Aquaporins and Their Regulation after Spinal Cord Injury




Abstract
1. Introduction–Human Aquaporins
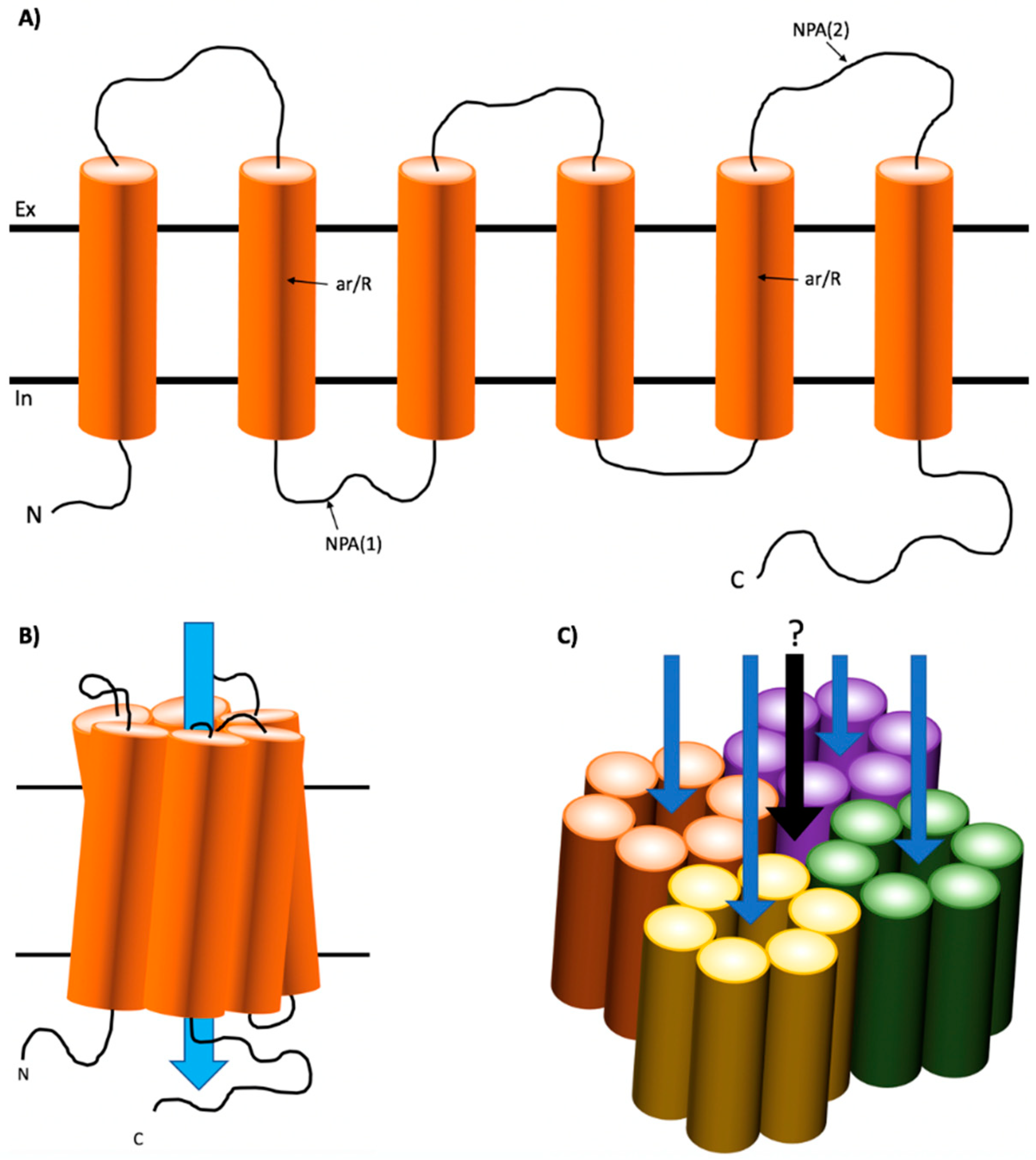
1.1. Structure of Mammalian AQPs
1.2. Localization and Functions of Human AQPs
1.3. Regulation of AQPs
2. AQPs in the CNS–Physiological Roles
2.1. AQP1
2.2. AQP4
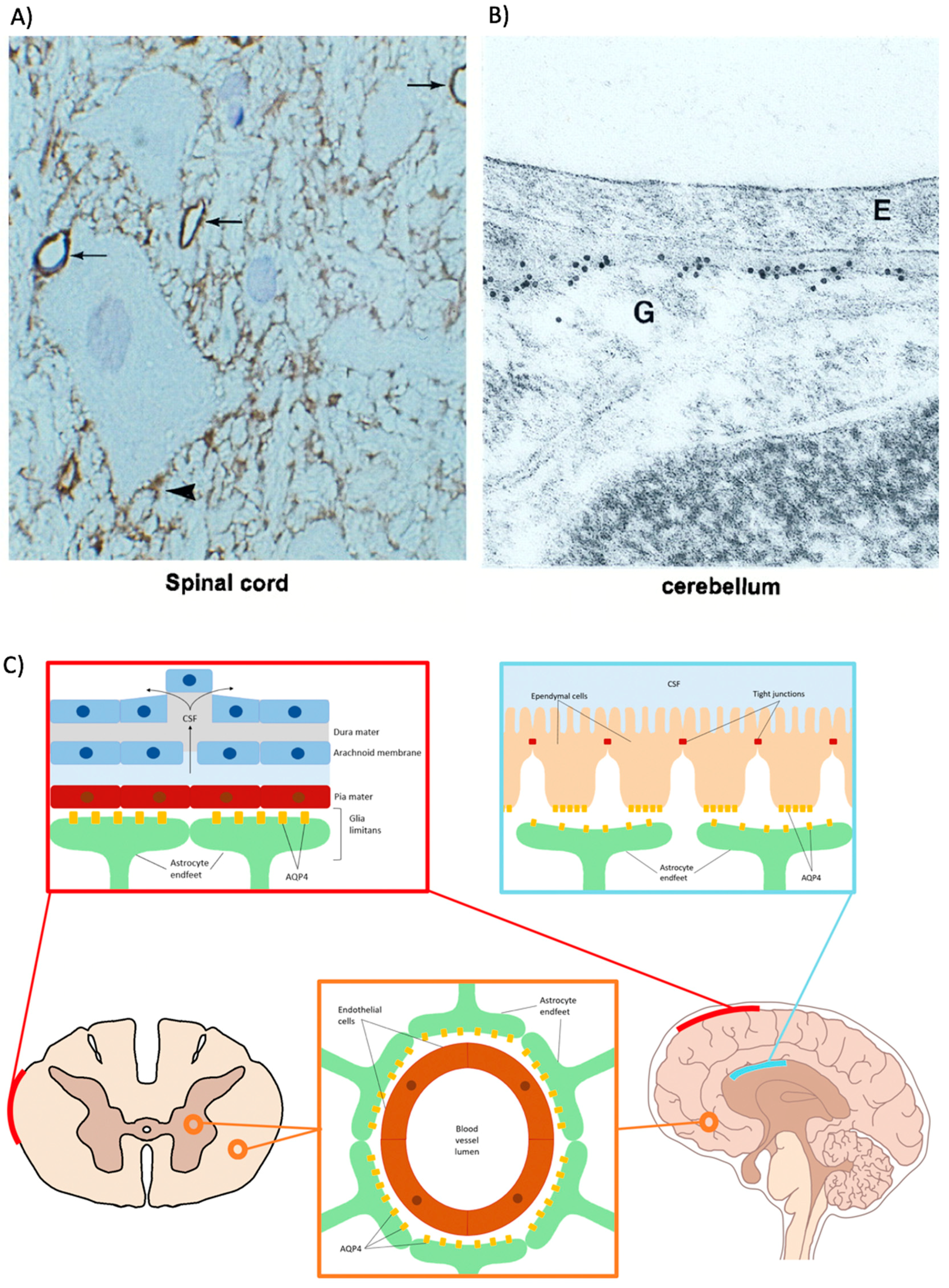
2.2.1. Astrocytes
2.2.2. Ependymal Cells
2.3. AQP9
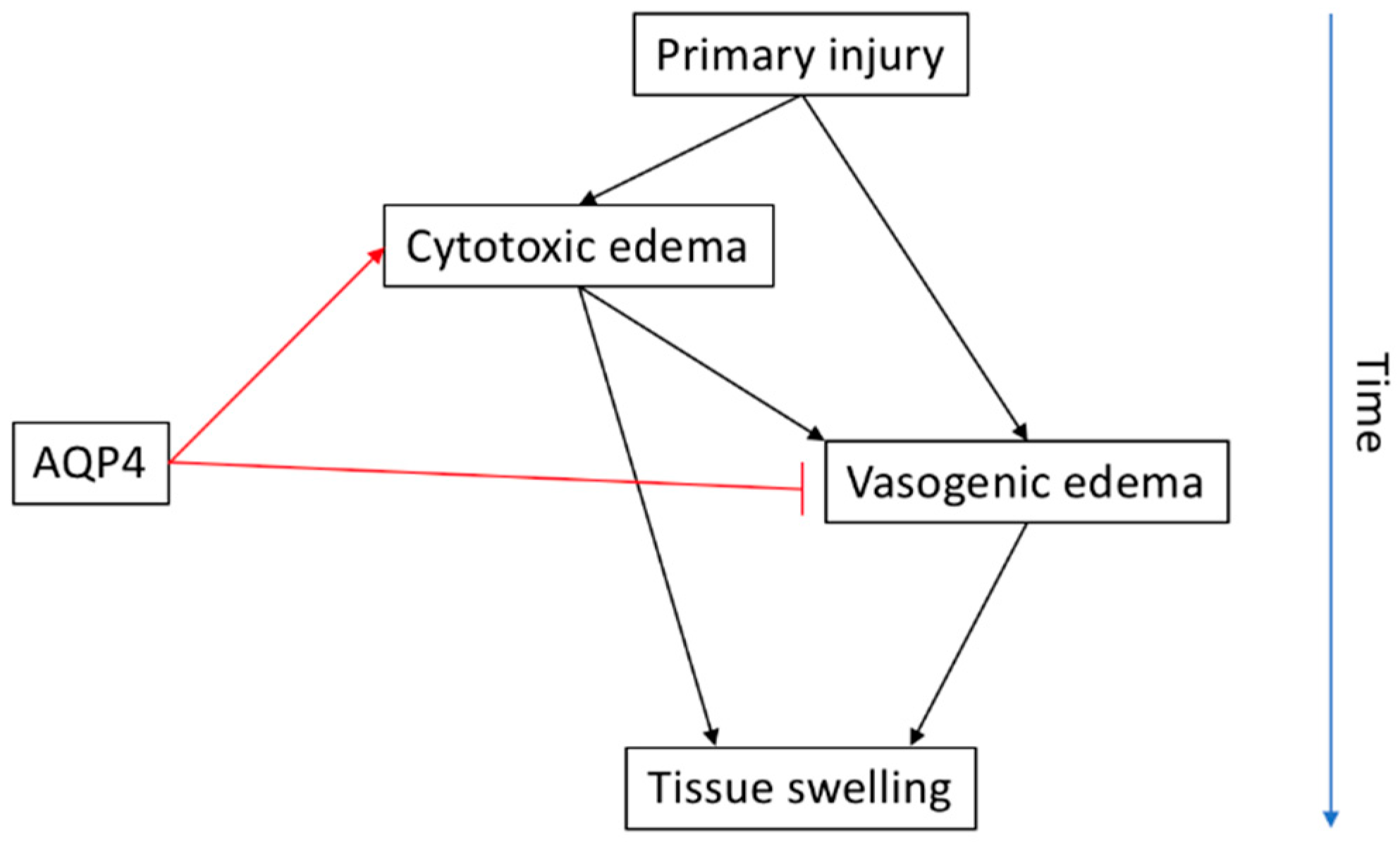
3. Spinal Cord Injury and Oedema
3.1. AQP1 in SCI
3.2. AQP4 in SCI
3.3. AQP9 in SCI
3.4. AQPs as a Clinical Target to Reduce Neurotraumatic Edema: Past, Present, and Future
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Solenov, E. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. AJP Cell Physiol. 2004, 286, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.D.F.; Zeuthen, T.; Chandy, G.; Wright, E.M. Cotransport of water by the Na+/glucose cotransporter. Proc. Natl. Acad. Sci. USA 1996, 93, 13367–13370. [Google Scholar] [CrossRef] [PubMed]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.A.S.; Mitra, A.K.A. Structure and function of aquaporin water channels. Am. J. Physiol. Ren. Physiol. 2000, 278, 13. [Google Scholar] [CrossRef] [PubMed]
- De Groot, B.L.; Hub, J.S.; Grubmüller, H. Dynamics and energetics of permeation through aquaporins. What Do we learn from molecular dynamics simulations? Handb. Exp. Pharmacol. 2009, 190, 57–76. [Google Scholar]
- Harries, W.E.C.; Akhavan, D.; Miercke, L.J.W.; Khademi, S.; Stroud, R.M. The channel architecture of aquaporin 0 at a 2.2-A. resolution. Proc. Natl. Acad. Sci. USA 2004, 101, 14045–14050. [Google Scholar] [CrossRef] [PubMed]
- Horsefield, R.; Nordén, K.; Fellert, M.; Backmark, A.; Törnroth-Horsefield, S.; Terwisscha van Scheltinga, A.C.; Kvassman, J.; Kjellbom, P.; Johanson, U.; Neutze, R. High-resolution x-ray structure of human aquaporin 5. Proc. Natl. Acad. Sci. USA 2008, 105, 13327–13332. [Google Scholar] [CrossRef] [PubMed]
- Magni, F.; Sarto, C.; Ticozzi, D.; Soldi, M.; Bosso, N.; Mocarelli, P.; Kienle, M.G. Proteomic knowledge of human aquaporins. Proteomics 2006, 6, 5637–5649. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; Preston, G.M.; Smith, B.L.; Jung, J.S.; Raina, S.; Moon, C.; Guggino, W.B.; Nielsen, S. Aquaporin CHIP: The archetypal molecular water channel. Am. J. Physiol. 1993, 265, F463–F476. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of Water Channels in Xenopus Oocytes Expressing Red Cell CHIP28 Protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Horner, A.; Zocher, F.; Preiner, J.; Ollinger, N.; Siligan, C.; Akimov, S.A.; Pohl, P. The mobility of single-file water molecules is governed by the number of H-bonds they may form with channel-lining residues. Sci. Adv. 2015, 1, e1400083. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Conner, M.T.; Bill, R.M.; Conner, A.C. Structural determinants of oligomerization of the aquaporin-4 channel. J. Biol. Chem. 2016, 291, 6858–6871. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Mitsuoka, K.; Hiral, T.; Walz, T.; Agre, P.; Heymann, J.B.; Engel, A.; Fujiyoshi, Y. Structural determinants of water permeation through aquaporin-1. Nature 2000, 407, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Libson, A.; Miercke, L.J.W.; Weitzman, C.; Nollert, P.; Krucinski, J.; Stroud, R.M. Structure of a glycerol-conducting channel and the basis for its selectivity. Science 2000, 290, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; King, L.S.; Yasui, M.; Guggino, W.B.; Ottersen, O.P.; Fujiyoshi, Y.; Engel, A.; Nielsen, S. Aquaporin water channels—From atomic structure to clinical medicine. J. Physiol. 2002, 542, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Bastien, D.A. Water transport in human aquaporin-4: Molecular dynamics (MD) simulations. Biochem. Biophys. Res. Commun. 2011, 412, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Jung, J.S.; Guggino, W.B.; Agre, P. The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J. Biol. Chem. 1993, 268, 17–20. [Google Scholar] [PubMed]
