Mesenchymal Stem Cells in the Adult Human Liver: Hype or Hope?

 , ,
, , 
Abstract
1. Introduction
2. Methods of Isolation of Human Liver MSCs
3. Morphology and Phenotype of Human Liver MSCs
3.1. Mesenchymal Stem/Stromal Cell Markers
3.2. Hematopoietic/Endothelial Cell Markers
3.3. Pluripotency Markers
3.4. Integrins and Adhesion Molecules
3.5. Cytokeratins and Hepatic Markers
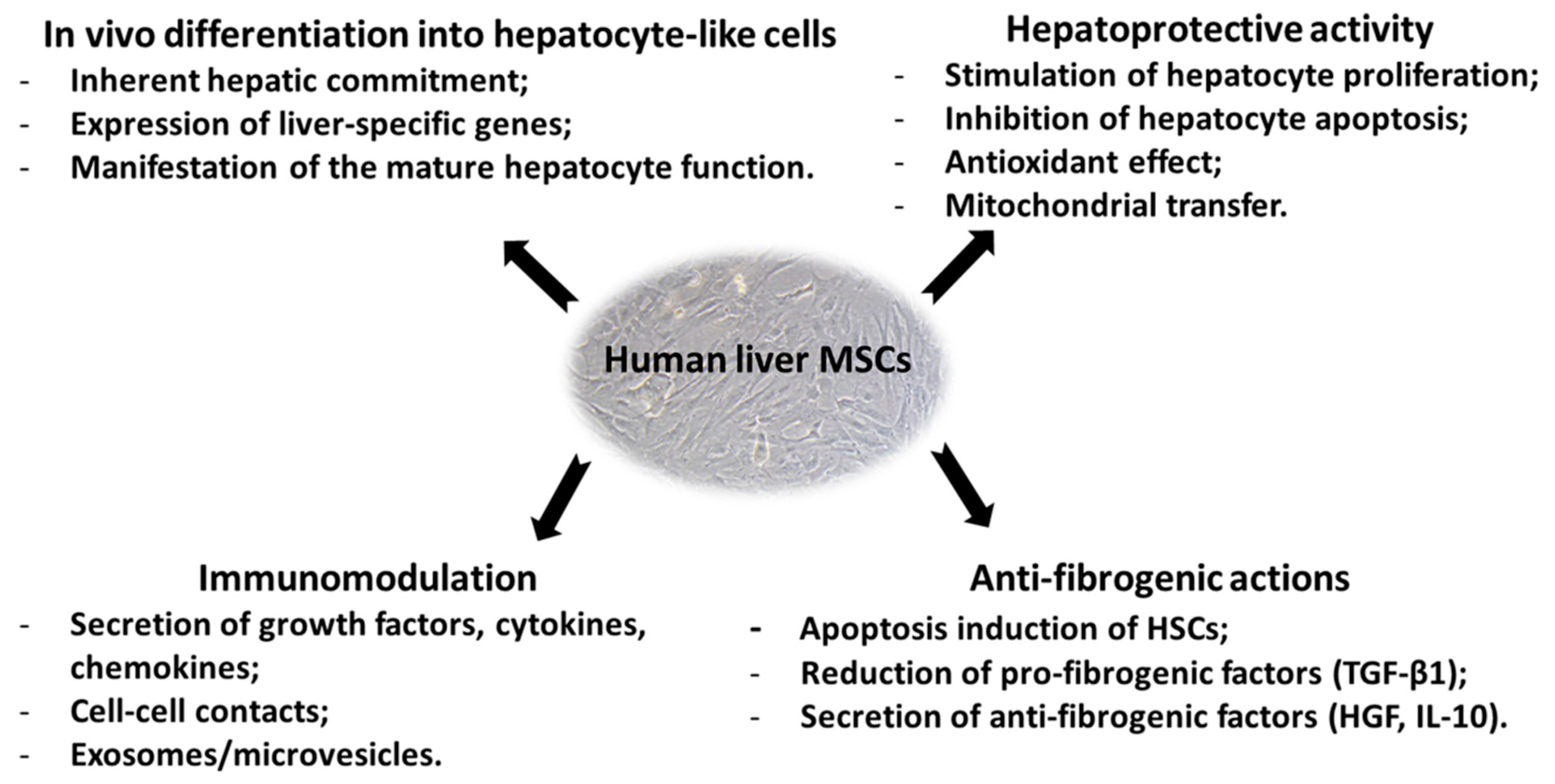
4. Properties and Therapeutic Potential of Human Liver MSCs
4.1. In Vitro and In Vivo Differentiation Potential
4.2. Immunomodulation Properties of Liver MSCs
4.3. Therapeutic Effects of Microvesicles/Exosomes and Conditioned Media Derived from Cultured Liver MSCs
4.4. Biodistribution of Liver MSCs after Transplantation
4.5. Therapeutic Effects of Liver MSCs
4.6. Clinical Trials of Human Liver MSCs
5. The Origin of Mesenchymal Stem Cells in the Liver
6. Conclusions
Funding
Conflicts of Interest
References
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [PubMed]
- Friedenstein, A.J. Stromal mechanisms of bone marrow: Cloning in vitro and retransplantation in vivo. Haematol. Blood Transfus. 1980, 25, 19–29. [Google Scholar] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Suva, D.; Garavaglia, G.; Menetrey, J.; Chapuis, B.; Hoffmeyer, P.; Bernheim, L.; Kindler, V. Non-hematopoietic human bone marrow contains long-lasting, pluripotential mesenchymal stem cells. J. Cell Physiol. 2004, 198, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. The mesengenic process. Clin. Plast. Surg. 1994, 21, 429–435. [Google Scholar] [PubMed]
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Further proof of the plasticity of adult stem cells and their role in tissue repair. J. Cell. Biol. 2003, 160, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Bertani, N.; Malatesta, P.; Volpi, G.; Sonego, P.; Perris, R. Neurogenic potential of human mesenchymal stem cells revisited: Analysis by immunostaining, time-lapse video and microarray. J. Cell Sci. 2005, 118, 3925–3936. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef]
- Bosch, J.; Houben, A.P.; Radke, T.F.; Stapelkamp, D.; Bünemann, E.; Balan, P.; Buchheiser, A.; Liedtke, S.; Kögler, G. Distinct differentiation potential of “MSC” derived from cord blood and umbilical cord: Are cord-derived cells true mesenchymal stromal cells? Stem Cells Dev. 2012, 21, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Rep. 2016, 6, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Baki, J.A.; Tapper, E.B. Contemporary Epidemiology of Cirrhosis. Curr. Treat Options Gastroenterol. 2019, 17, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Matas, A.J.; Sutherland, D.E.; Steffes, M.W.; Mauer, S.M.; Sowe, A.; Simmons, R.L.; Najarian, J.S. Hepatocellular transplantation for metabolic deficiencies: Decrease of plasma bilirubin in Gunn rats. Science 1976, 192, 892–894. [Google Scholar] [CrossRef]
- Iansante, V.; Mitry, R.R.; Filippi, C.; Fitzpatrick, E.; Dhawan, A. Human hepatocyte transplantation for liver disease: Current status and future perspectives. Pediatr. Res. 2018, 83, 232–240. [Google Scholar] [CrossRef]
- Wang, L.J.; Chen, Y.M.; George, D.; Smets, F.; Sokal, E.M.; Bremer, E.G.; Soriano, H.E. Engraftment assessment in human and mouse liver tissue after sex-mismatched liver cell transplantation by real-time quantitative PCR for Y chromosome sequences. Liver Transpl. 2002, 8, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, Z.; Xu, R.; Lin, H.; Fu, J.; Zou, Z.; Zhang, A.; Shi, J.; Chen, L.; Lv, S.; et al. Human mesenchymal stem cell transfusion is safe and improves liver function in acute-on-chronic liver failure patients. Stem Cells Transl. Med. 2012, 1, 725–731. [Google Scholar] [CrossRef]
- El-Ansary, M.; Abdel-Aziz, I.; Mogawer, S.; Abdel-Hamid, S.; Hammam, O.; Teaema, S.; Wahdan, M. Phase II trial: Undifferentiated versus differentiated autologous mesenchymal stem cells transplantation in Egyptian patients with HCV induced liver cirrhosis. Stem Cell Rev. 2012, 8, 972–981. [Google Scholar] [CrossRef]
- Najimi, M.; Khuu, D.N.; Lysy, P.A.; Jazouli, N.; Abarca, J.; Sempoux, C.; Sokal, E.M. Adult-derived human liver mesenchymal-like cells as a potential progenitor reservoir of hepatocytes? Cell Transpl. 2007, 16, 717–728. [Google Scholar] [CrossRef]
- Herrera, M.B.; Bruno, S.; Buttiglieri, S.; Tetta, C.; Gatti, S.; Deregibus, M.C.; Bussolati, B.; Camussi, G. Isolation and characterization of a stem cell population from adult human liver. Stem Cells 2006, 24, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Fouraschen, S.M.; Kaya, F.S.; Verstegen, M.M.; Pescatori, M.; Stubbs, A.P.; van Ijcken, W.; van der Sloot, A.; Smits, R.; Kwekkeboom, J.; et al. Mobilization of hepatic mesenchymal stem cells from human liver grafts. Liver Transpl. 2011, 17, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.B.; Fonsato, V.; Gatti, S.; Deregibus, M.C.; Sordi, A.; Cantarella, D.; Calogero, R.; Bussolati, B.; Tetta, C.; Camussi, G. Human liver stem cell-derived microvesicles accelerate hepatic regeneration in hepatectomized rats. J. Cell Mol. Med. 2010, 14, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Demirkiran, A.; Bosma, B.M.; Kok, A.; Baan, C.C.; Metselaar, H.J.; Ijzermans, J.N.; Tilanus, H.W.; Kwekkeboom, J.; van der Laan, L.J. Allosuppressive donor CD4+CD25+ regulatory T cells detach from the graft and circulate in recipients after liver transplantation. J. Immunol. 2007, 178, 6066–6072. [Google Scholar] [CrossRef] [PubMed]
- Bosma, B.M.; Metselaar, H.J.; Mancham, S.; Boor, P.P.; Kusters, J.G.; Kazemier, G.; Tilanus, H.W.; Kuipers, E.J.; Kwekkeboom, J. Characterization of human liver dendritic cells in liver grafts and perfusates. Liver Transpl. 2006, 12, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Aerts-Kaya, F.S.F.; Kazemier, G.; Kwekkeboom, J.; Metselaar, H.J.; Tilanus, H.W.; Janssen, H.L.A.; Wagemaker, G.; van der Laan, L.J.W. Human liver hematopoietic stem cells have a multi-potent and self-renewal capacity and may function as local progenitors for liver resident leukocytes. The International Liver Transplantation Society: 15th Annual International Congress. Liver Transpl. 2009, 15, S99. [Google Scholar]
- Lee, J.H.; Park, H.J.; Jang, I.K.; Kim, H.E.; Lee, D.H.; Park, J.K.; Lee, S.K.; Yoon, H.H. In vitro differentiation of human liver-derived stem cells with mesenchymal characteristics into immature hepatocyte-like cells. Transplant. Proc. 2014, 46, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, H.J.; Kim, Y.A.; Lee, D.H.; Noh, J.K.; Kwon, C.H.; Jung, S.M.; Lee, S.K. The phenotypic characteristic of liver-derived stem cells from adult human deceased donor liver. Transplant. Proc. 2012, 44, 1110–1112. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, I.V.; Kholodenko, R.V.; Manukyan, G.V.; Burunova, V.V.; Yarygin, K.N. Mesenchymal-Epithelial Transition in Culture of Stromal Progenitor Cells Isolated from the Liver of a Patient with Alcoholic Cirrhosis. Bull. Exp. Biol. Med. 2016, 162, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Kellner, J.; Sivajothi, S.; McNiece, I. Differential properties of human stromal cells from bone marrow, adipose, liver and cardiac tissues. Cytotherapy 2015, 17, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.C.; Over, P.; Turner, M.E.; Thompson, R.L.; Foka, H.G.; Chen, W.C.; Péault, B.; Gridelli, B.; Schmelzer, E. Perivascular mesenchymal progenitors in human fetal and adult liver. Stem Cells Dev. 2012, 21, 3258–3269. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, A.; Abdelsamed, M.A.; Abdelrahman, A.; Nabil, A.; Fatah, A.A.; Hozayen, W.; Shiha, G. Markers for the Characterization of Liver Mesenchymal Stem Cell. Int. J. Cell Sci. Mol. Biol. 2019, 6, 001–006. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Kholodenko, R.V.; Manukyan, G.V.; Yarygin, K.N. The hepatic differentiation of adult and fetal liver stromal cells in vitro. Biomed. Khim. 2016, 62, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Cesselli, D.; Bergamin, N.; Marcon, P.; Rigo, S.; Puppato, E.; D’Aurizio, F.; Verardo, R.; Piazza, S.; Pignatelli, A.; et al. Multipotent cells can be generated in vitro from several adult human organs (heart, liver, and bone marrow). Blood 2007, 110, 3438–3446. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, Dj.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Dollet, P.E.; Ravau, J.; André, F.; Najimi, M.; Sokal, E.; Lombard, C. Comprehensive Screening of Cell Surface Markers Expressed by Adult-Derived Human Liver Stem/Progenitor Cells Harvested at Passage 5: Potential Implications for Engraftment. Stem Cells Int. 2016, 2016, 9302537. [Google Scholar] [CrossRef] [PubMed]
