.; Navarra, M.; Cavaliere, R.; Traggiai, E.; Gattorno, M.; et al. The immune inhibitory receptor LAIR-1 is highly expressed by plasmacytoid dendritic cells and acts complementary with NKp44 to control IFNalpha production. PLoS ONE 2010, 5, e15080. [Google Scholar] [CrossRef] [PubMed]
- Courtney, P.A.; Crockard, A.D.; Williamson, K.; Irvine, A.E.; Kennedy, R.J.; Bell, A.L. Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: Relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann. Rheum. Dis. 1999, 58, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.; Roake, W.; Brown, S.; Young, P.; Naik, H.; Wordsworth, P.; Isenberg, D.A.; Reid, K.B.; Eggleton, P. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 1543–1556. [Google Scholar] [CrossRef]
- Alves, C.M.; Marzocchi-Machado, C.M.; Louzada-Junior, P.; Azzolini, A.E.; Polizello, A.C.; de Carvalho, I.F.; Lucisano-Valim, Y.M. Superoxide anion production by neutrophils is associated with prevalent clinical manifestations in systemic lupus erythematosus. Clin. Rheumatol. 2008, 27, 701–708. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The balancing act of neutrophils. Cell Host Microbe 2014, 15, 526–536. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Zykova, S.N.; Tveita, A.A.; Rekvig, O.P. Renal Dnase1 enzyme activity and protein expression is selectively shut down in murine and human membranoproliferative lupus nephritis. PLoS ONE 2010, 5, e12096. [Google Scholar] [CrossRef]
- Davis, J.C., Jr.; Manzi, S.; Yarboro, C.; Rairie, J.; McInnes, I.; Averthelyi, D.; Sinicropi, D.; Hale, V.G.; Balow, J.; Austin, H.; et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus 1999, 8, 68–76. [Google Scholar] [CrossRef]
- Steevels, T.A.M.; Lebbink, R.J.; Westerlaken, G.H.A.; Coffer, P.J.; Meyaard, L. Signal Inhibitory Receptor on Leukocytes-1 Is a Novel Functional Inhibitory Immune Receptor Expressed on Human Phagocytes. J. Immunol. 2010, 184, 4741. [Google Scholar] [CrossRef] [PubMed]
- Van Avondt, K.; Fritsch-Stork, R.; Derksen, R.H.; Meyaard, L. Ligation of signal inhibitory receptor on leukocytes-1 suppresses the release of neutrophil extracellular traps in systemic lupus erythematosus. PLoS ONE 2013, 8, e78459. [Google Scholar] [CrossRef] [PubMed]
- Van Avondt, K.; van der Linden, M.; Naccache, P.H.; Egan, D.A.; Meyaard, L. Signal inhibitory receptor on leukocytes-1 limits the formation of neutrophil extracellular traps, but preserves intracellular bacterial killing. J. Immunol. 2016, 196, 3686–3694. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shiratori, I.; Uehori, J.; Ikawa, M.; Arase, H. Neutrophil infiltration during inflammation is regulated by PILRα via modulation of integrin activation. Nat. Immunol. 2013, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Caplazi, P.; Zhang, J.; Mazloom, A.; Kummerfeld, S.; Quinones, G.; Senger, K.; Lesch, J.; Peng, I.; Sebrell, A. PILRα negatively regulates mouse inflammatory arthritis. J. Immunol. 2014, 193, 860–870. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Molecule | Expression | Ligand/Receptor | Possible Targeted Cells in SLE |
|---|---|---|---|
| CD80 and CD86 | APCs | CD28 | T cells |
| B7h | APCs | ICOS | T cells |
| OX40L | APCs | OX40 | T cells |
| SLAMF6 | T cells, B cells, and NK cells | SLAMF6 | T cells |
| CD137L | APCs | CD137 | T cells |
| CD40L | T cells | CD40 | B cells |
| Molecule | Expression | Ligand/Receptor | Possible Targeted Cells in SLE |
|---|---|---|---|
| CD80 and CD86 | APCs | CTLA4 | T cells |
| PD-L1 and PD-L2 | APCs | PD-1 | T cells and B cells |
| VSIG-3 | Unknown | VISTA | T cells |
| VISTA | APCs and T cells | VISTA receptor | T cells |
| CD200 | B cells, eosinophils, pDCs and a subset of T cells | CD200R1 | T cells, DCs, and neutrophils |