- Eriksson, U.K.; Fischer, G.; Friemann, R.; Enkavi, G.; Tajkhorshid, E.; Neutze, R. Subangstrom resolution x-ray structure details aquaporin-water interactions. Science 2013, 340, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Beitz, E.; Wu, B.; Holm, L.M.; Schultz, J.E.; Zeuthen, T. Point mutations in the aromatic/arginine region in aquaporin 1 allow passage of urea, glycerol, ammonia, and protons. Proc. Natl. Acad. Sci. USA 2006, 103, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Zeidel, M.L.; Ambudkar, S.V.; Smith, B.L.; Agre, P. Reconstitution of Functional Water Channels in Liposomes Containing Purified Red Cell CHIP28 Protein. Biochemistry 1992, 31, 7436–7440. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Van Hoek, A.N.; Yeager, M.; Verkman, A.S.; Mitra, A.K. Three-dimensional organization of a human water channel. Nature 1997, 387, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schulten, K.; Tajkhorshid, E. What makes an aquaporin a glycerol channel? A comparative study of AqpZ and GlpF. Structure 2005, 13, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.F.; Egea, P.F.; Robles-Colmenares, Y.; O’Connell, J.D.; Stroud, R.M. Architecture and selectivity in aquaporins: 2.5 Å X-ray structure of aquaporin Z. PLoS Biol. 2003, 1, e72. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Bron, P.; Ranchy, G.; Duchesne, L.; Cavalier, A.; Rolland, J.P.; Raguénès-Nicol, C.; Hubert, J.F.; Haase, W.; Delamarche, C. Aquaglyceroporins, one channel for two molecules. Biochim. Biophys. Acta Bioenerg. 2002, 1555, 181–186. [Google Scholar] [CrossRef]
- Ishibashi, K.; Kuwahara, M.; Gu, Y.; Tanaka, Y.; Marumo, F.; Sasaki, S. Cloning and functional expression of a new aquaporin (AQP9) abundantly expressed in the peripheral leukocytes permeable to water and urea, but not to glycerol. Biochem. Biophys. Res. Commun. 1998, 244, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, V.; Ferroni, S. Water transport between CNS compartments: Functional and molecular interactions between aquaporins and ion channels. Neuroscience 2010, 168, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.-H.; Callies, C.; Rojek, A.; Nielsen, S.; Cooper, T.G. Aquaporin Isoforms Involved in Physiological Volume Regulation of Murine Spermatozoa1. Biol. Reprod. 2009, 80, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Mola, M.G.; Sparaneo, A.; Gargano, C.D.; Spray, D.C.; Svelto, M.; Frigeri, A.; Scemes, E.; Nicchia, G.P. The speed of swelling kinetics modulates cell volume regulation and calcium signaling in astrocytes: A different point of view on the role of aquaporins. Glia 2016, 64, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Deen, P.; Verdijk, M.; Knoers, N.; Wieringa, B.; Monnens, L.; van Os, C.; van Oost, B. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science 1994, 264, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Nejsum, L.N.; Kwon, T.-H.; Jensen, U.B.; Fumagalli, O.; Frøkiaer, J.; Krane, C.M.; Menon, A.G.; King, L.S.; Agre, P.C.; Nielsen, S. Functional requirement of aquaporin-5 in plasma membranes of sweat glands. Proc. Natl. Acad. Sci. USA 2002, 99, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2004, 19, 76–78. [Google Scholar] [CrossRef] [PubMed]