- Berardis, S.; Lombard, C.; Evraerts, J.; El Taghdouini, A.; Rosseels, V.; Sancho-Bru, P.; Lozano, J.J.; van Grunsven, L.; Sokal, E.; Najimi, M. Gene expression profiling and secretome analysis differentiate adult-derived human liver stem/progenitor cells and human hepatic stellate cells. PLoS ONE 2014, 9, e86137. [Google Scholar] [CrossRef]
- Li, J.; Xin, J.; Zhang, L.; Wu, J.; Jiang, L.; Zhou, Q.; Li, J.; Guo, J.; Cao, H.; Li, L. Human hepatic progenitor cells express hematopoietic cell markers CD45 and CD109. Int. J. Med. Sci. 2013, 11, 65–79. [Google Scholar] [CrossRef]
- Young, H.E.; Steele, T.A.; Bray, R.A.; Detmer, K.; Blake, L.W.; Lucas, P.W.; Black, A.C., Jr. Human pluripotent and progenitor cells display cell surface cluster differentiation markers CD10, CD13, CD56, and MHC class-I. Proc. Soc. Exp. Biol. Med. 1999, 221, 63–71. [Google Scholar] [CrossRef]
- Musina, R.A.; Bekchanova, E.S.; Sukhikh, G.T. Comparison of mesenchymal stem cells obtained from different human tissues. Bull. Exp. Biol. Med. 2005, 139, 504–509. [Google Scholar] [CrossRef]
- Petrovic, N.; Schacke, W.; Gahagan, J.R.; O’Conor, C.A.; Winnicka, B.; Conway, R.E.; Mina-Osorio, P.; Shapiro, L.H. CD13/APN regulates endothelial invasion and filopodia formation. Blood 2007, 110, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; McAuliffe, B.; Subramani, J.; Basu, S.; Shapiro, L.H. CD13 regulates dendritic cell cross-presentation and T cell responses by inhibiting receptor-mediated antigen uptake. J. Immunol. 2012, 188, 5489–5499. [Google Scholar] [CrossRef] [PubMed]
- Pazhanisamy, S. Stem Cell Markers. Mater. Methods 2013, 3. [Google Scholar] [CrossRef]
- Calloni, R.; Cordero, E.A.; Henriques, J.A.; Bonatto, D. Reviewing and updating the major molecular markers for stem cells. Stem Cells Dev. 2013, 22, 1455–1476. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Subramani, J.; Ghosh, M.; Denninger, J.K.; Takeda, K.; Fong, G.H.; Carlson, M.E.; Shapiro, L.H. CD13 promotes mesenchymal stem cell-mediated regeneration of ischemic muscle. Front. Physiol. 2014, 4, 402. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Petrovic, N.; Okamoto, Y.; Shapiro, L.H. The angiogenic regulator CD13/APN is a transcriptional target of Ras signaling pathways in endothelial morphogenesis. Blood 2003, 101, 1818–1826. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, I.V.; Konieva, A.A.; Kholodenko, R.V.; Yarygin, K.N. Molecular mechanisms of migration and homing of intravenously transplanted mesenchymal stem cells. J. Regen. Med. Tissue Eng. 2013, 2. [Google Scholar] [CrossRef]
- Konieva, A.A.; Kholodenko, I.V.; Shragina, O.A.; Kholodenko, R.V.; Burunova, V.V.; Bibaeva, L.V.; Yarygin, K.N.; Yarygin, V.N. Functional properties of mesenchymal stem cells labeled with magnetic microparticles in vitro and analysis of their distribution after transplantation. Bull. Exp. Biol. Med. 2010, 150, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, V.; Garg, A.; Davies, N.; Bartlett, D.C.; Youster, J.; Beard, H.; Kavanagh, D.P.; Kalia, N.; Frampton, J.; Lalor, P.F.; et al. Human mesenchymal stem cells are recruited to injured liver in a β1-integrin and CD44 dependent manner. Hepatology 2012, 56, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; McAvoy, E.F.; Lam, F.; Gill, V.; de la Motte, C.; Savani, R.C.; Kubes, P. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 2008, 205, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bennett, S.A.; Wang, L. Role of E-cadherin and other cell adhesion molecules in survival and differentiation of human pluripotent stem cells. Cell Adh. Migr. 2012, 6, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Niessen, C.M.; Leckband, D.; Yap, A.S. Tissue organization by cadherin adhesion molecules: Dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev. 2011, 91, 691–731. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Westbroek, W.; Verloes, A.; Bloemen, N.; Bracke, M.; Gespach, C.; Bruyneel, E.; Mareel, M. Critical role of N-cadherin in myofibroblast invasion and migration in vitro stimulated by colon-cancer-cell-derived TGF-β or wounding. J. Cell Sci. 2004, 117, 4691–4703. [Google Scholar] [CrossRef] [PubMed]
- Akitaya, T.; Bronner-Fraser, M. Expression of cell adhesion molecules during initiation and cessation of neural crest cell migration. Dev. Dyn. 1992, 194, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Radice, G.L.; Rayburn, H.; Matsunami, H.; Knudsen, K.A.; Takeichi, M.; Hynes, R.O. Developmental defects in mouse embryos lacking N-cadherin. Dev. Biol. 1997, 181, 64–78. [Google Scholar] [CrossRef] [PubMed]
- García-Castro, M.I.; Vielmetter, E.; Bronner-Fraser, M. N-Cadherin, a cell adhesion molecule involved in establishment of embryonic left-right asymmetry. Science 2000, 288, 1047–1051. [Google Scholar] [CrossRef]
- Kashima, T.; Nakamura, K.; Kawaguchi, J.; Takanashi, M.; Ishida, T.; Aburatani, H.; Kudo, A.; Fukayama, M.; Grigoriadis, A.E. Overexpression of cadherins suppresses pulmonary metastasis of osteosarcoma in vivo. Int. J. Cancer 2003, 104, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnie, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.S.; Cole, F.; Gaio, U.; Takaesu, G.; Zhang, W.; Kang, J.S. Close encounters: Regulation of vertebrate skeletal myogenesis by cell–cell contact. J. Cell Sci. 2005, 118, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, K.; Arai, F.; Yoshihara, H.; Iwasaki, H.; Nakamura, Y.; Gomei, Y.; Suda, T. Knockdown of N-cadherin suppresses the long-term engraftment of hematopoietic stem cells. Blood 2010, 116, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Matsumoto, Y.; Fukushi, J.; Matsunobu, T.; Endo, M.; Okada, S.; Iura, K.; Kamura, S.; Fujiwara, T.; Iida, K.; et al. Cadherin-11 regulates the metastasis of Ewing sarcoma cells to bone. Clin. Exp. Metastasis 2015, 32, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Row, S.; Liu, Y.; Alimperti, S.; Agarwal, S.K.; Andreadis, S.T. Cadherin-11 is a novel regulator of extracellular matrix synthesis and tissue mechanics. J. Cell Sci. 2016, 129, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Madarampalli, B.; Watts, G.F.M.; Panipinto, P.M.; Nguygen, H.N.; Brenner, M.B.; Noss, E.H. Interactions between cadherin-11 and platelet-derived growth factor receptor-alpha signaling link cell adhesion and proliferation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1516–1524. [Google Scholar] [CrossRef]
- Hinz, B.; Pittet, P.; Smith-Clerc, J.; Chaponnier, C.; Meister, J.J. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol. Biol. Cell 2004, 15, 4310–4320. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, A.; Watkins, M.; Grimston, S.; Salazar, V.; Donsante, C.; Mbalaviele, G.; Radice, G.L.; Civitelli, R. N-cadherin and cadherin 11 modulate postnatal bone growth and osteoblast differentiation by distinct mechanisms. J. Cell Sci. 2010, 123, 2640–2648. [Google Scholar] [CrossRef]
