Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress

 , , and
, , and
Abstract
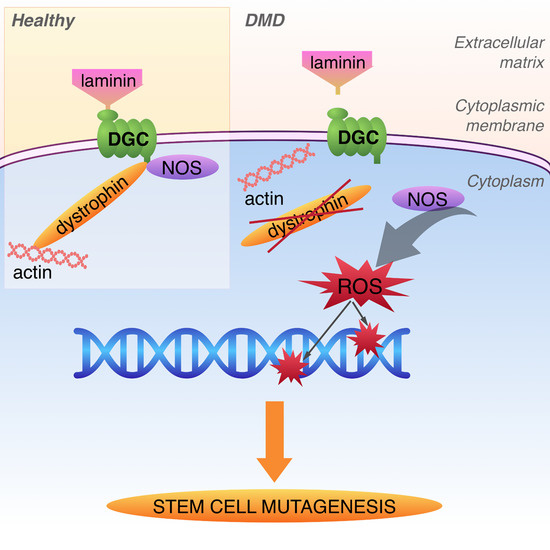
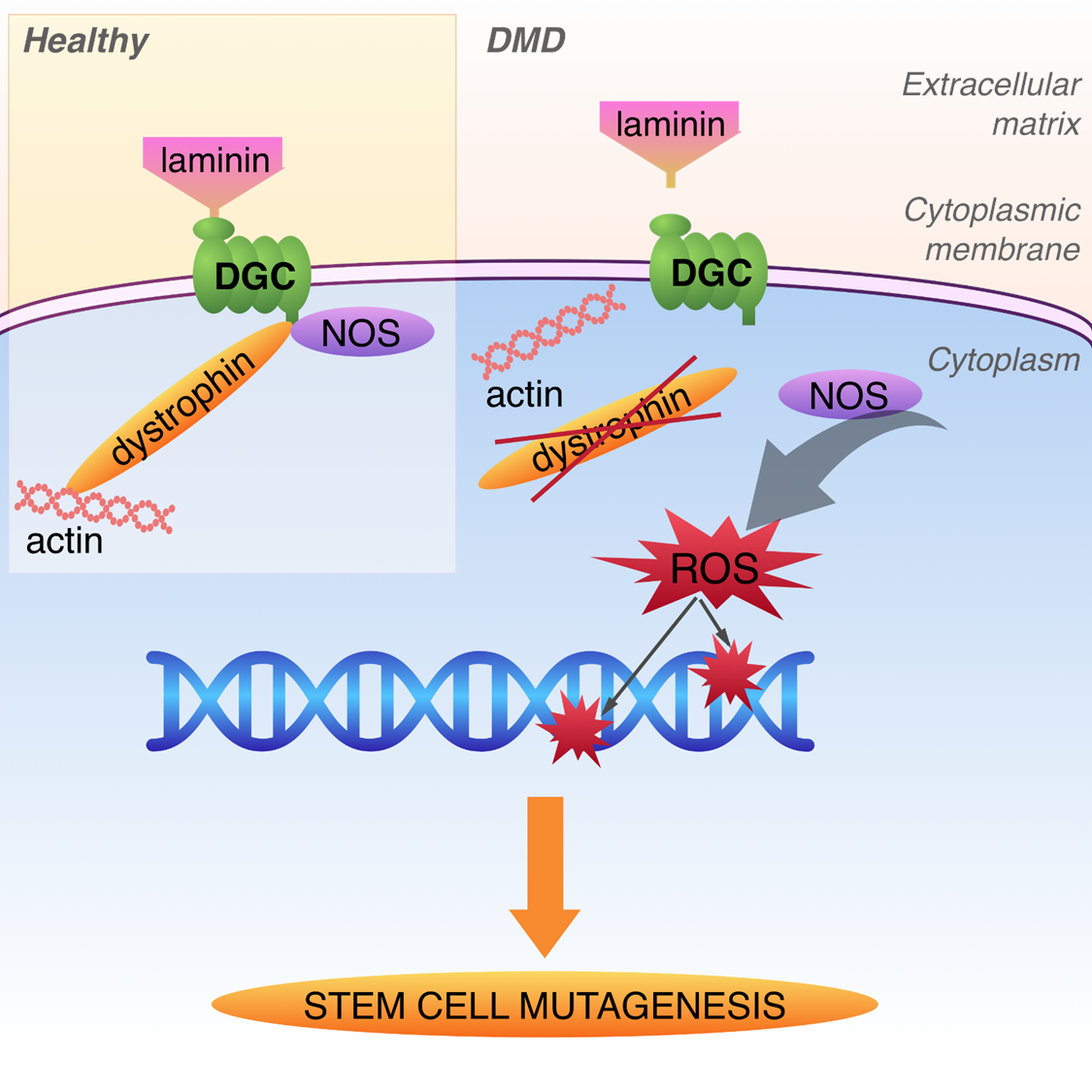{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Control Cell Lines and Cultivation
2.2. Tissue Processing, Reprogramming and Identification
2.3. CRISPR/Cas9 DMD hESC Clone Generation
2.4. Fluorescent Labeling
2.5. Inhibition Conditions
2.6. Mutation Frequency
2.7. Western Blot
2.8. Reverse Transcription PCR
2.9. Statistical Analysis
3. Results
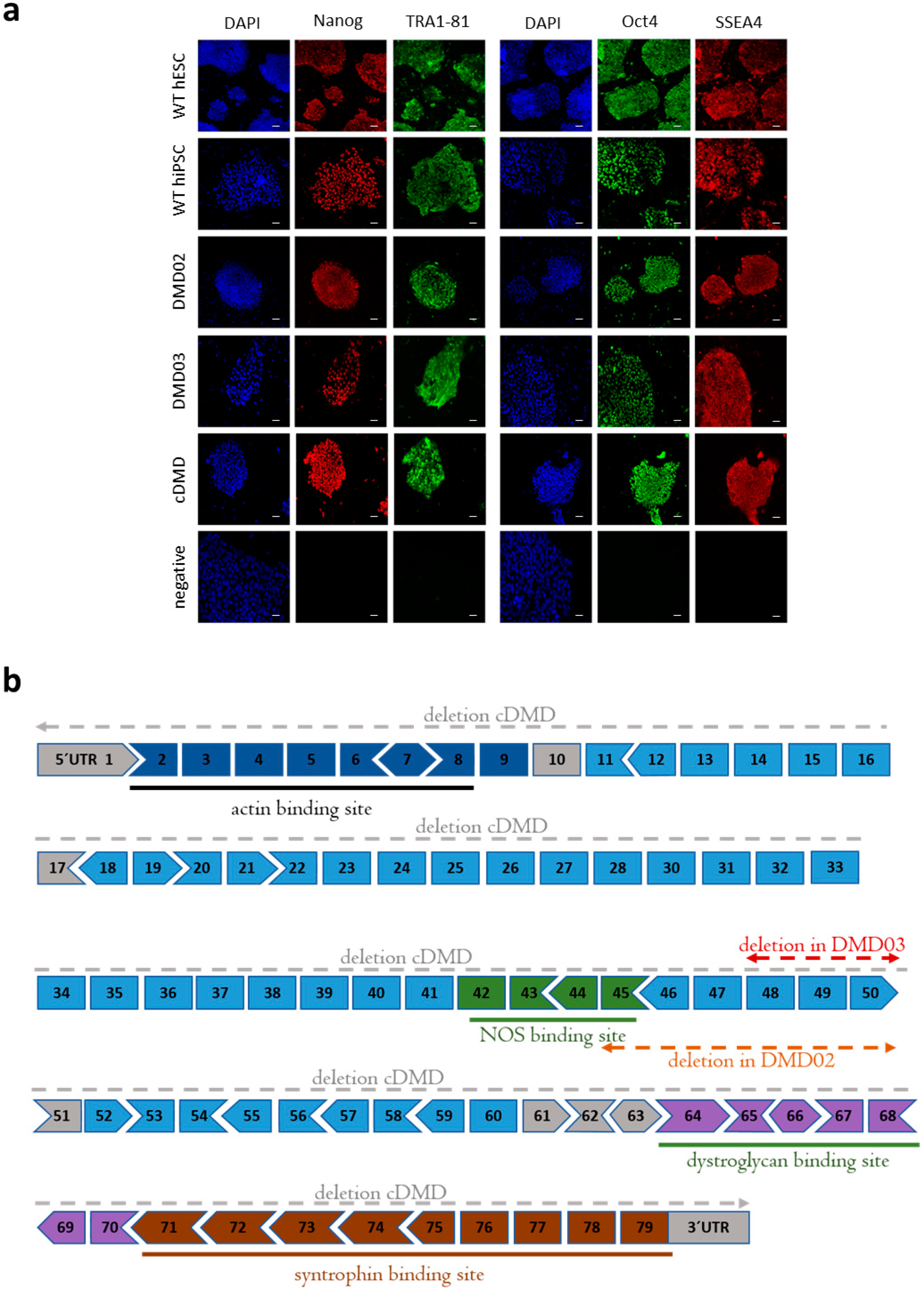
3.1. Model DMD hPSC Cell Lines Harboring Dystrophin Mutation Are Pluripotent
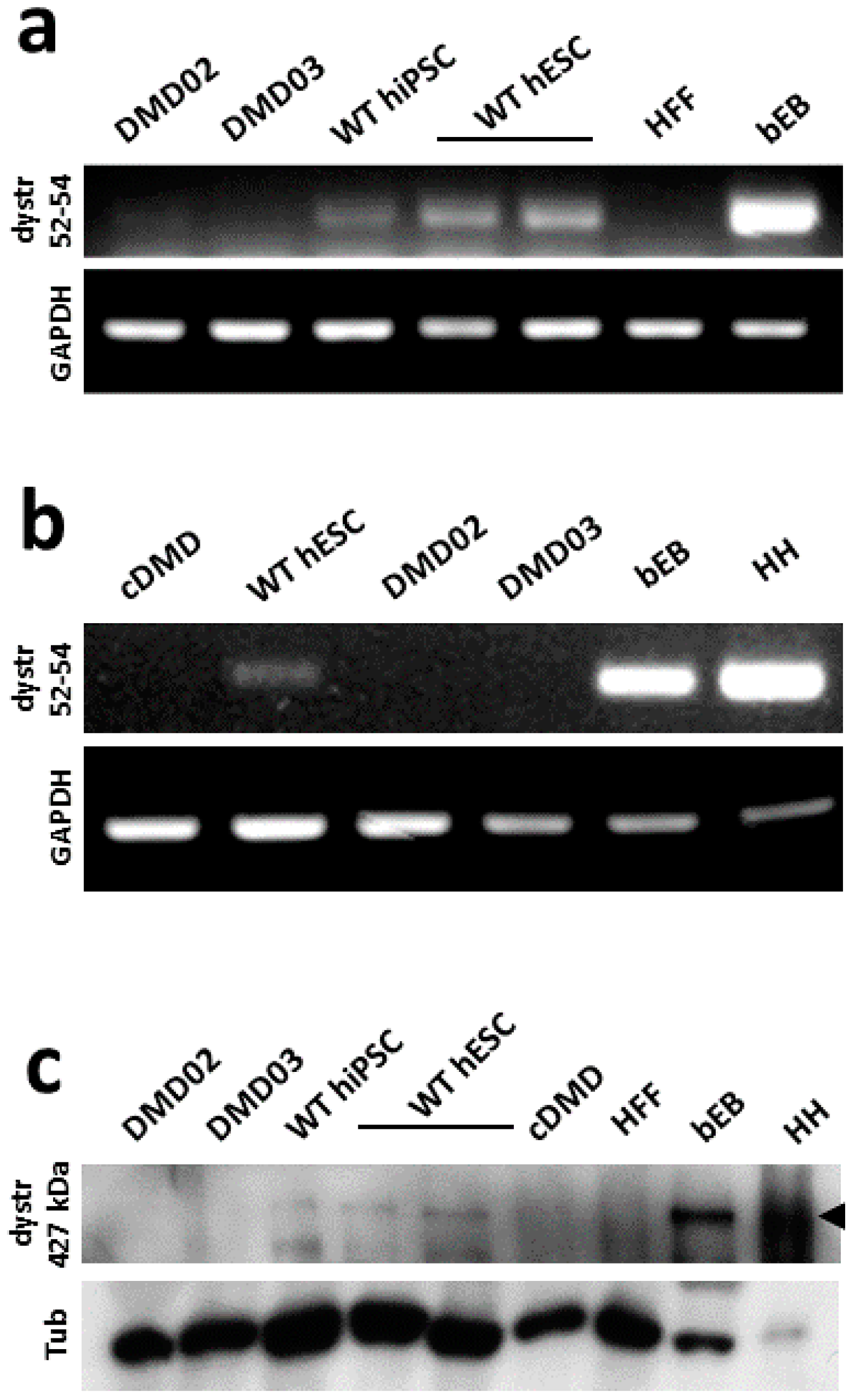
3.2. WT hPSC, But Not DMD hPSC, Express High Molecular Weight Isoform of Dystrophin
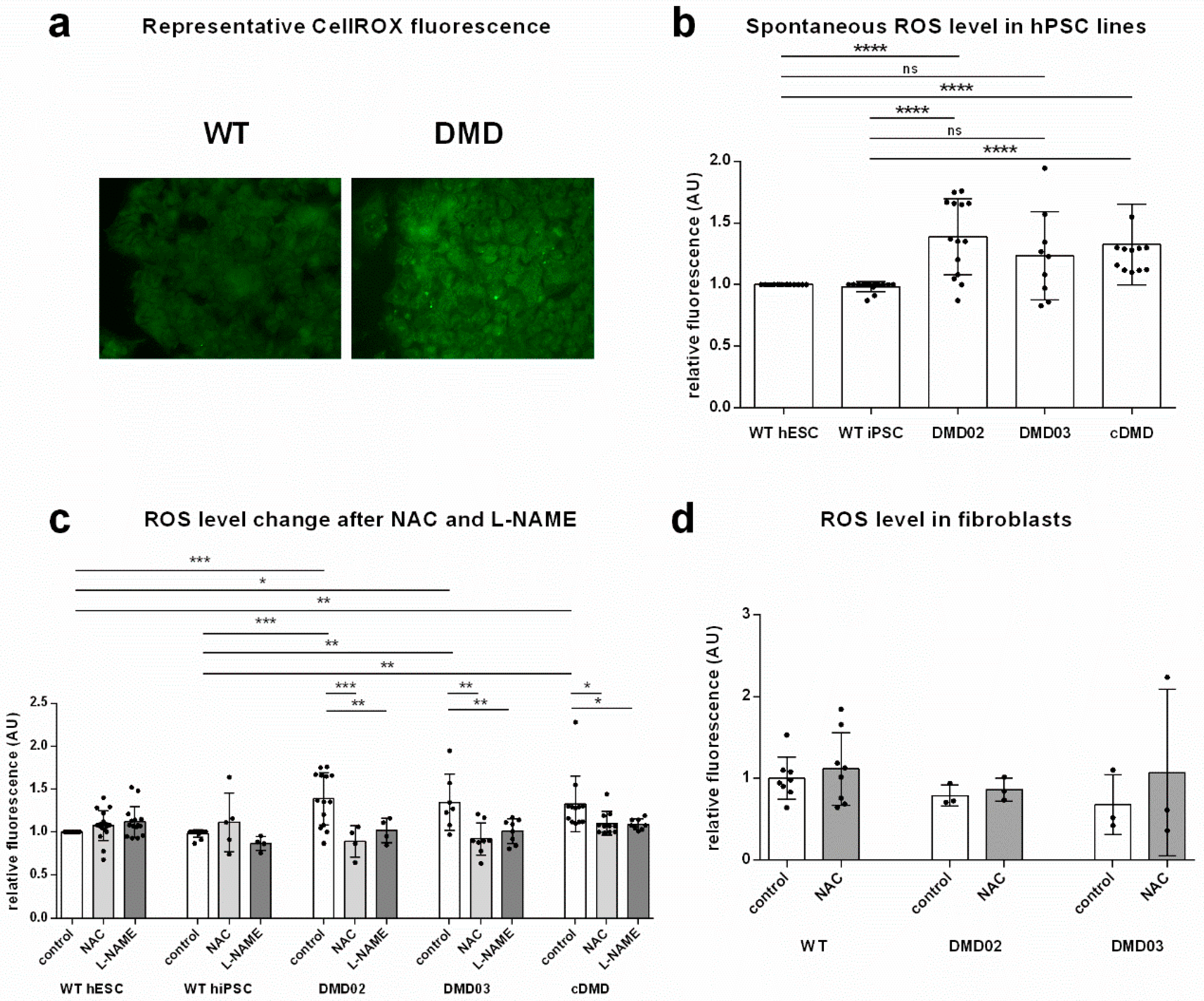
3.3. Dystrophin Defect Leads to Elevated Oxidative Stress in DMD hPSC
3.4. Inhibition of NOS Activity Led to Decrease in ROS Production in DMD hPSC But Not in WT hPSC
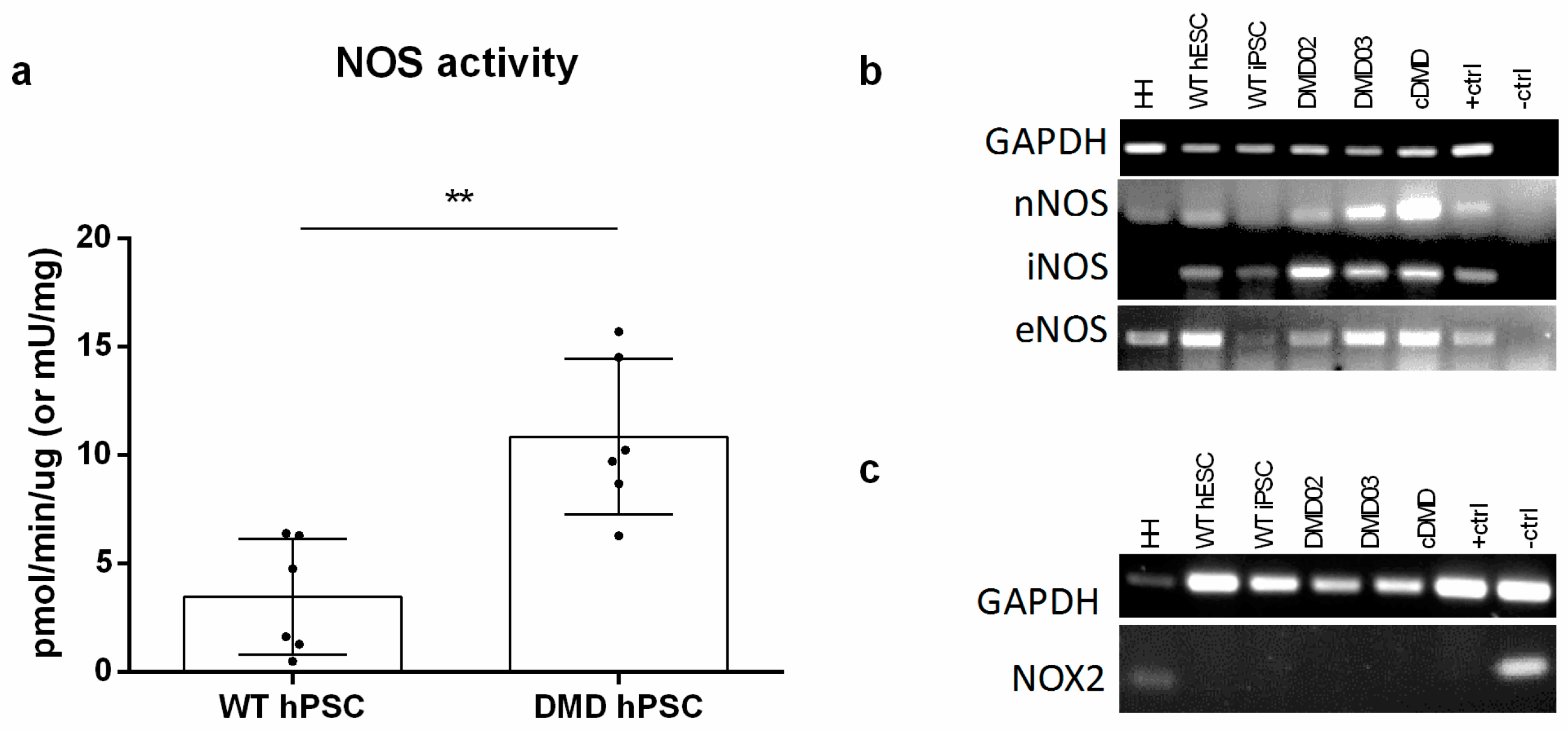
3.5. NOS Activity Is Elevated in DMD hPSC Lines
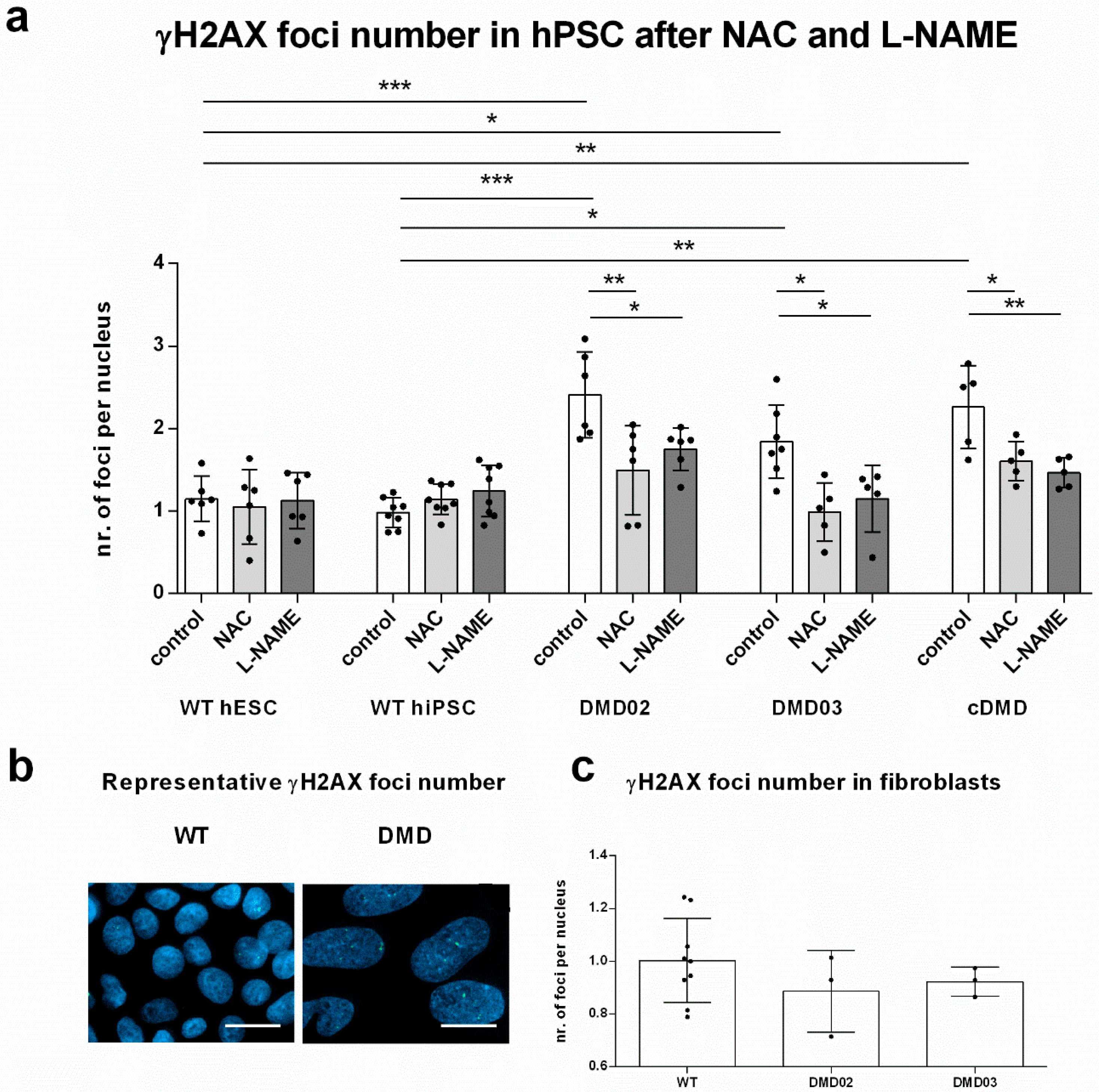
3.6. Elevated ROS Leads to Elevated DNA Damage in DMD hPSC
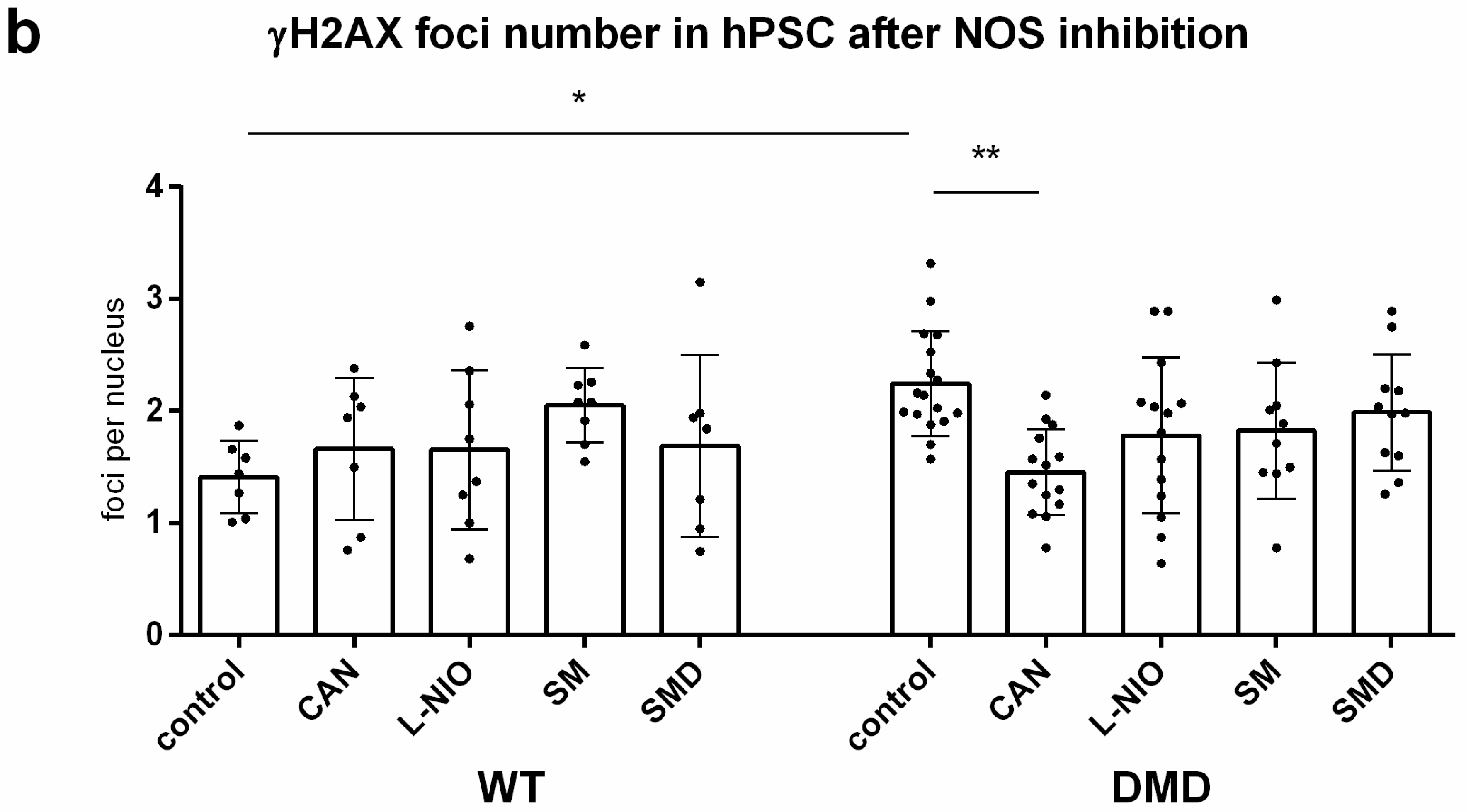
3.7. NOS Activity Causes ROS Release Which Leads to DNA Damage in DMD hPSC
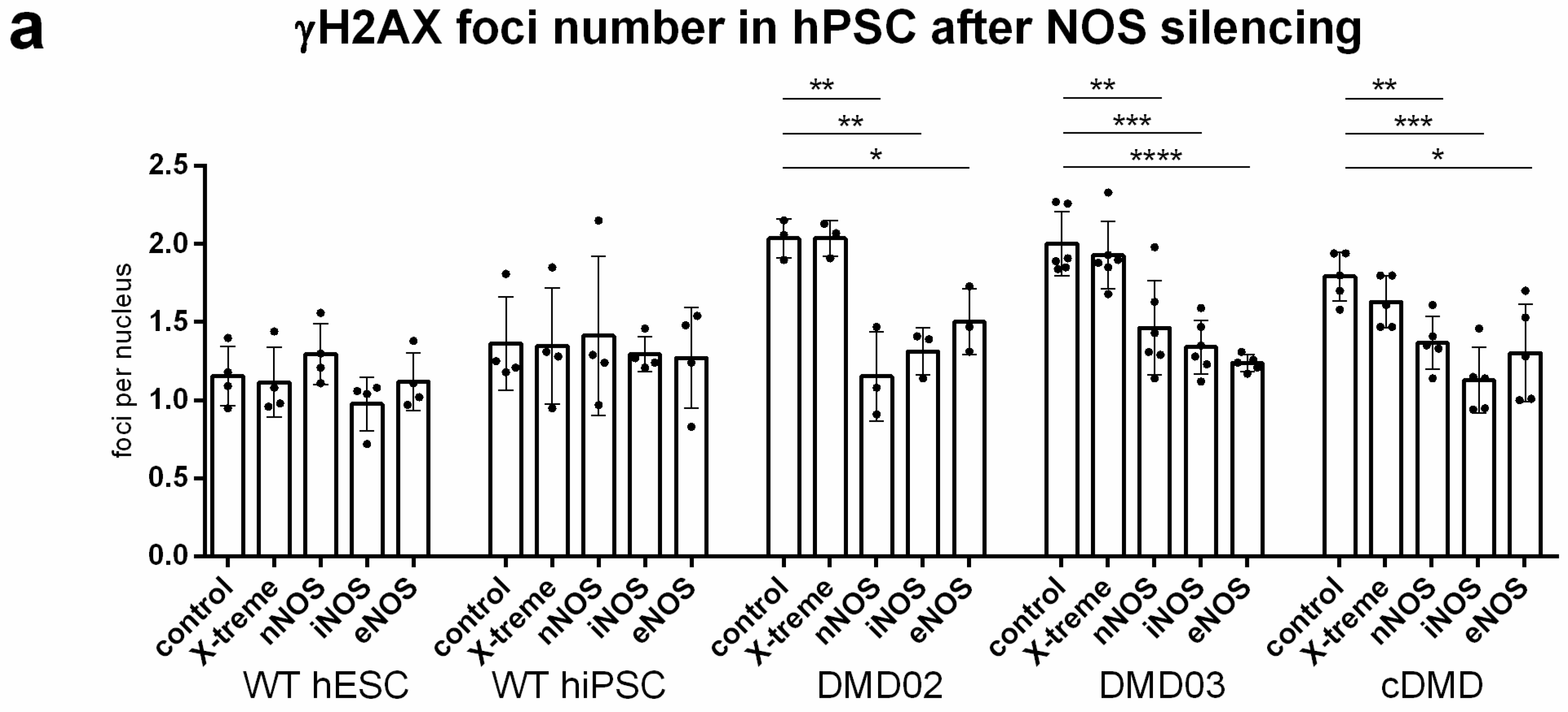
3.8. DNA Damage Increase in DMD hPSC Is Caused by nNOS, iNOS and eNOS Isoforms of NOS
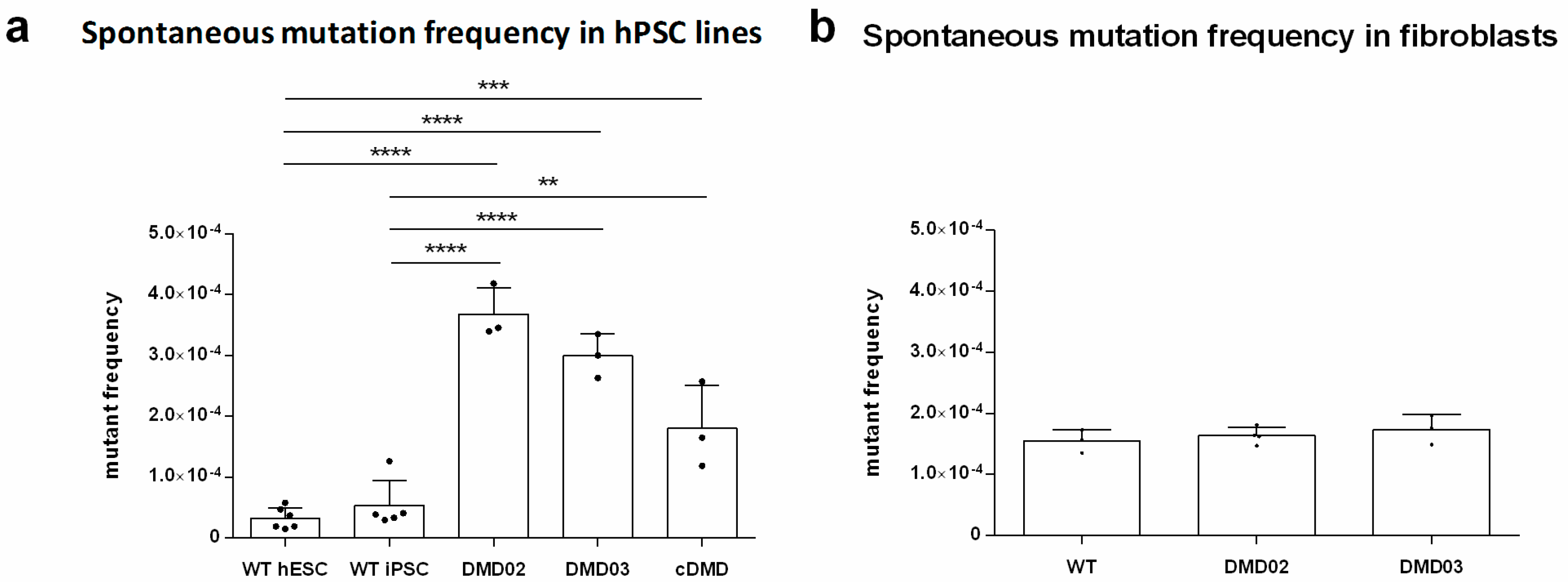
3.9. Dystrophin Defect Leads to Compromised Genomic Stability of hPSC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Tidball, J.G.; Albrecht, D.E.; Lokensgard, B.E.; Spencer, M.J. Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci. 1995, 108 (Pt 6), 2197–2204. [Google Scholar] [PubMed]
- Mikhaĭlov, V.M.; Komarov, S.A.; Nilova, V.K.; Shteĭn, G.I.; Baranov, V.S. Ultrastructural and morphometrical analysis of apoptosis stages in cardiomyocytes of MDX mice. Tsitologiia 2001, 43, 729–737. [Google Scholar] [PubMed]
- Finsterer, J.; Stöllberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chenard, A.A.; Becane, H.M.; Tertrain, F.; de Kermadec, J.M.; Weiss, Y.A. Ventricular arrhythmia in Duchenne muscular dystrophy: Prevalence, significance and prognosis. Neuromuscul. Disord. 1993, 3, 201–206. [Google Scholar] [CrossRef]
- Himmrich, E.; Popov, S.; Liebrich, A.; Rosocha, S.; Zellerhoff, C.; Nowak, B.; Przibille, O. Hidden intracardiac conduction disturbances and their spontaneous course in patients with progressive muscular dystrophy. Z. Kardiol. 2000, 89, 592–598. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [CrossRef]