- De Nadal, E.; Ammerer, G.; Posas, F. Controlling gene expression in response to stress. Nat. Rev. Genet. 2011, 12, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Sobue, K.; Miyachi, T.; Inagaki, M.; Miura, Y.; Katsuya, H.; Asai, K. Differential regulation of aquaporin expression in astrocytes by protein kinase C. Mol. Brain Res. 2001, 95, 110–116. [Google Scholar] [CrossRef]
- Bedford, J.J. Aquaporin Expression in Normal Human Kidney and in Renal Disease. J. Am. Soc. Nephrol. 2003, 14, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, Y. Aquaporin expression in normal and pathological skeletal muscles: A brief review with focus on AQP4. J. Biomed. Biotechnol. 2010, 2010, 731569. [Google Scholar] [CrossRef] [PubMed]
- Nesic, O.; Lee, J.; Ye, Z.; Unabia, G.C.; Rafati, D.; Hulsebosch, C.E.; Perez-Polo, J.R. Acute and chronic changes in aquaporin 4 expression after spinal cord injury. Neuroscience 2006, 143, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Törnroth-Horsefield, S.; Wang, Y.; Hedfalk, K.; Johanson, U.; Karlsson, M.; Tajkhorshid, E.; Neutze, R.; Kjellbom, P. Structural mechanism of plant aquaporin gating. Nature 2006, 439, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Walz, T.; Fujiyoshi, Y.; Engel, A. The AQP structure and functional implications. Handb. Exp. Pharmacol. 2009, 31–56. [Google Scholar] [CrossRef]
- Alberga, D.; Nicolotti, O.; Lattanzi, G.; Nicchia, G.P.; Frigeri, A.; Pisani, F.; Benfenati, V.; Mangiatordi, G.F. A new gating site in human aquaporin-4: Insights from molecular dynamics simulations. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Sasaki, S. Trafficking mechanism of water channel aquaporin-2. Biol. Cell 2005, 97, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, K.; Sasaki, S.; Marumo, F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J. Biol. Chem. 1997, 272, 14800–14804. [Google Scholar] [CrossRef] [PubMed]
- Katsura, T.; Gustafson, C.E.; Ausiello, D.A.; Brown, D. Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am. J. Physiol. 1997, 272, F817–F822. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Patil, R.V. Protein kinase A-dependent phosphorylation of aquaporin-1. Biochem. Biophys. Res. Commun. 2000, 273, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Wax, M.B.; Patil, R.V. Regulation of aquaporin-4 water channels by phorbol ester-dependent protein phosphorylation. J. Biol. Chem. 1998, 273, 6001–6004. [Google Scholar] [CrossRef] [PubMed]
- Zelenina, M.; Zelenin, S.; Bondar, A.A.; Brismar, H.; Aperia, A. Water permeability of aquaporin-4 is decreased by protein kinase C. and dopamine. Am. J. Physiol. Renal Physiol. 2002, 283, F309–F318. [Google Scholar] [CrossRef] [PubMed]
- Morest, D.K.; Silver, J. Precursors of neurons, neuroglia, and ependymal cells in the CNS: What are they? Where are they from? How do they get where they are going? Glia 2003, 43, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, C. Neuron-glia interactions: With a little help from glia. Nat. Rev. Neurosci. 2010, 11, 152–153. [Google Scholar] [CrossRef]