- Alimperti, S.; You, H.; George, T.; Agarwal, S.K.; Andreadis, S.T. Cadherin-11 regulates both mesenchymal stem cell differentiation into smooth muscle cells and the development of contractile function in vivo. J. Cell Sci. 2014, 127, 2627–2638. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Agarwal, S.K. Integrins and cadherins as therapeutic targets in fibrosis. Front. Pharmacol. 2014, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Fonsato, V.; Herrera, M.B.; Buttiglieri, S.; Gatti, S.; Camussi, G.; Tetta, C. Use of a rotary bioartificial liver in the differentiation of human liver stem cells. Tissue Eng. Part C Methods 2010, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, H.J.; Kim, Y.A.; Lee, D.H.; Noh, J.K.; Kwon, C.H.; Jung, S.M.; Lee, S.K. Differentiation and major histocompatibility complex antigen expression in human liver-derived stem cells. Transplant. Proc. 2012, 44, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Rolandsson Enes, S.; Åhrman, E.; Palani, A.; Hallgren, O.; Bjermer, L.; Malmström, A.; Scheding, S.; Malmström, J.; Westergren-Thorsson, G. Quantitative proteomic characterization of lung-MSC and bone marrow-MSC using DIA-mass spectrometry. Sci. Rep. 2017, 7, 9316. [Google Scholar] [CrossRef] [PubMed]
- Campard, D.; Lysy, P.A.; Najimi, M.; Sokal, E.M. Native umbilical cord matrix stem cells express hepatic markers and differentiate into hepatocyte-like cells. Gastroenterology 2008, 134, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Khodabandeh, Z.; Vojdani, Z.; Talaei-Khozani, T.; Jaberipour, M.; Hosseini, A.; Bahmanpour, S. Comparison of the Expression of Hepatic Genes by Human Wharton’s Jelly Mesenchymal Stem Cells Cultured in 2D and 3D Collagen Culture Systems. Iran J. Med. Sci. 2016, 41, 28–36. [Google Scholar]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar]
- Kannt, A.; Pfenninger, A.; Teichert, L.; Tönjes, A.; Dietrich, A.; Schön, M.R.; Klöting, N.; Blüher, M. Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia 2015, 58, 799–808. [Google Scholar] [CrossRef]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef]
- Sartini, D.; Muzzonigro, G.; Milanese, G.; Pierella, F.; Rossi, V.; Emanuelli, M. Identification of nicotinamide N-methyltransferase as a novel tumor marker for renal clear cell carcinoma. J. Urol. 2006, 176, 2248–2254. [Google Scholar] [CrossRef]
- Roessler, M.; Rollinger, W.; Palme, S.; Hagmann, M.L.; Berndt, P.; Engel, A.M.; Schneidinger, B.; Pfeffer, M.; Andres, H.; Karl, J.; et al. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin. Cancer Res. 2005, 11, 6550–6557. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.H.; Cho, B.I.; Kim, Y.N.; Kim, J.W.; Park, S.T.; Lee, C.W. Overexpression of nicotinamide N-methyltransferase in gastric cancer tissues and its potential post-translational modification. Exp. Mol. Med. 2006, 38, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Hosseini, M.; Liu, X.; Kisseleva, T.; Brenner, D.A. Human hepatic stellate cell isolation and characterization. J. Gastroenterol. 2018, 53, 6–17. [Google Scholar] [CrossRef] [PubMed]
- El-Kehdy, H.; Sargiacomo, C.; Fayyad-Kazan, M.; Fayyad-Kazan, H.; Lombard, C.; Lagneaux, L.; Sokal, E.; Najar, M.; Najimi, M. Immunoprofiling of Adult-Derived Human Liver Stem/Progenitor Cells: Impact of Hepatogenic Differentiation and Inflammation. Stem Cells Int. 2017, 2017, 2679518. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Müller-Marbach, A.; Ale-Agha, N.; Keitel, V.; Klonowski-Stumpe, H.; Häussinger, D. CD133+ hepatic stellate cells are progenitor cells. Biochem. Biophys. Res. Commun. 2007, 352, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Viñas, O.; Bataller, R.; Sancho-Bru, P.; Ginès, P.; Berenguer, C.; Enrich, C.; Nicolás, J.M.; Ercilla, G.; Gallart, T.; Vives, J.; et al. Human hepatic stellate cells show features of antigen-presenting cells and stimulate lymphocyte proliferation. Hepatology 2003, 38, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Amati, E.; Perbellini, O.; Rotta, G.; Bernardi, M.; Chieregato, K.; Sella, S.; Rodeghiero, F.; Ruggeri, M.; Astori, G. High-throughput immunophenotypic characterization of bone marrow- and cord blood-derived mesenchymal stromal cells reveals common and differentially expressed markers: Identification of angiotensin-converting enzyme (CD143) as a marker differentially expressed between adult and perinatal tissue sources. Stem Cell Res. Ther. 2018, 9, 10. [Google Scholar]
- McLeod, C.M.; Mauck, R.L. On the origin and impact of mesenchymal stem cell heterogeneity: New insights and emerging tools for single cell analysis. Eur. Cell Mater. 2017, 34, 217–231. [Google Scholar] [CrossRef]
- Han, Z.C.; Du, W.J.; Han, Z.B.; Liang, L. New insights into the heterogeneity and functional diversity of human mesenchymal stem cells. Biomed. Mater Eng. 2017, 28, S29–S45. [Google Scholar] [CrossRef]
- Seshi, B.; Kumar, S.; King, D. Multilineage gene expression in human bone marrow stromal cells as evidenced by single-cell microarray analysis. Blood Cells Mol. Dis. 2003, 31, 268–285. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Q.; Zhang, K.; Hu, M.; Wang, Y.; Du, L.; Lin, L.; Li, S.; Sorokin, L.; Melino, G.; et al. Single cell transcriptomic analysis of human mesenchymal stem cells reveals limited heterogeneity. Cell Death Dis. 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiang, Q.; Xu, F.; Huang, J.; Yu, N.; Zhang, Q.; Long, X.; Zhou, Z. Single-cell RNA-seq of cultured human adipose-derived mesenchymal stem cells. Sci. Data 2019, 6, 190031. [Google Scholar] [CrossRef] [PubMed]
- Porretti, L.; Cattaneo, A.; Colombo, F.; Lopa, R.; Rossi, G.; Mazzaferro, V.; Battiston, C.; Svegliati-Baroni, G.; Bertolini, F.; Rebulla, P.; et al. Simultaneous characterization of progenitor cell compartments in adult human liver. Cytometry A 2010, 77, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, I.V.; Kholodenko, R.V.; Manukyan, G.V.; Lupatov, A.Y.; Yarygin, K.N. Isolation of Induced Pluripotent Cells from Stromal Liver Cells of Patients with Alcoholic Cirrhosis. Bull. Exp. Biol. Med. 2017, 163, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Paganelli, M.; Nyabi, O.; Sid, B.; Evraerts, J.; El Malmi, I.; Heremans, Y.; Dollé, L.; Benton, C.; Calderon, P.B.; van Grunsven, L.; et al. Downregulation of Sox9 expression associates with hepatogenic differentiation of human liver mesenchymal stem/progenitor cells. Stem Cells Dev. 2014, 23, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Khuu, D.N.; Scheers, I.; Ehnert, S.; Jazouli, N.; Nyabi, O.; Buc-Calderon, P.; Meulemans, A.; Nussler, A.; Sokal, E.; Najimi, M. In vitro differentiated adult human liver progenitor cells display mature hepatic metabolic functions: A potential tool for in vitro pharmacotoxicological testing. Cell Transpl. 2011, 20, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Nyabi, O.; Najimi, M.; Fauvart, M.; Sokal, E. Adult human liver mesenchymal progenitor cells express phenylalanine hydroxylase. J. Pediatr. Endocrinol. Metab. 2014, 27, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Khuu, D.N.; Nyabi, O.; Maerckx, C.; Sokal, E.; Najimi, M. Adult human liver mesenchymal stem/progenitor cells participate in mouse liver regeneration after hepatectomy. Cell Transpl. 2013, 22, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Yarygin, K.N.; Lupatov, A.Y.; Sukhikh, G.T. Modulation of Immune Responses by Mesenchymal Stromal Cells. Bull. Exp. Biol. Med. 2016, 161, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yamamoto, Y.; Xiao, Z.; Ochiya, T. The Immunomodulatory Functions of Mesenchymal Stromal/Stem Cells Mediated via Paracrine Activity. J. Clin. Med. 2019, 8, 1025. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Minas, K.; Liversidge, J. Is the CD200/CD200 receptor interaction more than just a myeloid cell inhibitory signal? Crit. Rev. Immunol. 2006, 26, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.N.; Wright, G.J.; Brooke, G.; Brown, M.H. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002, 23, 285–290. [Google Scholar] [CrossRef]
- Moreaux, J.; Hose, D.; Reme, T.; Jourdan, E.; Hundemer, M.; Legouffe, E.; Moine, P.; Bourin, P.; Moos, M.; Corre, J.; et al. CD200 is a new prognostic factor in multiple myeloma. Blood 2006, 108, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, R.M. CD200:CD200R-Mediated Regulation of Immunity. ISRN Immunol. 2012, 2012. [Google Scholar] [CrossRef]