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in Duchenne muscular dystrophy: Pathogenesis and therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef]
- Heslop, L.; Morgan, J.E.; Partridge, T.A. Evidence for a myogenic stem cell that is exhausted in dystrophic muscle. J. Cell Sci. 2000, 113 Pt 12, 2299–2308. [Google Scholar]
- Webster, C.; Blau, H.M. Accelerated age-related decline in replicative life-span of Duchenne muscular dystrophy myoblasts: Implications for cell and gene therapy. Somat. Cell Mol. Genet. 1990, 16, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Clarac, F.; Massion, J.; Smith, A.M. Duchenne, Charcot and Babinski, three neurologists of La Salpetrière Hospital, and their contribution to concepts of the central organization of motor synergy. J. Physiol. Paris 2009, 103, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Des Rosiers, C.; Petrof, B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; McNally, E.M. The Dystrophin Complex: Structure, function and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [PubMed]
- Massouridès, E.; Polentes, J.; Mangeot, P.-E.; Mournetas, V.; Nectoux, J.; Deburgrave, N.; Nusbaum, P.; Leturcq, F.; Popplewell, L.; Dickson, G.; et al. Dp412e: A novel human embryonic dystrophin isoform induced by BMP4 in early differentiated cells. Skelet Muscle 2015, 5, 40. [Google Scholar] [CrossRef]
- Dick, E.; Kalra, S.; Anderson, D.; George, V.; Ritso, M.; Laval, S.H.; Barresi, R.; Aartsma-Rus, A.; Lochmüller, H.; Denning, C. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013, 22, 2714–2724. [Google Scholar] [CrossRef]
- Janke, A.; Upadhaya, R.; Snow, W.M.; Anderson, J.E. A new look at cytoskeletal NOS-1 and β-dystroglycan changes in developing muscle and brain in control and mdx dystrophic mice. Dev. Dyn. 2013, 242, 1369–1381. [Google Scholar] [CrossRef]
- Ramachandran, J.; Schneider, J.S.; Crassous, P.-A.; Zheng, R.; Gonzalez, J.P.; Xie, L.-H.; Beuve, A.; Fraidenraich, D.; Peluffo, R.D. Nitric Oxide Signaling Pathway in Duchenne Muscular Dystrophy Mice: Upregulation of L-arginine Transporters. Biochem. J. 2013, 449, 133–142. [Google Scholar] [CrossRef]
- Li, D.; Yue, Y.; Lai, Y.; Hakim, C.H.; Duan, D. Nitrosative stress elicited by nNOSμ delocalization inhibits muscle force in dystrophin-null mice. J. Pathol. 2011, 223, 88–98. [Google Scholar] [CrossRef]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, B.; John, M.; Klatt, P.; Böhme, E.; Mayer, B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J. 1992, 281, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, M.; Zeiger, U.; Förderer, M.; Brinkmeier, H.; Fink, R.H.A. Cardiac and respiratory dysfunction in Duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev. 2013, 33, 1174–1213. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Kyrychenko, V.; Badr, M.A.; Ikeda, Y.; Sadoshima, J.; Shirokova, N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 2015, 108, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Olguín, C.; Altamirano, F.; Valladares, D.; López, J.R.; Allen, P.D.; Jaimovich, E. Altered ROS production, NF-κB activation and interleukin-6 gene expression induced by electrical stimulation in dystrophic mdx skeletal muscle cells. Biochim. Biophys. Acta 2015, 1852, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The role of oxidative stress in skeletal muscle injury and regeneration: Focus on antioxidant enzymes. J. Muscle Res. Cell. Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Sciorati, C.; Staszewsky, L.; Zambelli, V.; Russo, I.; Salio, M.; Novelli, D.; Di Grigoli, G.; Moresco, R.M.; Clementi, E.; Latini, R. Ibuprofen plus isosorbide dinitrate treatment in the mdx mice ameliorates dystrophic heart structure. Pharmacol. Res. 2013, 73, 35–43. [Google Scholar] [CrossRef]
- Straub, V.; Rafael, J.A.; Chamberlain, J.S.; Campbell, K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997, 139, 375–385. [Google Scholar] [CrossRef]
- Farini, A.; Meregalli, M.; Belicchi, M.; Battistelli, M.; Parolini, D.; D’Antona, G.; Gavina, M.; Ottoboni, L.; Constantin, G.; Bottinelli, R.; et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. J. Pathol. 2007, 213, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Lu, Q.L.; Pastoret, C.; Partridge, T.; Bou-Gharios, G. T-cell-dependent fibrosis in the mdx dystrophic mouse. Lab. Investig. 2000, 80, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-kB pathway in Duchenne muscular dystrophy: Relation to age. Acta Myol. 2011, 30, 16–23. [Google Scholar] [PubMed]
- Lu, A.; Poddar, M.; Tang, Y.; Proto, J.D.; Sohn, J.; Mu, X.; Oyster, N.; Wang, B.; Huard, J. Rapid depletion of muscle progenitor cells in dystrophic mdx/utrophin-/- mice. Hum. Mol. Genet. 2014, 23, 4786–4800. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Aoto, T.; Binh, N.T.; Okamoto, N.; Tanimura, S.; Wakayama, T.; Iseki, S.; Hara, E.; Masunaga, T.; Shimizu, H.; et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 2009, 137, 1088–1099. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef]
- Rübe, C.E.; Fricke, A.; Widmann, T.A.; Fürst, T.; Madry, H.; Pfreundschuh, M.; Rübe, C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS ONE 2011, 6, e17487. [Google Scholar] [CrossRef]
