Individual Copy Number of Ribosomal Genes as a Factor of Mental Retardation and Autism Risk and Severity


Abstract
:1. Introduction
2. Hypothesis
3. Evaluation of Hypothesis
4. Predictions from the Hypothesis and Direct Testing
4.1. Testing on Patients
4.2. Testing on Animal Models
5. Discussion
Funding
Acknowledgments
Conflicts of Interest
References
- Simons Foundation Autism Research Initiative Base. Available online: https://www.sfari.org/resource/sfari-base/ (accessed on 25 September 2019).
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson-Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Bassell, G.J.; Klann, E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015, 16, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Mamcarz, M.; Mullins, C.; Choudhury, A.; Boyle, R.G.; Smith, D.G.; Walker, D.W.; Klann, E. Targeting Translation Control with p70 S6 Kinase 1 Inhibitors to Reverse Phenotypes in Fragile X Syndrome Mice. Neuropsychopharmacology 2016, 41, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. Translational homeostasis via eIF4E and 4E-BP1. Mol. Cell 2012, 46, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Bear, M.F. The autistic neuron: Troubled translation? Cell 2008, 135, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Laggerbauer, B.; Ostareck, D.; Keidel, E.M.; Ostareck-Lederer, A.; Fischer, U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 2001, 10, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Joseph, S. Fragile X mental retardation protein: A paradigm for translational control by RNA-binding proteins. Biochimie 2015, 114, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef]
- Chang, S.; Bray, S.M.; Li, Z.; Zarnescu, D.C.; He, C.; Jin, P.; Warren, S.T. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 2008, 4, 256–263. [Google Scholar] [CrossRef]
- Bey, A.L.; Jiang, Y.H. Overview of mouse models of autism spectrum disorders. Curr. Protoc. Pharmacol. 2014, 66, 5.66.1–5.66.26. [Google Scholar]
- Chen, J.; Lei, L.; Tian, L.; Hou, F.; Roper, C.; Ge, X.; Zhao, Y.; Chen, Y.; Dong, Q.; Tanguay, R.L.; et al. Developmental and behavioral alterations in zebrafish embryonically exposed to valproic acid (VPA): An aquatic model for autism. Neurotoxicol. Teratol. 2018, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Gumbinger, C.; Rohsbach, C.B.; Schulze-Bonhage, A.; Korinthenberg, R.; Zentner, J.; Häffner, M.; Fauser, S. Focal cortical dysplasia: A genotype-phenotype analysis of polymorphisms and mutations in the TSC genes. Epilepsia 2009, 50, 1396–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, L.A.; Blute, M.L.; Ryu, J.H.; Swensen, S.J.; Malek, R.S. Renal angiomyolipoma in association with pulmonary lymphangioleiomyomatosis: Forme fruste of tuberous sclerosis? Urology 1993, 41, 440–444. [Google Scholar] [CrossRef]
- Kwiatkowska, J.; Wigowska-Sowinska, J.; Napierala, D.; Slomski, R.; Kwiatkowski, D.J. Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N. Engl. J. Med. 1999, 340, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Moavero, R.; Roberto, D.; Graziola, F. Genotype/Phenotype Correlations in Tuberous Sclerosis Complex. Semin. Pediatr. Neurol. 2015, 22, 259–273. [Google Scholar] [CrossRef]
- Byers, H.M.; Jensen, D.M.; Glass, I.A.; Bennett, J.T. Minimal mosaicism, maximal phenotype: Discordance between clinical and molecular findings in two patients with tuberous sclerosis. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 374–378. [Google Scholar] [CrossRef]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2012, 493, 371–377. [Google Scholar] [CrossRef]