| CD155 | DCs or macrophages | TIGIT | T cells and NK cells |
| Galectin-9 | Cytoplasmic expression in most cell types. | TIM-3 | T cells and macrophages |
| B7S1 | APCs | B7S1 receptor | T cells |
| BTNL2 | T cells, B cells, and macrophages | BTNL2 receptor | T cells |
| Unknown | APCs | B7S3 | T cells |
| Sialic acid | Siglec-2/CD22 | B cells | |
| Immune complexes | FCγRIIB | B cells | |
| Collagen (C1qCLR) | LAIR-1 | B cells, DCs, and macrophages | |
| Asialo-galactosyl-oligosaccharide | BDCA2 | pDCs | |
| HLA-G | Monocytes and trophoblasts | ILT-4 | Myeloid cells, including monocytes, macrophages, dendritic cells, and granulocytes. |
| HLA-G | Monocytes and trophoblasts | ILT-2 | T cells, B cells, DCs, and NK cells |
| VSTM1-L | SIRL-1 | Neutrophils | |
| Sialylated surface protein | PILR-α | Neutrophils |
| Medication | Target | Phase/Outcome | Clinical Trials.gov ID |
|---|---|---|---|
| Abatacept | CD80 and CD86 | Phase III—terminated | NCT00430677 |
| Abatacept | CD80 and CD86 | Phase II—failed to meet endpoint | NCT00119678 |
| Abatacept | CD80 and CD86 | Phase II—failed to meet endpoint | NCT00774852 |
| Abatacept | CD80 and CD86 | Phase II—recruiting | NCT02270957 |
| Abatacept | CD80 and CD86 | Phase II—recruiting | NCT02429934 |
| BMS-931699 | CD28 | Phase II—failed to meet endpoint | NCT02265744 |
| AMG557 | ICOSL | Phase I—acceptable safety profile | NCT00774943 |
| JNJ-61610588 | VISTA | Phase I—terminated | NCT02671955 |
| CFZ533 | CD40 | Phase II—recruiting | NCT03656562 |
| BG9588 | CD40L | Phase II—terminated | Boumpas DT, et al. Arthritis Rheum. 2003. |
| IDEC-131 | CD40L | Phase II—failed to meet endpoint | Kalunian KC, et al. Arthritis Rheum. 2002. |
| Dapirolizumab Pegol | CD40L | Phase II—unpublished | NCT02804763 |
| Anti-CD40L | CD40L | Phase II—terminated | NCT00001789 |
| Epratuzumab | CD22 | Phase III—unpublished | NCT01408576 |
| Epratuzumab | CD22 | Phase III—terminated | NCT00111306 |
| Epratuzumab | CD22 | Phase III—terminated | NCT00383214 |
| Epratuzumab | CD22 | Phase III—withdrawn | NCT00382837 |
| Epratuzumab | CD22 | Phase III—failed to meet endpoint | NCT01262365 |
| Epratuzumab | CD22 | Phase III—failed to meet endpoint | NCT01261793 |
| Epratuzumab | CD22 | Phase II—unpublished | NCT01534403 |
| Epratuzumab | CD22 | Phase II—encouraging | NCT00624351 |
| Epratuzumab | CD22 | Phase II—encouraging | NCT00660881 |
| Epratuzumab | CD22 | Phase II—encouraging | NCT00383513 |
| Epratuzumab | CD22 | Phase II—terminated | NCT00113971 |
| Epratuzumab | CD22 | Phase I/II—acceptable safety profile | NCT01449071 |
| Epratuzumab | CD22 | Phase I—unpublished | NCT00011908 |
| BIIB059 | BDCA2 | Phase II—active, not recruiting | NCT02847598 |
| BIIB059 | BDCA2 | Phase I—acceptable safety profile | NCT02106897 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, K.-L.; Wu, M.-Y.; Wang, C.-H.; Wang, C.-W.; Hung, S.-I.; Chung, W.-H.; Chen, C.-B. The Role of Immune Checkpoint Receptors in Regulating Immune Reactivity in Lupus. Cells 2019, 8, 1213. https://doi.org/10.3390/cells8101213
Lu K-L, Wu M-Y, Wang C-H, Wang C-W, Hung S-I, Chung W-H, Chen C-B. The Role of Immune Checkpoint Receptors in Regulating Immune Reactivity in Lupus. Cells. 2019; 8(10):1213. https://doi.org/10.3390/cells8101213
Chicago/Turabian StyleLu, Kun-Lin, Ming-Ying Wu, Chi-Hui Wang, Chuang-Wei Wang, Shuen-Iu Hung, Wen-Hung Chung, and Chun-Bing Chen. 2019. "The Role of Immune Checkpoint Receptors in Regulating Immune Reactivity in Lupus" Cells 8, no. 10: 1213. https://doi.org/10.3390/cells8101213
APA StyleLu, K.-L., Wu, M.-Y., Wang, C.-H., Wang, C.-W., Hung, S.-I., Chung, W.-H., & Chen, C.-B. (2019). The Role of Immune Checkpoint Receptors in Regulating Immune Reactivity in Lupus. Cells, 8(10), 1213. https://doi.org/10.3390/cells8101213