- Miyagoe-Suzuki, Y.; Nishiyama, T.; Nakamura, M.; Narita, A.; Takemura, F.; Masuda, S.; Minami, N.; Murayama, K.; Komaki, H.; Goto, Y.-I.; et al. Induction of Pluripotent Stem Cells from a Manifesting Carrier of Duchenne Muscular Dystrophy and Characterization of Their X-Inactivation Status. Stem Cells Int. 2017, 2017, 7906843. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lim, H.; Estrellas, K.; Mula, J.; Cohen, T.V.; Zhang, Y.; Donnelly, C.J.; Richard, J.-P.; Kim, Y.J.; Kim, H.; et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep. 2016, 15, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Naito, A.T.; Lee, J.-K.; Kitazume-Taneike, R.; Ito, M.; Yamaguchi, T.; Nakata, R.; Sumida, T.; Okada, K.; Nakagawa, A.; et al. Generation of Induced Pluripotent Stem Cells From Patients With Duchenne Muscular Dystrophy and Their Induction to Cardiomyocytes. Int. Heart J. 2016, 57, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Spaltro, G.; Vigorelli, V.; Casalnuovo, F.; Spinelli, P.; Castiglioni, E.; Rovina, D.; Paganini, S.; Di Segni, M.; Nigro, P.; Gervasini, C.; et al. Derivation of the Duchenne muscular dystrophy patient-derived induced pluripotent stem cell line lacking DMD exons 49 and 50 (CCMi001DMD-A-3, ∆49, ∆50). Stem Cell Res. 2017, 25, 128–131. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- International Stem Cell Initiative; Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar] [PubMed]
- Krutá, M.; Šeneklová, M.; Raška, J.; Salykin, A.; Zerzánková, L.; Pešl, M.; Bártová, E.; Franek, M.; Baumeisterová, A.; Košková, S.; et al. Mutation frequency dynamics in HPRT locus in culture-adapted human embryonic stem cells and induced pluripotent stem cells correspond to their differentiated counterparts. Stem Cells Dev. 2014, 23, 2443–2454. [Google Scholar] [CrossRef]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef]
- Šimara, P.; Tesařová, L.; Padourová, S.; Koutná, I. Generation of human induced pluripotent stem cells using genome integrating or non-integrating methods. Folia Biol. (Praha) 2014, 60 (Suppl. 1), 85–89. [Google Scholar]
- Dvorak, P.; Dvorakova, D.; Koskova, S.; Vodinska, M.; Najvirtova, M.; Krekac, D.; Hampl, A. Expression and potential role of fibroblast growth factor 2 and its receptors in human embryonic stem cells. Stem Cells 2005, 23, 1200–1211. [Google Scholar] [CrossRef]