- Santini, E.; Huynh, T.N.; MacAskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; Kaphzan, H.; Klann, E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2012, 493, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Buffington, S.A.; Huang, W.; Costa-Mattioli, M. Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 2014, 37, 17–38. [Google Scholar] [CrossRef]
- Klauck, S.M.; Felder, B.; Kolb-Kokocinski, A.; Schuster, C.; Chiocchetti, A.; Schupp, I.; Wellenreuther, R.; Schmötzer, G.; Poustka, F.; Breitenbach-Koller, L.; et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol. Psychiatry 2006, 11, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocchetti, A.; Pakalapati, G.; Duketis, E.; Wiemann, S.; Poustka, A.; Poustka, F.; Klauck, S.M. Mutation and expression analyses of the ribosomal protein gene RPL10 in an extended German sample of patients with autism spectrum disorder. Am. J. Med. Genet. A 2011, 155, 1472–1475. [Google Scholar] [CrossRef] [PubMed]
- Alsop, R.J.; Soomro, A.; Zhang, Y.; Pieterse, M.; Fatona, A.; Dej, K.; Rheinstädter, M.C. Structural Abnormalities in the Hair of a Patient with a Novel Ribosomopathy. PLoS ONE 2016, 11, e0149619. [Google Scholar] [CrossRef] [PubMed]
- Paolini, N.A.; Attwood, M.; Sondalle, S.B.; Vieira, C.M.D.S.; van Adrichem, A.M.; di Summa, F.M.; O’Donohue, M.F.; Gleizes, P.E.; Rachuri, S.; Briggs, J.W.; et al. A Ribosomopathy Reveals Decoding Defective Ribosomes Driving Human Dysmorphism. Am. J. Hum. Genet. 2017, 100, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porokhovnik, L.N.; Lyapunova, N.A. Dosage effects of human ribosomal genes (rDNA) in health and disease. Chromosome Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.G.; Branco, A.T.; Yu, S.; Lemos, B. Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat. Commun. 2014, 5, 4850. [Google Scholar] [CrossRef] [Green Version]
- Larson, D.E.; Zahradka, P.; Sells, B.H. Control points in eucaryotic ribosome biogenesis. Biochem. Cell Biol. 1991, 69, 5–22. [Google Scholar] [CrossRef]
- Likhoshvai, V.A.; Kogai, V.V.; Fadeev, S.I.; Khlebodarova, T.M. Chaos and Hyperchaos in a Model of Ribosome Autocatalytic Synthesis. Sci. Rep. 2016, 6, 38870. [Google Scholar] [CrossRef] [Green Version]
- Von der Haar, T. A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst. Biol. 2008, 2, 87. [Google Scholar] [CrossRef]
- Perry, R.P. Balanced production of ribosomal proteins. Gene 2007, 401, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Bhugra, D. The global prevalence of schizophrenia. Plos Med. 2005, 2, e151. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Chestkov, I.V.; Jestkova, E.M.; Ershova, E.S.; Golimbet, V.E.; Lezheiko, T.V.; Kolesina, N.Y.; Porokhovnik, L.N.; Lyapunova, N.A.; Izhevskaya, V.L.; Kutsev, S.I.; et al. Abundance of ribosomal RNA gene copies in the genomes of schizophrenia patients. Schizophr. Res. 2018, 197, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Curran, L.K.; Newschaffer, C.J.; Lee, L.C.; Crawford, S.O.; Johnston, M.V.; Zimmerman, A.W. Behaviors associated with fever in children with autism spectrum disorders. Pediatrics 2007, 120, e1386–e1392. [Google Scholar] [CrossRef] [PubMed]
- Beyond Health® vitamin and supplement supplier’s corporate website. Available online: http://www.beyondhealthnews.com/wpnews/index.php/2013/03/far-infrared-saunas-help-reverse-autism/ (accessed on 25 September 2019).
- Forbes Magazine, Issue November 5. 2012. Available online: https://www.forbes.com/sites/emilywillingham/2012/11/05/we-can-now-add-forced-sweating-to-the-faux-autism-treatment-list/#362969e0369f (accessed on 25 September 2019).