- Pontikoglou, C.; Langonné, A.; Ba, M.A.; Varin, A.; Rosset, P.; Charbord, P.; Sensébé, L.; Deschaseaux, F. CD200 expression in human cultured bone marrow mesenchymal stem cells is induced by pro-osteogenic and pro-inflammatory cues. J. Cell Mol. Med. 2016, 20, 655–665. [Google Scholar] [CrossRef]
- Pietilä, M.; Lehtonen, S.; Tuovinen, E.; Lähteenmäki, K.; Laitinen, S.; Leskelä, H.V.; Nätynki, A.; Pesälä, J.; Nordström, K.; Lehenkari, P. CD200 positive human mesenchymal stem cells suppress TNF-alpha secretion from CD200 receptor positive macrophage-like cells. PLoS ONE 2012, 7, e31671. [Google Scholar] [CrossRef]
- Taner, T.; Gustafson, M.; Hansen, M.; Dietz, A.; Stegall, M. Liver Mesenchymal Stem Cells Inhibit T Cell Alloresponses. Am. J. Transplant. 2017, 17, A36. [Google Scholar]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Mutt, S.J.; Karhu, T.; Lehtonen, S.; Lehenkari, P.; Carlberg, C.; Saarnio, J.; Sebert, S.; Hyppönen, E.; Järvelin, M.R.; Herzig, K.H. Inhibition of cytokine secretion from adipocytes by 1,25-dihydroxyvitamin D₃ via the NF-κB pathway. FASEB J. 2012, 26, 4400–4407. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liang, X.; Zhang, Y.; Yi, L.; Shum, H.C.; Chen, Q.; Chan, B.P.; Fan, H.; Liu, Z.; Tergaonkar, V.; et al. Rap1 deficiency-provoked paracrine dysfunction impairs immunosuppressive potency of mesenchymal stem cells in allograft rejection of heart transplantation. Cell Death Dis. 2018, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- de Mare-Bredemeijer, E.L.; Mancham, S.; Verstegen, M.M.; de Ruiter, P.E.; van Gent, R.; O’Neill, D.; Tilanus, H.W.; Metselaar, H.J.; de Jonge, J.; Kwekkeboom, J.; et al. Human graft-derived mesenchymal stromal cells potently suppress alloreactive T-cell responses. Stem Cells Dev. 2015, 24, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Raicevic, G.; Najar, M.; Najimi, M.; El Taghdouini, A.; van Grunsven, L.A.; Sokal, E.; Toungouz, M. Influence of inflammation on the immunological profile of adult-derived human liver mesenchymal stromal cells and stellate cells. Cytotherapy 2015, 17, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Raicevic, G.; Rouas, R.; Najar, M.; Stordeur, P.; Boufker, H.I.; Bron, D.; Martiat, P.; Goldman, M.; Nevessignsky, M.T.; Lagneaux, L. Inflammation modifies the pattern and the function of Toll-like receptors expressed by human mesenchymal stromal cells. Hum. Immunol. 2010, 71, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Crompot, E.; Raicevic, G.; Sokal, E.M.; Najimi, M.; Lagneaux, L. Cytokinome of adult-derived human liver stem/progenitor cells: Immunological and inflammatory features. Hepatobiliary Surg. Nutr. 2018, 7, 331–344. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Kholodenko, R.V.; Lupatov, A.Y.; Yarygin, K.N. Cell Therapy as a Tool for Induction of Immunological Tolerance after Liver Transplantation. Bull. Exp. Biol. Med. 2018, 165, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Cantaluppi, V. Role of stem-cell-derived microvesicles in the paracrine action of stem cells. Biochem. Soc. Trans. 2013, 41, 283–287. [Google Scholar] [CrossRef]
- Fonsato, V.; Collino, F.; Herrera, M.B.; Cavallari, C.; Deregibus, M.C.; Cisterna, B.; Bruno, S.; Romagnoli, R.; Salizzoni, M.; Tetta, C.; et al. Human liver stem cell-derived microvesicles inhibit hepatoma growth in SCID mice by delivering antitumor microRNAs. Stem Cells 2012, 30, 1985–1998. [Google Scholar] [CrossRef]
- Wu, J.; Qu, Z.; Fei, Z.W.; Wu, J.H.; Jiang, C.P. Role of stem cell-derived exosomes in cancer. Oncol. Lett. 2017, 13, 2855–2866. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, X.; Tan, Y.; Li, Q.; Ma, J.; Wang, G. Mesenchymal Stem Cell Derived Exosomes in Cancer Progression, Metastasis and Drug Delivery: A Comprehensive Review. J. Cancer 2018, 9, 3129–3137. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Deregibus, M.C.; Bruno, S.; Sterpone, L.; Aghemo, G.; Viltono, L.; Tetta, C.; Camussi, G. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS ONE 2010, 5, e11803. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, C.; Fonsato, V.; Herrera, M.B.; Bruno, S.; Tetta, C.; Camussi, G. Role of Lefty in the anti tumor activity of human adult liver stem cells. Oncogene 2013, 32, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, A.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Chae, Y.K.; Matsangou, M.; Hendrix, M.J.C.; Giles, F. Nodal Signaling as a Developmental Therapeutics Target in Oncology. Mol. Cancer Ther. 2017, 16, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Chen, Z.; Zheng, M.; Liu, Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp. Mol. Med. 2017, 49, e346. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38, S38–S53. [Google Scholar] [CrossRef]
- Altamirano-Barrera, A.; Barranco-Fragoso, B.; Méndez-Sánchez, N. Management strategies for liver fibrosis. Ann. Hepatol. 2017, 16, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Schon, H.T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Alfaifi, M.; Eom, Y.W.; Newsome, P.N.; Baik, S.K. Mesenchymal stromal cell therapy for liver diseases. J. Hepatol. 2018, 68, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.T.; Peng, X.B.; Fang, X.Q.; Li, J.F.; Chen, H.; Mao, X.R. Human umbilical cord mesenchymal stem cells inhibit proliferation of hepatic stellate cells in vitro. Int. J. Mol. Med. 2018, 41, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.O.; Jun, B.G.; Baik, S.K.; Kim, M.Y.; Kwon, S.O. Inhibition of hepatic stellate cells by bone marrow-derived mesenchymal stem cells in hepatic fibrosis. Clin. Mol. Hepatol. 2015, 21, 141–149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Najimi, M.; Berardis, S.; El-Kehdy, H.; Rosseels, V.; Evraerts, J.; Lombard, C.; El Taghdouini, A.; Henriet, P.; van Grunsven, L.; Sokal, E.M. Human liver mesenchymal stem/progenitor cells inhibit hepatic stellate cell activation: In vitro and in vivo evaluation. Stem Cell Res. Ther. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.B.; Fonsato, V.; Bruno, S.; Grange, C.; Gilbo, N.; Romagnoli, R.; Tetta, C.; Camussi, G. Human liver stem cells improve liver injury in a model of fulminant liver failure. Hepatology 2013, 57, 311–319. [Google Scholar] [CrossRef]
- Fouraschen, S.M.; Pan, Q.; de Ruiter, P.E.; Farid, W.R.; Kazemier, G.; Kwekkeboom, J.; Ijzermans, J.N.; Metselaar, H.J.; Tilanus, H.W.; de Jonge, J.; et al. Secreted factors of human liver-derived mesenchymal stem cells promote liver regeneration early after partial hepatectomy. Stem Cells Dev. 2012, 21, 2410–2419. [Google Scholar] [CrossRef]
- Maerckx, C.; Tondreau, T.; Berardis, S.; van Pelt, J.; Najimi, M.; Sokal, E. Human liver stem/progenitor cells decrease serum bilirubin in hyperbilirubinemic Gunn rat. World J. Gastroenterol. 2014, 20, 10553–10563. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef]
- Ayatollahi, M.; Hesami, Z.; Jamshidzadeh, A.; Gramizadeh, B. Antioxidant Effects of Bone Marrow Mesenchymal Stem Cell against Carbon Tetrachloride-Induced Oxidative Damage in Rat Livers. Int. J. Organ Transplant. Med. 2014, 5, 166–173. [Google Scholar]
- Jiang, W.; Tan, Y.; Cai, M.; Zhao, T.; Mao, F.; Zhang, X.; Xu, W.; Yan, Z.; Qian, H.; Yan, Y. Human Umbilical Cord MSC-Derived Exosomes Suppress the Development of CCl4-Induced Liver Injury through Antioxidant Effect. Stem Cells Int. 2018, 2018, 6079642. [Google Scholar] [CrossRef]
- Klein, D.; Steens, J.; Wiesemann, A.; Schulz, F.; Kaschani, F.; Röck, K.; Yamaguchi, M.; Wirsdörfer, F.; Kaiser, M.; Fischer, J.W.; et al. Mesenchymal Stem Cell Therapy Protects Lungs from Radiation-Induced Endothelial Cell Loss by Restoring Superoxide Dismutase 1 Expression. Antioxid. Redox Signal. 2017, 26, 563–582. [Google Scholar] [CrossRef]
- Nightingale, H.; Kemp, K.; Gray, E.; Hares, K.; Mallam, E.; Scolding, N.; Wilkins, A. Changes in expression of the antioxidant enzyme SOD3 occur upon differentiation of human bone marrow-derived mesenchymal stem cells in vitro. Stem Cells Dev. 2012, 21, 2026–2035. [Google Scholar] [CrossRef]