- Pesl, M.; Acimovic, I.; Pribyl, J.; Hezova, R.; Vilotic, A.; Fauconnier, J.; Vrbsky, J.; Kruzliak, P.; Skladal, P.; Kara, T.; et al. Forced aggregation and defined factors allow highly uniform-sized embryoid bodies and functional cardiomyocytes from human embryonic and induced pluripotent stem cells. Heart Vessels 2014, 29, 834–846. [Google Scholar] [CrossRef]
- Eiselleova, L.; Peterkova, I.; Neradil, J.; Slaninova, I.; Hampl, A.; Dvorak, P. Comparative study of mouse and human feeder cells for human embryonic stem cells. Int. J. Dev. Biol. 2008, 52, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Lerou, P.H.; Zhao, R.; Huo, H.; Daley, G.Q. Generation of human-induced pluripotent stem cells. Nat. Protoc. 2008, 3, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Available online: https://www.nature.com/articles/nmeth.2089 (accessed on 27 June 2018).
- Kunova, M.; Matulka, K.; Eiselleova, L.; Salykin, A.; Kubikova, I.; Kyrylenko, S.; Hampl, A.; Dvorak, P. Adaptation to robust monolayer expansion produces human pluripotent stem cells with improved viability. Stem Cells Transl. Med. 2013, 2, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Post-hoc Power Calculator. Available online: http://clincalc.com/Stats/Power.aspx (accessed on 22 August 2018).
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.L.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Mack, D.L.; Moreno, C.M.; Strande, J.L.; Mathieu, J.; Shi, Y.; Markert, C.D.; Wang, Z.; Liu, G.; Lawlor, M.W.; et al. Dystrophin-deficient cardiomyocytes derived from human urine: New biologic reagents for drug discovery. Stem Cell Res. 2014, 12, 467–480. [Google Scholar] [CrossRef]
- Acimovic, I.; Vilotic, A.; Pesl, M.; Lacampagne, A.; Dvorak, P.; Rotrekl, V.; Meli, A.C. Human Pluripotent Stem Cell-Derived Cardiomyocytes as Research and Therapeutic Tools. Available online: https://www.hindawi.com/journals/bmri/2014/512831/ (accessed on 18 June 2018).
- Hockemeyer, D.; Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.H.; Sun, N.; Sanchez-Freire, V.; Lee, A.S.; Almeida, P.; Hu, S.; Jan, T.; Wilson, K.D.; Leong, D.; Rosenberg, J.; et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J. Clin. Investig. 2011, 121, 1217–1221. [Google Scholar] [CrossRef]
- Cassano, M.; Berardi, E.; Crippa, S.; Toelen, J.; Barthelemy, I.; Micheletti, R.; Chuah, M.; Vandendriessche, T.; Debyser, Z.; Blot, S.; et al. Alteration of cardiac progenitor cell potency in GRMD dogs. Cell Transplant. 2012, 21, 1945–1967. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Tang, Y.; Lu, A.; Takayama, K.; Usas, A.; Wang, B.; Weiss, K.; Huard, J. The role of Notch signaling in muscle progenitor cell depletion and the rapid onset of histopathology in muscular dystrophy. Hum. Mol. Genet. 2015, 24, 2923–2937. [Google Scholar] [CrossRef] [PubMed]