- Ropper, A.H. Hyperosmolar Therapy for Raised Intracranial Pressure. N. Engl. J. Med. 2012, 367, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Uldall, M.; Bhatt, D.K.; Kruuse, C.; Juhler, M.; Jansen-Olesen, I.; Jensen, R.H. Choroid plexus aquaporin 1 and intracranial pressure are increased in obese rats: Towards an idiopathic intracranial hypertension model? Int. J. Obes. 2017, 41, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K.; Binder, D.K.; Yang, B.; Schecter, S.; Verkman, A.S.; Manley, G.T. Expression of aquaporin water channels in mouse spinal cord. Neuroscience 2004, 127, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Lasbennes, F.; Magistretti, P.J.; Regli, L. Aquaporins in Brain: Distribution, Physiology, and Pathophysiology. J. Cereb. Blood Flow Metab. 2002, 22, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, J.; Nielsen, S. Distribution of sodium transporters and aquaporin-1 in the human choroid plexus. Am. J. Physiol. Cell Physiol. 2006, 291, C59–C67. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.D.; Mazario, J.; Skinner, K.; Basbaum, A.I. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain 2007, 131, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.I.; Tabunoki, H.; Yamamura, T.; Arima, K.; Konno, H. Human astrocytes express aquaporin-1 and aquaporin-4 in vitro and in vivo. Neuropathology 2007, 27, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Bering, E.A. Choroid plexus and arterial pulsation of cerebrospinal fluid: Demonstration of the choroid plexuses as a cerebrospinal fluid pump. Arch. Neurol. Psychiatry 1955, 73, 165–172. [Google Scholar] [CrossRef]
- Boassa, D.; Stamer, W.D.; Yool, A.J. Ion Channel Function of Aquaporin-1 Natively Expressed in Choroid Plexus. J. Neurosci. 2006, 26, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K.; Watanabe, H.; Yan, D.; Verkman, A.S.; Manley, G.T. Impaired pain sensation in mice lacking Aquaporin-1 water channels. Biochem. Biophys. Res. Commun. 2006, 341, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Brunet, J.F.; Grollimund, L.; Hamou, M.F.; Magistretti, P.J.; Villemure, J.G.; Regli, L. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir. Suppl. 2003, 86, 495–498. [Google Scholar] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Bell, B.A.; Krishna, S. Increased aquaporin 1 water channel expression inhuman brain tumours. Br. J. Cancer 2002, 87, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Mccoy, E.; Sontheimer, H. MAPK induces AQP1 expression in astrocytes following injury. Glia 2010, 58, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Oklinski, M.K.; Lim, J.S.; Choi, H.J.; Oklinska, P.; Skowronski, M.T.; Kwon, T.H. Immunolocalization of Water Channel Proteins AQP1 and AQP4 in Rat Spinal Cord. J. Histochem. Cytochem. 2014, 62, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Arciénega, I.I.; Brunet, J.F.; Bloch, J.; Badaut, J. Cell locations for AQP1, AQP4 and 9 in the non-human primate brain. Neuroscience 2010, 167, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Pérez-Gracia, E.; Espinosa, J.C.; Pumarola, M.; Torres, J.M.; Ferrer, I. Increased expression of water channel aquaporin 1 and aquaporin 4 in Creutzfeldt-Jakob disease and in bovine spongiform encephalopathy-infected bovine-PrP transgenic mice. Acta Neuropathol. 2006, 112, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Hsu, M.S.; Seldin, M.M.; Binder, D.K. Expression of the astrocyte water channel aquaporin-4 in the mouse brain. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Yasumura, T.; Hudson, C.S.; Agre, P.; Nielsen, S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl. Acad. Sci. USA 1998, 95, 11981–11986. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Vitellaro-Zuccarello, L.; Mazzetti, S.; Bosisio, P.; Monti, C.; De Biasi, S. Distribution of aquaporin 4 in rodent spinal cord: Relationship with astrocyte markers and chondroitin sulfate proteoglycans. Glia 2005, 51, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Landis, D.M.D.; Reese, T.S. Arrays of particles in freeze-fractured astrocytic membranes. J. Cell Biol. 1974, 60, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Verbavatz, J.M.; Ma, T.; Gobin, R.; Verkman, A.S. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J. Cell Sci. 1997, 110, 2855–2860. [Google Scholar] [PubMed]