- Redondo, J.; Sarkar, P.; Kemp, K.; Heesom, K.J.; Wilkins, A.; Scolding, N.J.; Rice, C.M. Dysregulation of Mesenchymal Stromal Cell Antioxidant Responses in Progressive Multiple Sclerosis. Stem Cells Transl. Med. 2018, 7, 748–758. [Google Scholar] [CrossRef]
- Valle-Prieto, A.; Conget, P.A. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. 2010, 19, 1885–1893. [Google Scholar] [CrossRef]
- Qi, X.; Ng, K.T.; Lian, Q.; Li, C.X.; Geng, W.; Ling, C.C.; Yeung, W.H.; Ma, Y.Y.; Liu, X.B.; Liu, H.; et al. Glutathione Peroxidase 3 Delivered by hiPSC-MSCs Ameliorated Hepatic IR Injury via Inhibition of Hepatic Senescence. Theranostics 2018, 8, 212–222. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Gao, F.; Han, S.; Cheah, K.S.; Tse, H.F.; Lian, Q. CRISPR/Cas9 Genome-Editing System in Human Stem Cells: Current Status and Future Prospects. Mol. Ther. Nucleic Acids 2017, 9, 230–241. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef]
- Li, C.; Cheung, M.K.H.; Han, S.; Zhang, Z.; Chen, L.; Chen, J.; Zeng, H.; Qiu, J. Mesenchymal stem cells and their mitochondrial transfer: A double-edged sword. Biosci. Rep. 2019, 39, BSR20182417. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; McConnell, M.J.; Grasso, C.; Bajzikova, M.; Kovarova, J.; Neuzil, J. Horizontal transfer of mitochondria between mammalian cells: Beyond co-culture approaches. Curr. Opin. Genet. Dev. 2016, 38, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Scheers, I.; Maerckx, C.; Khuu, D.N.; Marcelle, S.; Decottignies, A.; Najimi, M.; Sokal, E. Adult-derived human liver progenitor cells in long-term culture maintain appropriate gatekeeper mechanisms against transformation. Cell Transplant. 2012, 21, 2241–2255. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M. Treating inborn errors of liver metabolism with stem cells: Current clinical development. J. Inherit. Metab. Dis. 2014, 37, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Stéphenne, X.; Ottolenghi, C.; Jazouli, N.; Clapuyt, P.; Lacaille, F.; Najimi, M.; de Lonlay, P.; Smets, F. Liver engraftment and repopulation by in vitro expanded adult derived human liver stem cells in a child with ornithine carbamoyltransferase deficiency. JIMD Rep. 2014, 13, 65–72. [Google Scholar]
- Defresne, F.; Tondreau, T.; Stéphenne, X.; Smets, F.; Bourgois, A.; Najimi, M.; Jamar, F.; Sokal, E.M. Biodistribution of adult derived human liver stem cells following intraportal infusion in a 17-year-old patient with glycogenosis type 1A. Nucl. Med. Biol. 2014, 41, 371–375. [Google Scholar] [CrossRef]
- Sokal, E.M.; Lombard, C.A.; Roelants, V.; Najimi, M.; Varma, S.; Sargiacomo, C.; Ravau, J.; Mazza, G.; Jamar, F.; Versavau, J.; et al. Biodistribution of Liver-Derived Mesenchymal Stem Cells After Peripheral Injection in a Hemophilia A Patient. Transplantation 2017, 101, 1845–1851. [Google Scholar] [CrossRef]
- Smets, F.; Dobbelaere, D.; McKiernan, P.; Dionisi-Vici, C.; Broué, P.; Jacquemin, E.; Lopes, A.I.; Gonçalves, I.; Mandel, H.; Pawlowska, J.; et al. Phase I/II Trial of Liver Derived Mesenchymal Stem Cells in Pediatric Liver Based Metabolic Disorders: A Prospective, Open Label, Multicenter, Partially Randomized, Safety Study of One Cycle of Heterologous Human Adult Liver-Derived Progenitor Cells (HepaStem®) in Urea Cycle Disorders and Crigler-Najjar Syndrome patients. Transplantation 2019. [Google Scholar] [CrossRef]
- Moll, G.; Ankrum, J.A.; Kamhieh-Milz, J.; Bieback, K.; Ringden, O.; Volk, H.D.; Geissler, S.; Reinke, P. Intravascular Mesenchymal Stromal/Stem Cell Therapy Product Diversification: Time for New Clinical Guidelines. Trends Mol. Med. 2019, 25, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, S.; Zhou, L.; Cai, J.; Tan, J.; Gao, X.; Zeng, Z.; Li, D. Thromboembolism Induced by Umbilical Cord Mesenchymal Stem Cell Infusion: A Report of Two Cases and Literature Review. Transplant. Proc. 2017, 49, 1656–1658. [Google Scholar] [CrossRef] [PubMed]
- Moll, G.; Ignatowicz, L.; Catar, R.; Luecht, C.; Sadeghi, B.; Hamad, O.; Jungebluth, P.; Dragun, D.; Schmidtchen, A.; Ringden, O. Dierent Procoagulant Activity of Therapeutic Mesenchymal Stromal Cells Derived from Bone Marrow and Placental Decidua. Stem Cells Dev. 2015, 24, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Christy, B.A.; Herzig, M.C.; Montgomery, R.K.; Delavan, C.; Bynum, J.A.; Reddoch, K.M.; Cap, A.P. Procoagulant activity of human mesenchymal stem cells. J. Trauma Acute Care Surg. 2017, 83, s164–s169. [Google Scholar] [CrossRef] [PubMed]
- Stephenne, X.; Vosters, O.; Najimi, M.; Beuneu, C.; Dung, K.N.; Wijns, W.; Goldman, M.; Sokal, E.M. Tissue factor-dependent procoagulant activity of isolated human hepatocytes: Relevance to liver cell transplantation. Liver Transplant. O. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2007, 13, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Stephenne, X.; Nicastro, E.; Eeckhoudt, S.; Hermans, C.; Nyabi, O.; Lombard, C.; Najimi, M.; Sokal, E. Bivalirudin in combination with heparin to control mesenchymal cell procoagulant activity. PLoS ONE 2012, 7, e42819. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Shi, B.; Chang, H.; Su, X.; Zhang, L.; Bi, C.; Shuai, Y.; Du, X.; Deng, Z.; Jin, Y. Heparin improves BMSC cell therapy: Anticoagulant treatment by heparin improves the safety and therapeutic effect of bone marrow-derived mesenchymal stem cell cytotherapy. Theranostics 2017, 7, 106–116. [Google Scholar] [CrossRef]
- Ettelaie, C.; Fountain, D.; Collier, M.E.; Elkeeb, A.M.; Xiao, Y.P.; Maraveyas, A. Low molecular weight heparin downregulates tissue factor expression and activity by modulating growth factor receptor-mediated induction of nuclear factor-kappaB. Biochim. Biophys. Acta 2011, 1812, 1591–1600. [Google Scholar] [CrossRef]
- Groeneveld, D.; Pereyra, D.; Veldhuis, Z.; Adelmeijer, J.; Ottens, P.; Kopec, A.K.; Starlinger, P.; Lisman, T.; Luyendyk, J.P. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood 2019, 133, 1245–1256. [Google Scholar] [CrossRef]
- Seeger, F.H.; Rasper, T.; Fischer, A.; Muhly-Reinholz, M.; Hergenreider, E.; Leistner, D.M.; Sommer, K.; Manavski, Y.; Henschler, R.; Chavakis, E.; et al. Heparin disrupts the CXCR4/SDF-1 axis and impairs the functional capacity of bone marrow-derived mononuclear cells used for cardiovascular repair. Circ. Res. 2012, 111, 854–862. [Google Scholar] [CrossRef]
- Coppin, L.; Najimi, M.; Bodart, J.; Rouchon, M.S.; van der Smissen, P.; Eeckhoudt, S.; Dahlqvist, G.; Castanares-Zapatero, D.; Komuta, M.; Brouns, S.L.; et al. Clinical Protocol to Prevent Thrombogenic Effect of Liver-Derived Mesenchymal Cells for Cell-Based Therapies. Cells 2019, 8, 846. [Google Scholar] [CrossRef]
- Frederik, N.; Thierry, G.; Pierre-François, L.; Luc, L.; Enev, H.L.; Victor, V.; Virginie, B.; Clerget-Chossat, N.; Sokal, E. GS-16-Safety and tolerability of liver-derived stem cells (HepaStem) infused in patients with acute-on-chronic liver failure or acute decompensation: A European phase I/IIa open-labelled study. J. Hepatol. 2019, 70, e83. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, I.V.; Yarygin, K.N. Cellular Mechanisms of Liver Regeneration and Cell-Based Therapies of Liver Diseases. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Farber, E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3’-methyl-4-dimethylaminoazobenzene. Cancer Res. 1956, 16, 142–148. [Google Scholar]
- Ogawa, K.; Minase, T.; Onhoe, T. Demonstration of glucose 6-phosphatase activity in the oval cells of rat liver and the significance of the oval cells in azo dye carcinogenesis. Cancer Res. 1974, 34, 3379–3386. [Google Scholar] [PubMed]
- De Vos, R.; Desmet, V. Ultrastructural characteristics of novel epithelial cell types identified in human pathologic liver specimens with chronic ductular reaction. Am. J. Pathol. 1992, 140, 1441–1450. [Google Scholar] [PubMed]