- Small, C.; Ramroop, J.; Otazo, M.; Huang, L.H.; Saleque, S.; Govind, S. An Unexpected Link Between Notch Signaling and ROS in Restricting the Differentiation of Hematopoietic Progenitors in Drosophila. Genetics 2014, 197, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Q.; Morita, Y.; Jiang, H.; Gross, A.; Lechel, A.; Hildner, K.; Guachalla, L.M.; Gompf, A.; Hartmann, D.; et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell 2012, 148, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Rossi, D.J. DNA-damage-induced differentiation in hematopoietic stem cells. Cell 2012, 148, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.A.; Allen, D.G. Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H846–H855. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. (Lond.) 2008, 586, 2003–2014. [Google Scholar] [CrossRef]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef]
- Messina, S.; Altavilla, D.; Aguennouz, M.; Seminara, P.; Minutoli, L.; Monici, M.C.; Bitto, A.; Mazzeo, A.; Marini, H.; Squadrito, F.; et al. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am. J. Pathol. 2006, 168, 918–926. [Google Scholar] [CrossRef]
- Davidson, S.M.; Duchen, M.R. Effects of NO on mitochondrial function in cardiomyocytes: Pathophysiological relevance. Cardiovasc. Res. 2006, 71, 10–21. [Google Scholar] [CrossRef]
- Hsie, A.W.; Recio, L.; Katz, D.S.; Lee, C.Q.; Wagner, M.; Schenley, R.L. Evidence for reactive oxygen species inducing mutations in mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 9616–9620. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Mockett, R.J.; Orr, W.C. Mechanisms of aging: An appraisal of the oxidative stress hypothesis. Free Radic. Biol. Med. 2002, 33, 575–586. [Google Scholar] [CrossRef]
- Douki, T.; Rivière, J.; Cadet, J. DNA tandem lesions containing 8-oxo-7,8-dihydroguanine and formamido residues arise from intramolecular addition of thymine peroxyl radical to guanine. Chem. Res. Toxicol. 2002, 15, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Krutá, M.; Bálek, L.; Hejnová, R.; Dobšáková, Z.; Eiselleová, L.; Matulka, K.; Bárta, T.; Fojtík, P.; Fajkus, J.; Hampl, A.; et al. Decrease in abundance of apurinic/apyrimidinic endonuclease causes failure of base excision repair in culture-adapted human embryonic stem cells. Stem Cells 2013, 31, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Kreutzer, D.A.; Essigmann, J.M. Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1998, 400, 99–115. [Google Scholar] [CrossRef]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.