- Smith, A.J.; Jin, B.J.; Ratelade, J.; Verkman, A.S. Aggregation state determines the localization and function of M1-and M23-aquaporin-4 in astrocytes. J. Cell Biol. 2014, 204, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of Select Forms of Spatial Memory and Neurotrophin-Dependent Synaptic Plasticity by Deletion of Glial Aquaporin-4. J. Neurosci. 2011, 31, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, O.; Laake, P.; Klungland, A.; Thoren, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [PubMed]
- 7Benfenati, V.; Caprini, M.; Dovizio, M.; Mylonakou, M.N.; Ferroni, S.; Ottersen, O.P.; Amiry-Moghaddam, M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, I.; Heinemann, U.; Hofmeier, G.; Lux, H.D. Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp. Brain Res. 1980, 40, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Patil, R.V.; Verkman, A.S. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Investig. Ophthalmol. Vis. Sci. 2002, 43, 573–579. [Google Scholar]
- Smith, A.J.; Verkman, A.S. Superresolution Imaging of Aquaporin-4 Cluster Size in Antibody-Stained Paraffin Brain Sections. Biophys. J. 2015, 109, 2511–2522. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Papadopoulos, M.C.; Liu, J.; Li, L.; Zhang, D.; Zhang, H.; Verkman, A.S.; Ma, T. Sporadic obstructive hydrocephalus in Aqp4 null mice. J. Neurosci. Res. 2009, 87, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, J.; Wang, X.; Li, W.; Chen, J.; Sun, H. Pretreatment with AQP4 and NKCC1 Inhibitors Concurrently Attenuated Spinal Cord Edema and Tissue Damage after Spinal Cord Injury in Rats. Front. Physiol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Petit, J.M.; Brunet, J.F.; Magistretti, P.J.; Charriaut-Marlangue, C.; Regli, L. Distribution of Aquaporin 9 in the adult rat brain: Preferential expression in catecholaminergic neurons and in glial cells. Neuroscience 2004, 128, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Mylonakou, M.N.; Petersen, P.H.; Rinvik, E.; Rojek, A.; Vladimarsdottir, E.; Zelenin, S.; Zeuthen, T.; Nielsen, S.; Ottersen, O.P.; Amiry-Moghaddam, M. Analysis of mice with targeted deletion of AQP9 gene provides conclusive evidence for expression of AQP9 in neurons. J. Neurosci. Res. 2009, 87, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; DiGiovanni, S.R.; Christensen, E.I.; Knepper, M.A.; Harris, H.W. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc. Natl. Acad. Sci. USA 1993, 90, 11663–11667. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.M.; Pop, V.; Spagnoli, D.; Ashwal, S.; Obenaus, A.; Badaut, J. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: Possible role in edema resolution? Neuroscience 2012, 222, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Arima, K.; Mizusawa, H.; Satoh, J. Close association of water channel AQP1 with amyloid-beta deposition in Alzheimer disease brains. Acta Neuropathol. 2008, 116, 247. [Google Scholar] [CrossRef] [PubMed]
- Umenishi, F.; Schrier, R.W. Identification and characterization of a novel hypertonicity-responsive element in the human aquaporin-1 gene. Biochem. Biophys. Res. Commun. 2002, 292, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef] [PubMed]