- Matthews, V.B.; Yeoh, G.C. Liver stem cells. IUBMB Life 2005, 57, 549–553. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef]
- de Souza, L.E.; Malta, T.M.; Kashima Haddad, S.; Covas, D.T. Mesenchymal Stem Cells and Pericytes: To What Extent Are They Related? Stem Cells Dev. 2016, 25, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Corselli, M.; Chen, C.W.; Sun, B.; Yap, S.; Rubin, J.P.; Péault, B. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev. 2011, 21, 1299–1308. [Google Scholar] [CrossRef]
- Kostallari, E.; Shah, V.H. Pericytes in the Liver. Adv. Exp. Med. Biol. 2019, 1122, 153–167. [Google Scholar]
- Asahina, K.; Tsai, S.Y.; Li, P.; Ishii, M.; Maxson, R.E., Jr.; Sucov, H.M.; Tsukamoto, H. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 2009, 49, 998–1011. [Google Scholar] [CrossRef]
- Asahina, K.; Zhou, B.; Pu, W.T.; Tsukamoto, H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 2011, 53, 983–995. [Google Scholar] [CrossRef]
- Sheng, G. The developmental basis of mesenchymal stem/stromal cells (MSCs). BMC Dev. Biol. 2015, 15, 44. [Google Scholar] [CrossRef]
- Miwa, H.; Era, T. Tracing the destiny of mesenchymal stem cells from embryo to adult bone marrow and white adipose tissue via Pdgfrα expression. Development 2018, 145. [Google Scholar] [CrossRef]
- Yang, L.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.; Vandongen, H.M.; Huang, J.; Alpini, G.D.; Diehl, A.M. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells 2008, 26, 2104–2113. [Google Scholar] [CrossRef]
- Kordes, C.; Sawitza, I.; Götze, S.; Herebian, D.; Häussinger, D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J. Clin. Investig. 2014, 124, 5503–5515. [Google Scholar] [CrossRef]
- Kordes, C.; Sawitza, I.; Götze, S.; Schumacher, E.; Häussinger, D. Beyond fibrosis: Stellate cells as liver stem cells. Z. Gastroenterol. 2015, 53, 1425–1431. [Google Scholar] [CrossRef]
- Shenoy, P.S.; Bose, B. Hepatic perivascular mesenchymal stem cells with myogenic properties. J. Tissue Eng. Regen. Med. 2018, 12, e1297–e1310. [Google Scholar] [CrossRef]
- Mansilla, E.; Marín, G.H.; Drago, H.; Sturla, F.; Salas, E.; Gardiner, C.; Bossi, S.; Lamonega, R.; Guzmán, A.; Nuñez, A.; et al. Bloodstream cells phenotypically identical to human mesenchymal bone marrow stem cells circulate in large amounts under the influence of acute large skin damage: New evidence for their use in regenerative medicine. Transplant. Proc. 2006, 38, 967–969. [Google Scholar] [CrossRef]
- Pedone, E.; Olteanu, V.A.; Marucci, L.; Muñoz-Martin, M.I.; Youssef, S.A.; de Bruin, A.; Cosma, M.P. Modeling Dynamics and Function of Bone Marrow Cells in Mouse Liver Regeneration. Cell Rep. 2017, 18, 107–121. [Google Scholar] [CrossRef][Green Version]
- Lee, S.G.; Moon, S.H.; Kim, H.J.; Lee, J.Y.; Park, S.J.; Chung, H.M.; Ha, T.Y.; Song, G.W.; Jung, D.H.; Park, H.; et al. Bone marrow-derived progenitor cells in de novo liver regeneration in liver transplant. Liver Transpl. 2015, 21, 1186–1194. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Jing, Y.; Zhang, S.; Zong, C.; Jiang, J.; Sun, K.; Li, R.; Gao, L.; Zhao, X.; et al. Contribution and Mobilization of Mesenchymal Stem Cells in a mouse model of carbon tetrachloride-induced liver fibrosis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Nishiyama, K.; Nakashima, H.; Ikarashi, M.; Kinoshita, M.; Nakashima, M.; Aosasa, S.; Seki, S.; Yamamoto, J. Mouse CD11b+Kupffer Cells Recruited from Bone Marrow Accelerate Liver Regeneration after Partial Hepatectomy. PLoS ONE 2015, 10, e0136774. [Google Scholar] [CrossRef]
- Ding, B.S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Wang, L. VEGF-sdf1 recruitment of CXCR7+ bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G739–G746. [Google Scholar] [CrossRef]
- Hernanda, P.Y.; Pedroza-Gonzalez, A.; van der Laan, L.J.; Bröker, M.E.; Hoogduijn, M.J.; Ijzermans, J.N.; Bruno, M.J.; Janssen, H.L.; Peppelenbosch, M.P.; Pan, Q. Tumor promotion through the mesenchymal stem cell compartment in human hepatocellular carcinoma. Carcinogenesis 2013, 34, 2330–2340. [Google Scholar] [CrossRef]
- Hernanda, P.Y.; Pedroza-Gonzalez, A.; van der Laan, L.J.W.; Hoogduijn, M.J.; Peppelenbosch, M.P.; Pan, Q. Mesenchymal stem/stromal cells exert trophic effect on colorectal cancer metastasis to the liver. J. Liver 2013, 2, 135. [Google Scholar] [CrossRef]
- Yan, X.L.; Jia, Y.L.; Chen, L.; Zeng, Q.; Zhou, J.N.; Fu, C.J.; Chen, H.X.; Yuan, H.F.; Li, Z.W.; Shi, L.; et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 2013, 57, 2274–2286. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, D.; Wu, W.; Wu, S.; Qian, J.; Hao, Y.; Yan, F.; Zhu, P.; Wu, J.; Huang, G.; et al. Mesenchymal Stem Cells Promote Hepatocarcinogenesis via lncRNA-MUF Interaction with ANXA2 and miR-34a. Cancer Res. 2017, 77, 6704–6716. [Google Scholar] [CrossRef]
- Garcia, M.G.; Bayo, J.; Bolontrade, M.F.; Sganga, L.; Malvicini, M.; Alaniz, L.; Aquino, J.B.; Fiore, E.; Rizzo, M.M.; Rodriguez, A.; et al. Hepatocellular carcinoma cells and their fibrotic microenvironment modulate bone marrow-derived mesenchymal stromal cell migration in vitro and in vivo. Mol. Pharm. 2011, 8, 1538–1548. [Google Scholar] [CrossRef]
- Bayo, J.; Fiore, E.; Aquino, J.B.; Malvicini, M.; Rizzo, M.; Peixoto, E.; Andriani, O.; Alaniz, L.; Piccioni, F.; Bolontrade, M.; et al. Increased migration of human mesenchymal stromal cells by autocrine motility factor (AMF) resulted in enhanced recruitment towards hepatocellular carcinoma. PLoS ONE 2014, 9, e95171. [Google Scholar] [CrossRef]
- Ling, C.C.; Ng, K.T.; Shao, Y.; Geng, W.; Xiao, J.W.; Liu, H.; Li, C.X.; Liu, X.B.; Ma, Y.Y.; Yeung, W.H.; et al. Posttransplant endothelial progenitor cell mobilization via CXCL10/CXCR3 signaling promotes liver tumor growth. J. Hepatol. 2014, 60, 103–109. [Google Scholar] [CrossRef]
- Hernanda, P.Y.; Pedroza-Gonzalez, A.; Sprengers, D.; Peppelenbosch, M.P.; Pan, Q. Multipotent mesenchymal stromal cells in liver cancer: Implications for tumor biology and therapy. Biochim. Biophys. Acta 2014, 1846, 439–445. [Google Scholar] [CrossRef]
- Ema, H.; Suda, T. Two anatomically distinct niches regulate stem cell activity. Blood 2012, 120, 2174–2181. [Google Scholar] [CrossRef]
- Dong, L.; Hao, H.; Han, W.; Fu, X. The role of the microenvironment on the fate of adult stem cells. Sci. China Life Sci. 2015, 58, 639–648. [Google Scholar] [CrossRef]
- Silberstein, L.; Goncalves, K.A.; Kharchenko, P.V.; Turcotte, R.; Kfoury, Y.; Mercier, F.; Baryawno, N.; Severe, N.; Bachand, J.; Spencer, J.A.; et al. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell 2016, 19, 530–543. [Google Scholar] [CrossRef]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014, 15, 605–618. [Google Scholar] [CrossRef]
- Luo, X.; Gupta, K.; Ananthanarayanan, A.; Wang, Z.; Xia, L.; Li, A.; Sakban, R.B.; Liu, S.; Yu, H. Directed Differentiation of Adult Liver Derived Mesenchymal Like Stem Cells into Functional Hepatocytes. Sci. Rep. 2018, 8, 2818. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Probe Name | Systematic Name | Gene Name | Description | Liver2 * | Liver3 * | ||
|---|---|---|---|---|---|---|---|
| Mesenchymal Stem Cell Markers | |||||||
| A_23_P24870 | NM_000610 | CD44 | Homo sapiens CD44 molecule (Indian blood group) | 22,165.98 | 22,147.97 | 15,653.33 | 15,673.40 |
| A_23_P36364 | NM_006288 | THY1 | Homo sapiens Thy-1 cell surface antigen; CD90 | 1672.59 | 1462.77 | 33,192.08 | 28,736.66 |
| A_23_P83328 | NM_000118 | ENG | Homo sapiens endoglin; CD105 | 1555.26 | 1482.65 | 2847.10 | 2417.84 |
| A_23_P150053 | NM_001613 | ACTA2 | Homo sapiens actin, alpha 2, smooth muscle, aorta; α-SMA | 25,339.22 | 24,698.05 | 34,689.60 | 31,747.88 |
| A_23_P161194 | NM_003380 | VIM | Homo sapiens vimentin | 64,899.19 | 61,665.47 | 65,568.88 | 57,384.63 |
| A_23_P103672 | NM_006617 | NES | Homo sapiens nestin | 1438.84 | 1371.23 | 1717.24 | 1488.00 |
| A_24_P119745 | NM_212482 | FN1 | Homo sapiens fibronectin 1 | 33,852.13 | 31,302.16 | 41,280.02 | 37,071.62 |