-C.; Henze, J.; Fletcher, B.S.; Gu, Z.; et al. Dystrophin Is a Tumor Suppressor in Human Cancers with Myogenic Programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.M.; Uddin, M.H.; Dysek, S.; Moser-Thier, K.; Pirker, C.; Höger, H.; Ambros, I.M.; Ambros, P.F.; Berger, W.; Bittner, R.E. DNA Damage, Somatic Aneuploidy, and Malignant Sarcoma Susceptibility in Muscular Dystrophies. PLoS Genet 2011, 7, e1002042. [Google Scholar] [CrossRef]
- Burkhalter, M.D.; Rudolph, K.L.; Sperka, T. Genome instability of ageing stem cells—Induction and defence mechanisms. Ageing Res. Rev. 2015, 23, 29–36. [Google Scholar] [CrossRef]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Feron, O.; Belhassen, L.; Kobzik, L.; Smith, T.W.; Kelly, R.A.; Michel, T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 1996, 271, 22810–22814. [Google Scholar] [CrossRef] [PubMed]
- Dudley, R.W.R.; Danialou, G.; Govindaraju, K.; Lands, L.; Eidelman, D.E.; Petrof, B.J. Sarcolemmal damage in dystrophin deficiency is modulated by synergistic interactions between mechanical and oxidative/nitrosative stresses. Am. J. Pathol. 2006, 168, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Kinugawa, S.; Huang, H.; Wang, Z.; Kaminski, P.M.; Wolin, M.S.; Hintze, T.H. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ. Res. 2005, 96, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Poláková, E.; Kang, C.; Pocsai, K.; Ullrich, N.D.; Niggli, E.; Shirokova, N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res. 2013, 97, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Altun, A.; Temiz, T.K.; Balcı, E.; Polat, Z.A.; Turan, M. Effects of tyrosine kinase inhibitor E7080 and eNOS inhibitor L-NIO on colorectal cancer alone and in combination. Chin. J. Cancer Res. 2013, 25, 572–584. [Google Scholar] [PubMed]
- Díaz-Troya, S.; Najib, S.; Sánchez-Margalet, V. eNOS, nNOS, cGMP and protein kinase G mediate the inhibitory effect of pancreastatin, a chromogranin A-derived peptide, on growth and proliferation of hepatoma cells. Regul. Pept. 2005, 125, 41–46. [Google Scholar] [CrossRef]
- Ouameur, A.A.; Tajmir-Riahi, H.-A. Structural Analysis of DNA Interactions with Biogenic Polyamines and Cobalt(III)hexamine Studied by Fourier Transform Infrared and Capillary Electrophoresis. J. Biol. Chem. 2004, 279, 42041–42054. [Google Scholar] [CrossRef]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef]
- Gratton, J.-P.; Bernatchez, P.; Sessa, W.C. Caveolae and caveolins in the cardiovascular system. Circ. Res. 2004, 94, 1408–1417. [Google Scholar] [CrossRef]