- Thrane, A.S.; Rappold, P.M.; Fujita, T.; Torres, A.; Bekar, L.K.; Takano, T.; Peng, W.; Wang, F.; Rangroo Thrane, V.; Enger, R.; et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc. Natl. Acad. Sci. USA 2011, 108, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Noell, S.; Fallier-Becker, P.; Beyer, C.; Kröger, S.; Mack, A.F.; Wolburg, H. Effects of agrin on the expression and distribution of the water channel protein aquaporin-4 and volume regulation in cultured astrocytes. Eur. J. Neurosci. 2007, 26, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Noell, S.; Wolburg-Buchholz, K.; MacK, A.; Fallier-Becker, P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist 2009, 15, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [PubMed]
- Camassa, L.M.A.; Lunde, L.K.; Hoddevik, E.H.; Stensland, M.; Boldt, H.B.; De Souza, G.A.; Ottersen, O.P.; Amiry-Moghaddam, M. Mechanisms underlying AQP4 accumulation in astrocyte endfeet. Glia 2015, 63, 2073–2091. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.S.; Gorelick-Feldman, D.A.; Davidson, K.G.V.; Yasumura, T.; Neely, J.D.; Agre, P.; Rash, J.E. Aquaporin-4 square array assembly: Opposing actions of M1 and M23 isoforms. Proc. Natl. Acad. Sci. USA 2003, 100, 13609–13614. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ederra, J.; Zhang, H.; Verkman, A.S. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 potassium channels in retinal M.??ller cells. J. Biol. Chem. 2007, 282, 21866–21872. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, I.; Dani, J.A.; De Biasi, M. The medial habenula and interpeduncular nucleus circuitry is critical in addiction, anxiety, and mood regulation. J. Neurochem. 2017, 142, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, C.S.; Wilson, J.R.; Nori, S.; Kotter, M.R.N.; Druschel, C.; Curt, A.; Fehlings, M.G. Traumatic spinal cord injury. Nat. Rev. Dis. Prim. 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Ding, H.; Zhou, H.; Wei, Z.; Liu, L.; Pan, D.; Feng, S. Epidemiology of worldwide spinal cord injury: A literature review. J. Neurorestoratol. 2017, 6, 1–9. [Google Scholar] [CrossRef]
- World Health Organization. International Spinal Cord Society. International Perspectives on Spinal Cord Injury; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Post, M.W.M.; van Leeuwen, C.M.C. Psychosocial issues in spinal cord injury: A review. Spinal Cord 2012, 50, 382–389. [Google Scholar] [CrossRef] [PubMed]
- French, D.D.; Campbell, R.R.; Sabharwal, S.; Nelson, A.L.; Palacios, P.A.; Gavin-Dreschnack, D. Health care costs for patients with chronic spinal cord injury in the Veterans Health Administration. J. Spinal Cord Med. 2007, 30, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Dryden, D.M.; Saunders, L.D.; Jacobs, P.; Schopflocher, D.P.; Rowe, B.H.; May, L.A.; Yiannakoulias, N.; Svenson, L.W.; Voaklander, D.C. Direct health care costs after traumatic spinal cord injury. J. Trauma 2005, 59, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, A.; Marcoux, J.; Radhakrishna, M.; Pelletier, J.; Goulet, B. The Role of Magnetic Resonance Imaging in the Management of Acute Spinal Cord Injury. J. Neurotrauma 2011, 28, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Miyanji, F.; Furlan, J.C.; Aarabi, B.; Arnold, P.M.; Fehlings, M.G. Acute Cervical Traumatic Spinal Cord Injury: MR Imaging Findings Correlated with Neurologic Outcome—Prospective Study with 100 Consecutive Patients 1. Radiology 2007, 243, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Flanders, A.E.; Spettell, C.M.; Friedman, D.P.; Marino, R.J.; Herbison, G.J. The relationship between the functional abilities of patients with cervical spinal cord injury and the severity of damage revealed by MR imaging. AJNR. Am. J. Neuroradiol. 1999, 20, 926–934. [Google Scholar] [PubMed]