- Lin, M.; Zhao, D.; Hrabovsky, A.; Pedrosa, E.; Zheng, D.; Lachman, H.M. Heat shock alters the expression of schizophrenia and autism candidate genes in an induced pluripotent stem cell model of the human telencephalon. Plos ONE 2014, 9, e94968. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F.; Purpura, D.P. Autism, fever, epigenetics and the locus coeruleus. Brain Res. Rev. 2008, 59, 388–392. [Google Scholar] [CrossRef]
- Di Salvo, E.; Casciaro, M.; Quartuccio, S.; Genovese, L.; Gangemi, S. Do Alarmins Have a Potential Role in Autism Spectrum Disorders Pathogenesis and Progression? Biomolecules 2018, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Dammert, M.A.; Hoppe, S.; Bierhoff, H.; Grummt, I. Heat shock represses rRNA synthesis by inactivation of TIF-IA and lncRNA-dependent changes in nucleosome positioning. Nucleic Acids Res. 2016, 44, 8144–8152. [Google Scholar] [CrossRef]
- Hald, Ø.H.; Olsen, L.; Gallo-Oller, G.; Elfman, L.H.M.; Løkke, C.; Kogner, P.; Sveinbjörnsson, B.; Flægstad, T.; Johnsen, J.I.; Einvik, C. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F.M. The bobbed locus. In The Genetics and Biology of Drosophila; Ashburner, M., Novitski, E., Eds.; Academic Press: New York, NY, USA, 1976; pp. 801–846. [Google Scholar]