- Michel, J.B.; Feron, O.; Sacks, D.; Michel, T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 1997, 272, 15583–15586. [Google Scholar] [CrossRef] [PubMed]
- Schilling, K.; Opitz, N.; Wiesenthal, A.; Oess, S.; Tikkanen, R.; Müller-Esterl, W.; Icking, A. Translocation of endothelial nitric-oxide synthase involves a ternary complex with caveolin-1 and NOSTRIN. Mol. Biol. Cell 2006, 17, 3870–3880. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Trewin, A.J.; Stojanovska, V.; Robinson, A.; Goodman, C.A.; Nurgali, K.; Betik, A.C.; Stepto, N.; Hayes, A.; McConell, G.K.; et al. Attempting to Compensate for Reduced Neuronal Nitric Oxide Synthase Protein with Nitrate Supplementation Cannot Overcome Metabolic Dysfunction but Rather Has Detrimental Effects in Dystrophin-Deficient mdx Muscle. Neurotherapeutics 2017, 14, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bierie, B.; Li, A.G.; Pathania, S.; Toomire, K.; Dimitrov, S.D.; Liu, B.; Gelman, R.; Giobbie-Hurder, A.; Feunteun, J.; et al. BRCA1/FANCD2/BRG1-Driven DNA Repair Stabilizes the Differentiation State of Human Mammary Epithelial Cells. Mol. Cell 2016, 63, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Pilzecker, B.; Buoninfante, O.A.; van den Berk, P.; Lancini, C.; Song, J.-Y.; Citterio, E.; Jacobs, H. DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E6875–E6883. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jelinkova, S.; Fojtik, P.; Kohutova, A.; Vilotic, A.; Marková, L.; Pesl, M.; Jurakova, T.; Kruta, M.; Vrbsky, J.; Gaillyova, R.; et al. Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells 2019, 8, 53. https://doi.org/10.3390/cells8010053
Jelinkova S, Fojtik P, Kohutova A, Vilotic A, Marková L, Pesl M, Jurakova T, Kruta M, Vrbsky J, Gaillyova R, et al. Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells. 2019; 8(1):53. https://doi.org/10.3390/cells8010053
Chicago/Turabian StyleJelinkova, Sarka, Petr Fojtik, Aneta Kohutova, Aleksandra Vilotic, Lenka Marková, Martin Pesl, Tereza Jurakova, Miriama Kruta, Jan Vrbsky, Renata Gaillyova, and et al. 2019. "Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress" Cells 8, no. 1: 53. https://doi.org/10.3390/cells8010053
APA StyleJelinkova, S., Fojtik, P., Kohutova, A., Vilotic, A., Marková, L., Pesl, M., Jurakova, T., Kruta, M., Vrbsky, J., Gaillyova, R., Valášková, I., Frák, I., Lacampagne, A., Forte, G., Dvorak, P., Meli, A. C., & Rotrekl, V. (2019). Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells, 8(1), 53. https://doi.org/10.3390/cells8010053