- Tator, C.H.; Fehlings, M.G. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J. Neurosurg. 1991, 75, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Gunnarson, E. Potassium Dependent Regulation of Astrocyte Water Permeability Is Mediated by cAMP Signaling. PLoS ONE 2012, 7, e34936. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Chan, P.H.; Longar, S. Alterations in Lipid Metabolism, Na+, K+-ATPase Activity, and Tissue Water Content of Spinal Cord Following Experimental Traumatic Injury. J. Neurochem. 1987, 48, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-K.; Xu, X.-M. Neuroprotection and its molecular mechanism following spinal cord injury. Neural Regen. Res. 2012, 7, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K. Water homeostasis in the brain: Basic concepts. Neuroscience 2004, 129, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Choi, H.B.; Tyson, J.R.; Malik, A.; Dissing-Olesen, L.; Lin, P.J.C.; Cain, S.M.; Cullis, P.R.; Snutch, T.P.; Macvicar, B.A. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell 2015, 161, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.D.; Betz, A.L.; Schielke, G.P.; Hoff, J.T. Transport of sodium from blood to brain in ischemic brain edema. Stroke 1987, 18, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.H.; Bingham, W.G., Jr.; Hunt, W.E. Ultrastructural blood-brain barrier alterations and edema formation in acute spinal cord trauma. J. Neurosurg. 1976, 44, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Noble, L.J.; Wrathall, J.R. Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res. 1989, 482, 57–66. [Google Scholar] [CrossRef]
- Leonard, A.V.; Vink, R. Reducing intrathecal pressure after traumatic spinal cord injury: A potential clinical target to promote tissue survival. Neural Regen. Res. 2015, 10, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins in endothelia. Kidney Int. 2006, 69, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- Nashmi, R.; Fehlings, M.G. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience 2001, 104, 235–251. [Google Scholar] [CrossRef]
- Sun, L.; Li, M.; Ma, X.; Feng, H.; Song, J.; Lv, C.; He, Y. Inhibition of HMGB1 reduces rat spinal cord astrocytic swelling and AQP4 expression after oxygen-glucose deprivation and reoxygenation via TLR4 and NF-ΚB signaling in an IL-6-dependent manner. J. Neuroinflamm. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. Greatly improved neurological outcome after spinal cord compression injury in AQP4-deficient mice. Brain 2008, 131, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Hsu, M.; Seldin, M.; Verkman, A.S.; Scharfman, H.E.; Binder, D.K. Protective role of aquaporin-4 water channels after contusion spinal cord injury. Ann. Neurol. 2010, 67, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, Y.J.; Gao, J.Y.; Li, X.M.; Kong, H.; Zhang, Y.P.; Xiao, M.; Shields, C.B.; Hu, G. Aquaporin-4 mitigates retrograde degeneration of rubrospinal neurons by facilitating edema clearance and glial scar formation after spinal cord injury in mice. Mol. Neurobiol. 2014, 49, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.; Barrachina, M.; Rodríguez, A.; Torrejón-Escribano, B.; Boada, M.; Hernández, I.; Sánchez, M.; Ferrer, I. Aquaporin expression in the cerebral cortex is increased at early stages of Alzheimer disease. Brain Res. 2007, 1128, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Okuda, M.; Asai, J.; Nagashima, G.; Itokawa, H.; Matsunaga, A.; Fujimoto, T.; Suzuki, T. Astrocytes co-express aquaporin-1, -4, and vascular endothelial growth factor in brain edema tissue associated with brain contusion. Acta Neurochir. Suppl. 2006, 398–401. [Google Scholar] [CrossRef]