- Bridges, C.B. Non-disjunction as a proof of the chromosome theory of heredity. Genetics 1916, 1, 1–52. [Google Scholar] [PubMed]
- Ritossa, F.; Atwood, K.C.; Spiegelman, S. A molecular explanation of the bobbed mutants of Drosophila as partial deficiencies of “ribosomal” DNA. Genetics 1966, 54, 819–834. [Google Scholar] [PubMed]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosopila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Kazama, H. Systems neuroscience in Drosophila: Conceptual and technical advantages. Neuroscience 2015, 296, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Maximino, C.; Silva, R.X.; da Silva, S.N.; Rodrigues, L.S.; Barbosa, H.; de Carvalho, T.S.; Leão, L.K.; Lima, M.G.; Oliveira, K.R.; Herculano, A.M. Non-mammalian models in behavioral neuroscience: Consequences for biological psychiatry. Front. Behav. Neurosci. 2015, 9, 233. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nichols, C.D. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharm. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; He, J.; Wang, S.; Wang, L.; Cheng, Y.; Zhong, Y. Inability to activate Rac1-dependent forgetting contributes to behavioral inflexibility in mutants of multiple autism-risk genes. Proc. Natl. Acad. Sci. USA 2016, 113, 7644–7649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Zhang, Z.C.; Han, J. Drosophila Studies on Autism Spectrum Disorders. Neurosci. Bull. 2017, 33, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Koemans, T.S.; Oppitz, C.; Donders, R.A.T.; van Bokhoven, H.; Schenck, A.; Keleman, K.; Kramer, J.M. Drosophila Courtship Conditioning As a Measure of Learning and Memory. J. Vis. Exp. 2017, 124, 55808. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H. Genetics of autism spectrum disorder: Current status and possible clinical applications. Exp. Neurobiol. 2015, 24, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Pramparo, T.; Pierce, K.; Lombardo, M.V.; Barnes, C.C.; Marinero, S.; Ahrens-Barbeau, C.; Murray, S.S.; Lopez, L.; Xu, R.; Courchesne, E. Prediction of autism by translation and immune/inflammation coexpressed genes in toddlers from pediatric community practice. JAMA Psychiatry 2015, 72, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Onore, C.; Yang, H.; Van de Water, J.; Ashwood, P. Dynamic Akt/mTOR signaling in children with autism spectrum disorder. Front. Pediatr. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR activation in autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sahin, M.; Sur, M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 2015, 350, aab3897. [Google Scholar] [CrossRef]
- Bou Khalil, R. Is there any place for macrolides in mood disorders? Med. Hypotheses 2012, 78, 86–87. [Google Scholar] [CrossRef]
- Kraushar, M.L.; Popovitchenko, T.; Volk, N.L.; Rasin, M.R. The frontier of RNA metamorphosis and ribosome signature in neocortical development. Int. J. Dev. Neurosci. 2016, 55, 131–139. [Google Scholar] [CrossRef] [Green Version]
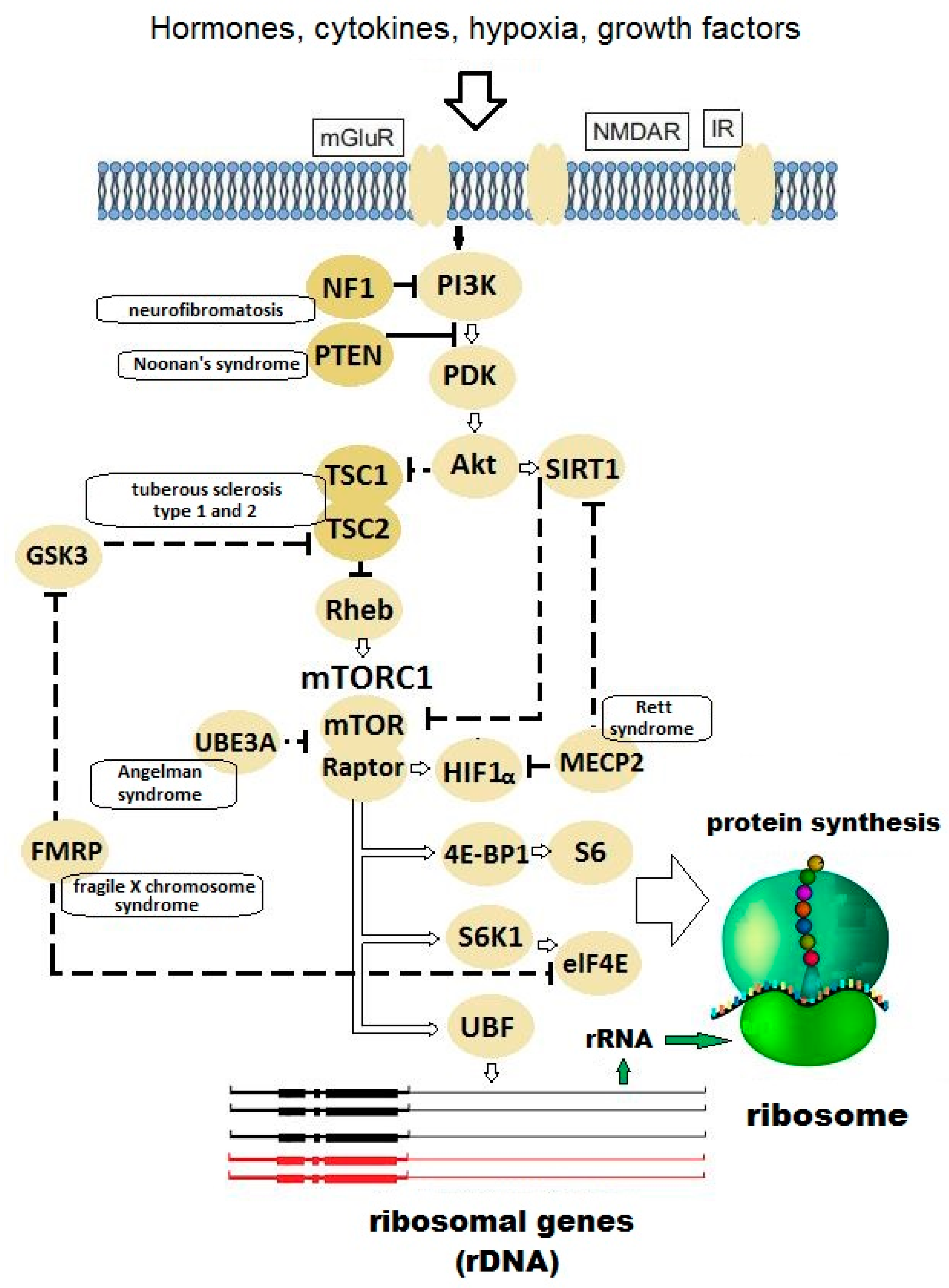

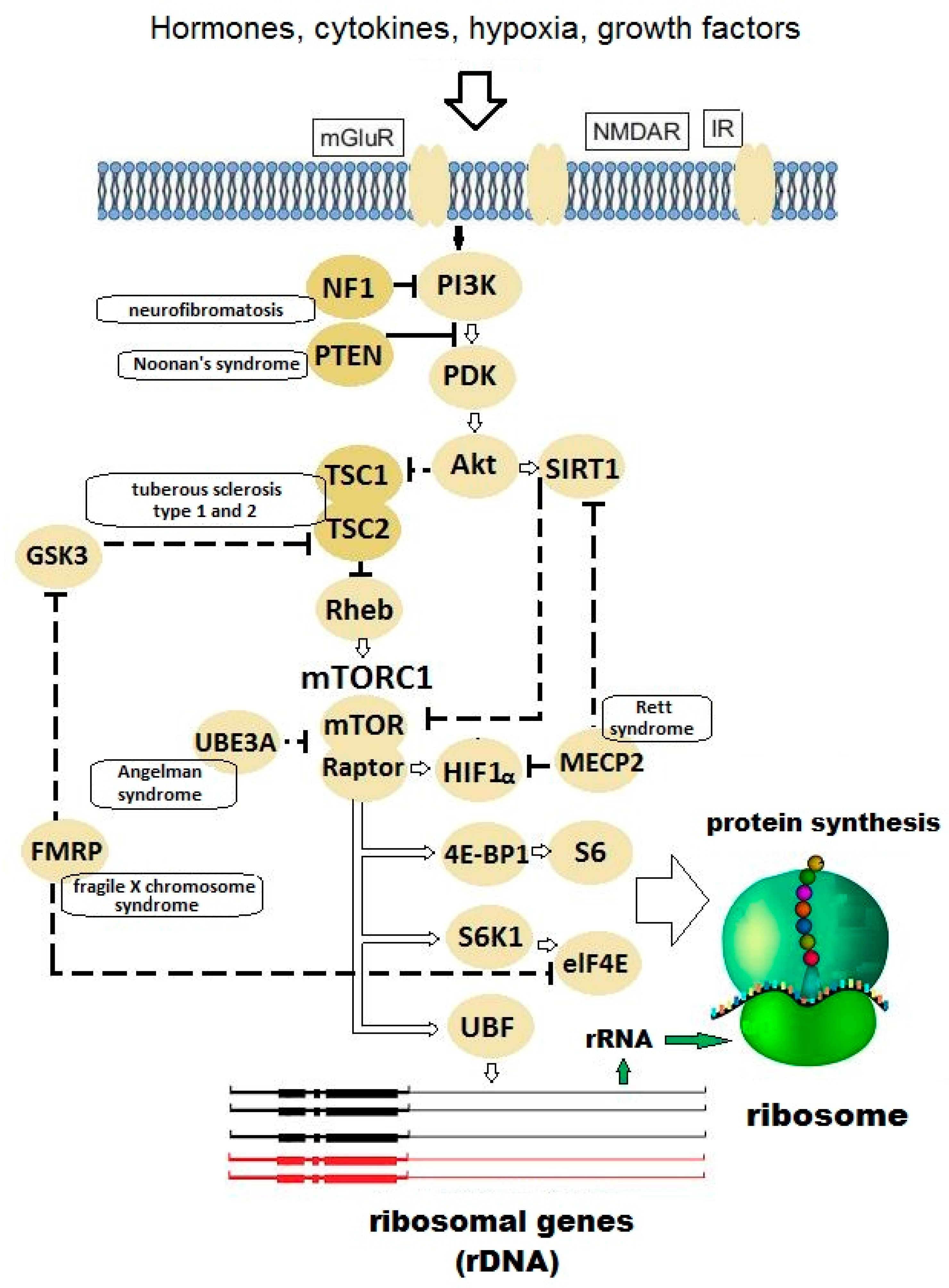{kind=link}
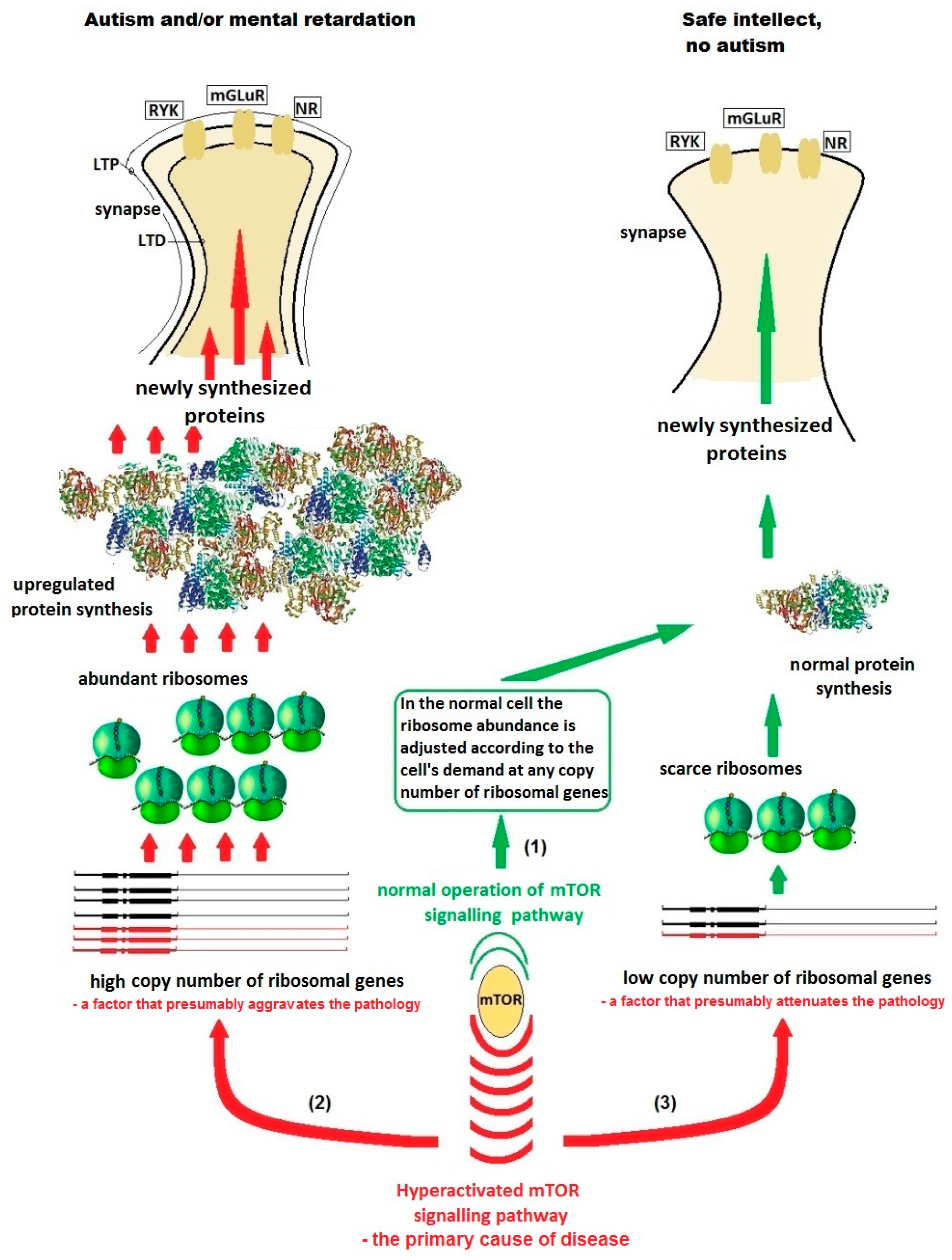
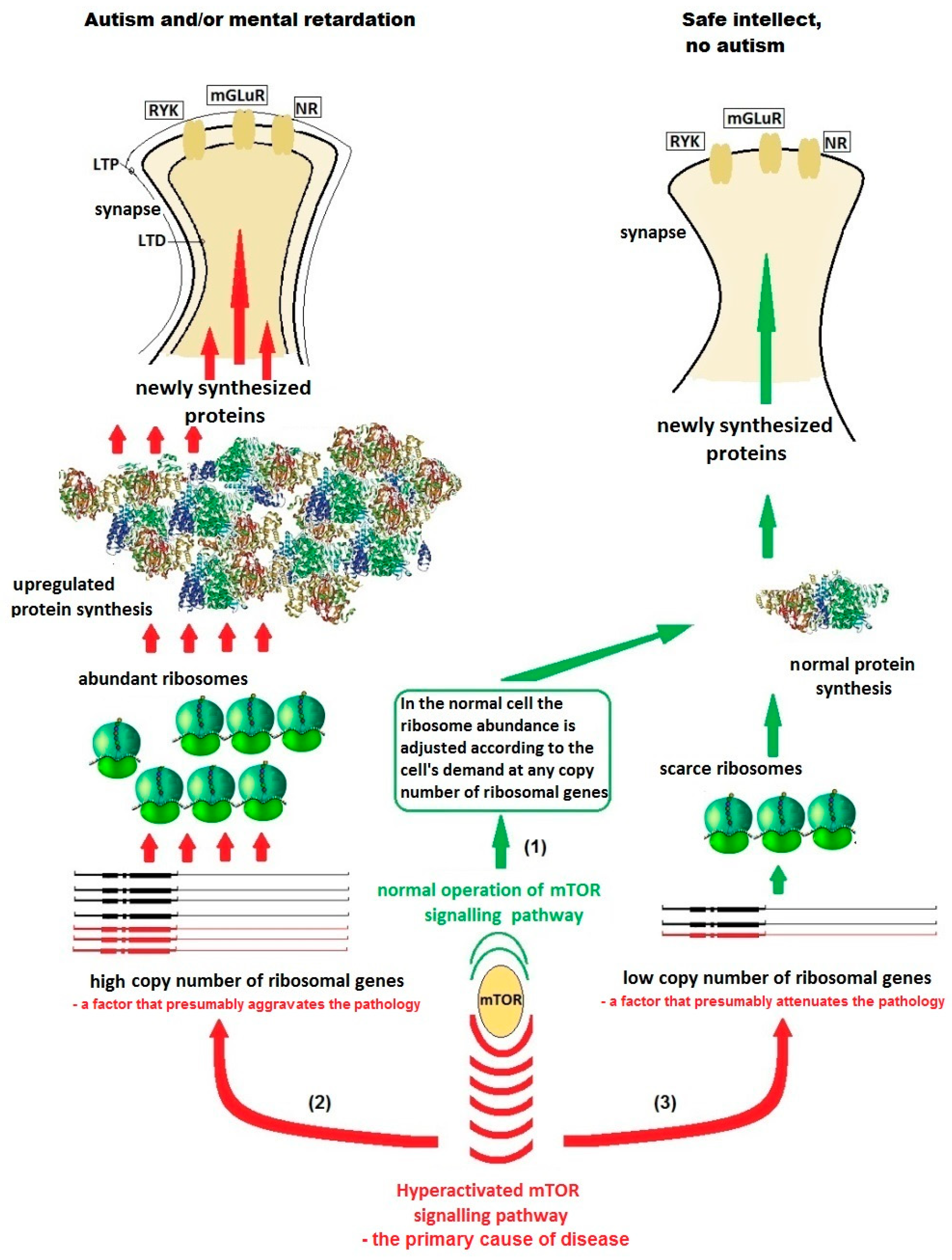{kind=link}
| Gene | Gene Function | Disease | Frequency of Autism in the Disease | Frequency of the Disease in All Autism Cases |
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 |
| FMR1 | Translation repressor | Fragile X syndrome | 15–30% | 2–5% |
| TSC1/2 | mTOR inhibitor | Tuberous sclerosis 1/2 | 25–60% | 1–4% |