| Pluripotent Markers | |||||||
| A_23_P204640 | NM_024865 | NANOG | Homo sapiens Nanog homeobox | 2.32 | 2.69 | 2.79 | 3.41 |
| A_23_P395582 | NM_174900 | ZFP42 | Homo sapiens ZFP42 zinc finger protein; REX-1 | 2.44 | 2.77 | 2.91 | 3.61 |
| A_23_P401055 | NM_003106 | SOX2 | Homo sapiens SRY-box 2 | 6.08 | 8.77 | 8.34 | 3.44 |
| A_23_P59138 | NM_002701 | POU5F1 | Homo sapiens POU class 5 homeobox 1; Oct3/4 | 313.82 | 315.20 | 36.68 | 40.28 |
| Integrins | |||||||
| A_23_P256334 | NM_181501 | ITGA1 | Homo sapiens integrin subunit alpha 1; CD49a or VLA-1 | 2206.52 | 2279.19 | 1514.58 | 1342.72 |
| A_32_P208076 | NM_002203 | ITGA2 | Homo sapiens integrin subunit alpha 2; CD49b or VLA-2 | 2257.31 | 2115.44 | 2226.08 | 2029.36 |
| A_23_P55251 | NM_002204 | ITGA3 | Homo sapiens integrin subunit alpha 3; CD49c or VLA-3 | 1045.91 | 974.42 | 2288.12 | 2026.87 |
| A_23_P56505 | NM_000885 | ITGA4 | Homo sapiens integrin subunit alpha 4; CD49d or VLA-4 | 628.68 | 628.96 | 709.31 | 667.34 |
| A_23_P36562 | NM_002205 | ITGA5 | Homo sapiens integrin subunit alpha 5; CD49e or VLA-5 | 1206.74 | 1296.71 | 1207.25 | 1142.43 |
| A_23_P210176 | NM_000210 | ITGA6 | Homo sapiens integrin subunit alpha 6; CD49f or VLA-6 | 526.16 | 587.86 | 908.29 | 907.40 |
| A_23_P206022 | NM_001004439 | ITGA11 | Homo sapiens integrin subunit alpha 11 | 1745.93 | 1722.10 | 8940.13 | 8343.51 |
| A_23_P50907 | NM_002210 | ITGAV | Homo sapiens integrin subunit alpha-V; CD51 | 6462.36 | 6587.11 | 7948.93 | 7574.29 |
| A_23_P128084 | NM_002206 | ITGA7 | Homo sapiens integrin subunit alpha 7 | 1268.72 | 1201.85 | 35,025.66 | 32,855.67 |
| A_23_P218375 | NM_002208 | ITGAE | Homo sapiens integrin subunit alpha E; CD103 | 3050.15 | 2959.34 | 2299.69 | 2459.11 |
| Cadherins | |||||||
| A_23_P38732 | NM_001792 | CDH2 | Homo sapiens cadherin 2 | 3149.20 | 3461.18 | 2933.76 | 2428.21 |
| A_23_P17593 | NM_001794 | CDH4 | Homo sapiens cadherin 4 | 906.85 | 865.44 | 294.76 | 261.65 |
| A_23_P214011 | NM_004932 | CDH6 | Homo sapiens cadherin 6 | 262.23 | 253.28 | 127.30 | 120.94 |
| A_23_P152305 | NM_001797 | CDH11 | Homo sapiens cadherin 11 | 6958.79 | 6745.47 | 6684.07 | 6513.42 |
| A_23_P40192 | NM_021248 | CDH22 | Homo sapiens cadherin 22 | 464.46 | 385.37 | 409.64 | 360.69 |
| A_23_P25790 | NM_022478 | CDH24 | Homo sapiens cadherin 24 | 5737.23 | 3785.29 | 5150.02 | 2650.23 |
| Liver-Specific Genes | |||||||
| A_23_P205531 | NM_001282192 | RNASE4 | Homo sapiens ribonuclease A family member 4 | 1136.06 | 1122.61 | 1057.69 | 931.28 |
| A_23_P127584 | NM_006169 | NNMT | Homo sapiens nicotinamide N-methyltransferase | 29,236.59 | 29,396.42 | 37,759.10 | 35,145.76 |
| A_24_P53976 | NM_002065 | GLUL | Homo sapiens glutamate-ammonia ligase | 852.81 | 794.77 | 543.40 | 525.56 |
| A_23_P120809 | NM_001288833 | GGT1 | Homo sapiens gamma-glutamyltransferase 1 | 687.35 | 622.17 | 2210.95 | 1819.73 |
| A_23_P209625 | NM_000104 | CYP1B1 | Homo sapiens cytochrome P450 family 1 subfamily B member 1 | 4169.50 | 4678.15 | 3127.02 | 3027.10 |
| A_23_P206110 | NM_000761 | CYP1A2 | Homo sapiens cytochrome P450 family 1 subfamily A member 2 | 1589.79 | 1231.16 | 1202.83 | 979.58 |
| A_23_P257834 | NM_000477 | ALB | Homo sapiens albumin | 2.29 | 2.69 | 2.71 | 4.04 |
| A_23_P58205 | NM_001134 | AFP | Homo sapiens alpha fetoprotein | 2.52 | 2.69 | 2.85 | 3.60 |
| A_23_P28761 | NM_001030004 | HNF4A | Homo sapiens hepatocyte nuclear factor 4 alpha | 4.49 | 4.69 | 3.15 | 3.91 |
| A_23_P359245 | NM_000245 | MET | Homo sapiens MET proto-oncogene, receptor tyrosine kinase | 8524.08 | 8927.24 | 4795.02 | 4726.29 |
| Immune Markers | |||||||
| A_23_P201758 | NM_002389 | CD46 | Homo sapiens CD46 molecule | 1161.59 | 1128.90 | 1383.52 | 1274.95 |
| A_23_P374862 | NM_000574 | CD55 | Homo sapiens CD55 molecule (Cromer blood group) | 1017.57 | 1029.88 | 513.73 | 507.53 |
| A_24_P141481 | NM_203330 | CD59 | Homo sapiens CD59 molecule (CD59 blood group) | 36,012.01 | 40,734.98 | 49,512.49 | 41,315.28 |
| A_23_P208293 | NM_001042724 | NECTIN2 | Homo sapiens nectin cell adhesion molecule 2; CD112 | 9739.88 | 10,486.87 | 10,514.83 | 9845.87 |
| A_23_P121480 | NM_001004196 | CD200 | Homo sapiens CD200 molecule | 50.63 | 48.11 | 73.49 | 67.06 |
| A_23_P256487 | NM_014143 | CD274 | Homo sapiens CD274 molecule; PD-L1 | 2114.17 | 2051.64 | 755.44 | 723.45 |
| Antioxidant Enzymes | |||||||
| A_23_P154840 | NM_000454 | SOD1 | Homo sapiens superoxide dismutase 1 | 51,374.15 | 51,358.13 | 55,628.91 | 52,328.87 |
| A_23_P254741 | NM_003102 | SOD3 | Homo sapiens superoxide dismutase 3 | 621.46 | 582.76 | 2163.03 | 2139.94 |
| A_23_P202658 | NM_000852 | GSTP1 | Homo sapiens glutathione S-transferase pi 1 | 68,476.57 | 71,676.05 | 62,832.28 | 59,208.49 |
| Mesenchymal Markers | ||
| CD13 | Membrane alanyl aminopeptidase | + [34,36,] |
| CD44 | Cell adhesion molecule; receptor for hyaluronic acid | + [20,21,22,23,33,36,71] |
| CD73 | Ecto-5’-nucleotidase | + [20,21,22,34,36,71] |
| CD90 | Thymocyte antigen (Thy-1) | + [20,21,22,23,33,34,36,71] |
| CD105 | Endoglin | + or ± [21,22,30,34,36,71] − [20] |
| CD146 | Melanoma cell adhesion molecule (MCAM) | + [71] ± [36] |
| CD166 | Activated leukocyte cell adhesion molecule (ALCAM) | + [22,36] |
| Vimentin | Type III intermediate filament | + [20] |
| α-SMA | Alpha-smooth muscle actin | + [20] |
| Fibronectin | Glycoprotein of the extracellular matrix | + [34,37] |
| Hematopoietic/Endothelial Markers | ||
| CD11b | Integrin alpha M (ITGAM) | − [20,23,27,33,36] |
| CD14 | Myeloid cell marker; co-receptor for lipopolysaccharide | − [20,23,27,33,36] |
| CD19 | B-lymphocyte antigen | − [20,23,27,33,36] |
| CD31 | Platelet endothelial cell adhesion molecule (PECAM-1) | − [20,23,27,33,36] |
| CD34 | Hematopoietic stem cell marker | − [20,23,27,33,36] |
| CD45 | Leukocyte common antigen | − [20,23,27,33,36] |
| CD79β | B-lymphocyte antigen | − [20,23,27,33,36] |
| CD117 | Mast/stem cell growth factor receptor (c-Kit) | − [20,23,27,33,36] |
| CD133 | Prominin-1; marker of hematopoietic stem cells, endothelial progenitor cells, cancer stem cells etc. | − [20,23,27,33,36] |
| CD144 | Vascular endothelial cadherin or cadherin-5 | − [20,23,27,33,36] |
| HLA-ABC | Major histocompatibility complex class I | + [20,23,27,33,36] |
| HLA-DR | Major histocompatibility complex class II | − [20,23,27,33,36] |
| Pluripotency Markers | ||
| SSEA-3 | Stage-specific embryonic antigen 3 | − [36] |
| SSEA-4 | Stage-specific embryonic antigen 4 | − [36] + [21] |
| Tra1-60 | Pluripotent stem cell markers | − [36] |
| Tra1-81 | Pluripotent stem cell markers | − [36] |
| NANOG | Transcription factor that maintain pluripotency | + [21,34] |
| OCT-4 | Homeodomain transcription factor of the POU family; involved in the self-renewal | + [21,34] |
| SOX2 | Transcription factor; essential for maintaining self-renewal and pluripotency | + [21] |
| REX1 | Pluripotency marker | + [34] |
| Integrins and Adhesion Molecules | ||
| CD29 | Integrin beta-1 (ITGB1) | + [6,20,21,22,23,28,36,71] |
| CD49a | Integrin alpha-1 (ITGA1) | + [34] ± [36,] |
| CD49b | Integrin alpha-2 (ITGA2) | + [20,28,34,36] |
| CD49c | Integrin alpha-3 (ITGA3) | + [28,34,36] |
| CD49d | Integrin alpha-4 (ITGA4) | − [36] |
| CD49e | Integrin alpha-5 (ITGA5) | + [20,28,34,36] |
| CD49f | Integrin alpha-6 (ITGA6) | ± [20] − [36] |
| CD51 | Integrin alpha-V (ITGAV) | + [36] |
| CD162 | Selectin P ligand (SELPLG) | − [36] |
| SSEA-1 | Sialyl LewisX (CD15s) | − [36] |
| Cadherin-11 | Integral membrane proteins that mediate calcium-dependent cell-cell adhesion | + [34] |
| Cytokeratins and Hepatic Markers | ||
| Albumin | Marker of mature hepatocytes | + [20,21] − [22,29] |
| α-fetoprotein | Marker of immature hepatocytes; highly expressed in the fetal liver; can be a sign of liver cancer, as well as noncancerous liver diseases | ± [20,21] − [29] |