| PTEN | PI3K/mTOR signaling pathway inhibitor | Cowden syndrome | No data | 1% |
| NF1 | Ras-GTPase activating protein | Neurofibromatosis 1 | 4% | 0–4% |
| MECP2 | Global translation repressor | Rett syndrome | 100% | 2% |
| UBE3A | E3 ubiquitin ligase | Angelman syndrome | 40% | 1% |
| NLGN3/4 | Synaptic adhesion | Familial ASD | No data | <1% |
| NRXN1 | Synaptic adhesion | Familial ASD | No data | <1% |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porokhovnik, L. Individual Copy Number of Ribosomal Genes as a Factor of Mental Retardation and Autism Risk and Severity. Cells 2019, 8, 1151. https://doi.org/10.3390/cells8101151
Porokhovnik L. Individual Copy Number of Ribosomal Genes as a Factor of Mental Retardation and Autism Risk and Severity. Cells. 2019; 8(10):1151. https://doi.org/10.3390/cells8101151
Chicago/Turabian StylePorokhovnik, Lev. 2019. "Individual Copy Number of Ribosomal Genes as a Factor of Mental Retardation and Autism Risk and Severity" Cells 8, no. 10: 1151. https://doi.org/10.3390/cells8101151
APA StylePorokhovnik, L. (2019). Individual Copy Number of Ribosomal Genes as a Factor of Mental Retardation and Autism Risk and Severity. Cells, 8(10), 1151. https://doi.org/10.3390/cells8101151