| Hepatocyte nuclear factor-4 (HNF-4) | Transcription factor that plays a critical role in the transcriptional regulation of genes involved in glucose metabolism in hepatocytes | + [20] − [29] |
| CYP3A4 | Cytochrome P450 3A4; it oxidizes small foreign organic molecules (xenobiotics) | + [20] |
| CYP1B1 | Cytochrome P450 1B1; it catalyzes estrogen hydroxylation and activates potential carcinogens | + [20] |
| Cytokeratin 7 | Type II cytoskeletal keratin; found on many glandular and transitional epithelia, hepatocytes, biliary epithelium etc. | − [20] |
| Cytokeratin 8 | Type II cytoskeletal keratin; expressed mainly by secretory epithelia | − [20] + [21] |
| Cytokeratin 18 | Type I cytoskeletal keratin; expressed in single layer epithelial tissues | − [20] ± [22,72] + [21] |
| Cytokeratin 19 | Type I cytoskeletal keratin; markers of cells of the epithelial origin | − [20,21] + [22] |
| c-Met | Hepatocyte growth factor receptor (HGFR) | ± [22] + [29] |
| Neuronal Markers | ||
| GFAP | Glial fibrillary acidic protein; an intermediate filament | − [37,83] |
| CD56 | Neural cell adhesion molecule (NCAM) | − [37,83] |
| NT-3 | Neurotrophin-3; neurotrophic factor in the nerve growth factor family | − [37,83] |
| CD271 | Low-affinity nerve growth factor receptor (NGFR) | − [36,37,83] |
| Nestin | Type VI intermediate filament protein; neuroectodermal stem cell marker | + [28] |
| References | Protocols | Results |
|---|---|---|
| [20] | 1. Iscove’s Modified Dulbecco’s Medium (IMDM) containing 20 ng/mL epidermal growth factor (EGF), 10 ng/mL basic fibroblast growth factor (bFGF) for 2 days. 2. IMDM containing 20 ng/mL hepatocyte growth factor (HGF), 10 ng/mL bFGF, nicotinamide 0.61 g/L, and 1% insulin-transferrin-selenium (ITS) premix for 10 days. 3. IMDM containing 20 ng/mL oncostatin M, 1 μM dexamethasone, and 1% ITS premix for 10 days. For each step, medium was changed every 3 days. | Morphology change from elongated to polygonal shape with granular cytoplasm; expression of liver-associated genes (TDO, TAP, CYP3A4, CYP2B6); storage of glycogen. |
| [21] | Liver MSCs were incubated under the condition of microgravity or in flasks coated with Matrigel in α-MEM/EBM 3:1, 12 mM Hepes, 2% FCS with hepatocyte growth factor (HGF) (10 ng/mL), and fibroblast growth factor 4 (FGF4) (10 ng/mL) for 15 days. | Change in the morphology from elongated to cuboid cells; positivity for cytochrome P450 activity; synthesis and release of albumin and urea; reduction of α-fetoprotein expression; increase in cytokeratin 8 and cytokeratin 18 expression. |
| [72] | Preinduction medium, consisting of IMDM supplemented with 2% FBS, 20 ng/mL EGF, and 10 ng/mL FGF-4 for 2 days before induction by a 2-step protocol (protocol A). Protocol A: Step-1 differentiation medium consisting of IMDM supplemented with 2% FBS, 10 ng/mL FGF-4, 20 ng/mL HGF, and 5 mol/L nicotinamide for 7 days. Step-2 maturation medium consisting of IMDM supplemented with 2% FBS, 10 ng/mL FGF-4, 20 ng/mL oncostatin M, 1 mol/L dexamethasone, 50 g/mL ITS premix, and 1 mol/L trichostatin A. Protocol B: Step-1 differentiation medium consisting of IMDM supplemented with 2% FBS, 10 ng/mL FGF-4, 20 ng/mL HGF, 5 mol/L nicotinamide, and 2.5 mmol/L sodium butyrate for 7 days. Step-2 maturation medium consisting of IMDM supplemented with 2% FBS, 10 ng/mL FGF-4, 20 ng/mL oncostatin M, 1 mol/L dexamethasone, 50 g/mL ITS premix, 1 mol/L trichostatin A, 2.5 mmol/L sodium butyrate, and 20 ng/mL HGF. Medium changes were performed twice weekly up to 21 days in both protocols. | Expression of mature hepatocyte markers, such as albumin, cytokeratin 18, and tryptophan 2,3-dioxygenase; albumin secretion. |
| [27] | Liver MSCs were seeded into fibronectin-coated plates. 1. DMEM (low glucose) containing 1% ITS premix, 10 ng/mL FGF-1, 10 ng/mL FGF-4, and 20 ng/mL HGF for 5 days. 2. DMEM (low glucose) containing 100 nM dexamethasone, 10 ng/mL FGF-4, 20 ng/mL HGF, 10 ng/mL oncostatin M, and 0.5% dimethyl sulfoxide on designated days. Medium was exchanged every 2 days. | The morphology was changed to a highly round or polygonal shape; expression of some hepatic markers, such as albumin, α1-antitrypsin, tryptophan 2,3-dioxygenase, and glutamine synthetase; urea production; CYP450 enzymatic functions were not detected. |
| [34] | Liver MSCs were seeded into fibronectin-coated dishes at high density and maintained for two weeks in a medium containing 0.5% FBS, 10 ng/mL FGF-4, and 20 ng/mL HGF. After this period, FGF-4 and HGF were substituted for 20 ng/mL oncostatin M for other 14 days. | The cell morphology was changed to a globular shape with an eccentric nucleus; expression of cytokeratins 18 and 19 and transcription factor GATA-4; glycogen storage, and albumin production; inducible cytochrome P450 activity. |
| [33] | 1. DMEM/F12 with 2 mM GlutaMAX supplemented with 0.5% FBS, 10 ng/mL HGF, and 100 ng/mL Activin A. 2. DMEM/F12 medium with 2 mM GlutaMAX containing 15 mM HEPES, 2 μg/mL insulin, 10 ng/mL FGF-4, 10 ng/mL HGF, 10 ng/mL oncostatin M and 10−7 M dexamethasone. In each medium, cells were cultivated for 10 days with medium change every 2 days. | The cell morphology was more flattened with a cuboidal epithelium-like shape and granular cytoplasm; prealbumin expression; reduction of α-fetoprotein expression; albumin secretion. |
| Status | Clinical Trials.gov Identifier | Study Title | Conditions | Interventions | Phase |
|---|---|---|---|---|---|
| Recruiting | NCT03884959 | A Prospective, Open-Label, Safety, and Efficacy Study of Infusions of HepaStem in Urea Cycle Disorders Pediatric Patients | Urea Cycle Disorder | HepaStem | II |
| Recruiting | NCT02946554 | Multicenter Phase II Safety and Preliminary Efficacy Study of 2 Dose Regimens of HepaStem in Patients with Acute-on-Chronic Liver Failure | Acute-On-Chronic Liver Failure | HepaStem | II |
| Recruiting | NCT03963921 | Multicenter, Open-Label, Safety and Tolerability Study of Ascending Doses of HepaStem in Patients with Cirrhotic and Pre-cirrhotic Non-alcoholic Steato-hepatitis (NASH) | Non-Alcoholic Steatohepatitis | HepaStem | I/II |
| Enrolling by invitation | NCT03343756 | HepaStem Long-Term Safety Registry (PROLONGSTEM) | Urea Cycle Disorder Crigler–Najjar Syndrome | HepaStem | |
| Enrolling by invitation | NCT03632148 | In Vitro Evaluation of the Effect of HepaStem in the Coagulation Activity in Blood of Patients with Liver Disease | Decompensated Cirrhosis | HepaStem | |
| Completed | NCT01765283 | A Prospective, Open-Label, Multicenter, Partially Randomized, Safety Study of One Cycle of Promethera HepaStem in Urea Cycle Disorders (UCD) and Crigler–Najjar Syndrome (CN) Pediatric Patients | Urea Cycle Disorders, Crigler–Najjar Syndrome | HepaStem | I/II |
| Completed | NCT02051049 | Long-term Safety Follow-up Study of Patients With Received Infusions of HepaStem | Urea Cycle Disorders Crigler–Najjar Syndrome | HepaStem | |
| Unknown | NCT02489292 | Prospective, Open-Label, Multicenter, Efficacy, and Safety Study of Several Infusions of HepaStem in Urea Cycle Disorders Pediatric Patients | Urea Cycle Disorders | HepaStem | II |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kholodenko, I.V.; Kurbatov, L.K.; Kholodenko, R.V.; Manukyan, G.V.; Yarygin, K.N. Mesenchymal Stem Cells in the Adult Human Liver: Hype or Hope? Cells 2019, 8, 1127. https://doi.org/10.3390/cells8101127
Kholodenko IV, Kurbatov LK, Kholodenko RV, Manukyan GV, Yarygin KN. Mesenchymal Stem Cells in the Adult Human Liver: Hype or Hope? Cells. 2019; 8(10):1127. https://doi.org/10.3390/cells8101127
Chicago/Turabian StyleKholodenko, Irina V., Leonid K. Kurbatov, Roman V. Kholodenko, Garik V. Manukyan, and Konstantin N. Yarygin. 2019. "Mesenchymal Stem Cells in the Adult Human Liver: Hype or Hope?" Cells 8, no. 10: 1127. https://doi.org/10.3390/cells8101127
APA StyleKholodenko, I. V., Kurbatov, L. K., Kholodenko, R. V., Manukyan, G. V., & Yarygin, K. N. (2019). Mesenchymal Stem Cells in the Adult Human Liver: Hype or Hope? Cells, 8(10), 1127. https://doi.org/10.3390/cells8101127