- Nesic, O.; Lee, J.; Unabia, G.C.; Johnson, K.; Ye, Z.; Vergara, L.; Hulsebosch, C.E.; Perez-Polo, J.R. Aquaporin 1—A novel player in spinal cord injury. J. Neurochem. 2008, 105, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Echevarría, M.; Muñoz-Cabello, A.M.; Sánchez-Silva, R.; Toledo-Aral, J.J.; López-Barneo, J. Development of cytosolic hypoxia and hypoxia-inducible factor stabilization are facilitated by aquaporin-1 expression. J. Biol. Chem. 2007, 282, 30207–30215. [Google Scholar] [CrossRef] [PubMed]
- Beggs, J.L.; Waggener, J.D. Vasogenic edema in the injured spinal cord: A method of evaluating the extent of blood-brain barrier alteration to horseradish peroxidase. Exp. Neurol. 1975, 49, 86–96. [Google Scholar] [CrossRef]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef]
- Cheriyan, T.; Ryan, D.J.; Weinreb, J.H.; Cheriyan, J.; Paul, J.C.; Lafage, V.; Kirsch, T.; Errico, T.J. Spinal cord injury models: A review. Spinal Cord 2014, 52, 588–595. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.; Monterrubio, A.; Ariza, J.; Martínez-Cerdeño, V. Calibrated Forceps Model of Spinal Cord Compression Injury. J. Vis. Exp. 2015, 98. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.M.; Li, J.J.; Sun, W.; Yang, D.G.; Yang, M.L.; Du, L.J.; Gu, R.; Gao, F.; Li, J.; Chu, H.Y.; et al. Myelotomy reduces spinal cord edema and inhibits aquaporin-4 and aquaporin-9 expression in rats with spinal cord injury. Spinal Cord 2015, 53, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Warms, C.A.; Turner, J.A.; Marshall, H.M.; Cardenas, D.D. Treatments for chronic pain associated with spinal cord injuries: Many are tried, few are helpful. Clin. J. Pain 2002, 18, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Bracken, M.B. Steroids for acute spinal cord injury. Cochrane Database Syst. Rev. 2012, 1, CD001046. [Google Scholar] [CrossRef]
- Hirano, Y.; Okimoto, N.; Kadohira, I.; Suematsu, M.; Yasuoka, K.; Yasui, M. Molecular mechanisms of how mercury inhibits water permeation through aquaporin-1: Understanding by molecular dynamics simulation. Biophys. J. 2010, 98, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Kondo, S.; Hara, S.; Morishita, Y. The evolutionary aspects of aquaporin family. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R566–R576. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.N.; Xie, L.L.; Liang, R.; Sun, X.L.; Fan, Y.; Hu, G. AQP4 Knockout Aggravates Ischemia/Reperfusion Injury in Mice. CNS Neurosci. Ther. 2012, 18, 388–394. [Google Scholar] [CrossRef]
- Tourdias, T.; Mori, N.; Dragonu, I.; Cassagno, N.; Boiziau, C.; Aussudre, J.; Brochet, B.; Moonen, C.; Petry, K.G.; Dousset, V. Differential aquaporin 4 expression during edema build-up and resolution phases of brain inflammation. J. Neuroinflamm. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.M.; Adami, A.; Pop, V.; Bellone, J.A.; Coats, J.S.; Hartman, R.E.; Ashwal, S.; Obenaus, A.; Badaut, J. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J. Cereb. Blood Flow Metab. 2013, 33, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef]
- Braughler, J.M.; Hall, E.D. Correlation of methylprednisolone levels in cat spinal cord with its effects on (Na+ + K+)-ATPase, lipid peroxidation, and alpha motor neuron function. J. Neurosurg. 1982, 56, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Gerndt, S.J.; Rodriguez, J.L.; Pawlik, J.W.; Taheri, P.A.; Wahl, W.L.; Micheals, A.J.; Papadopoulos, S.M. Consequences of high-dose steroid therapy for acute spinal cord injury. J. Trauma 1997, 42, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Shao, J.; Qi, H.H.; Song, D.W.; Zhu, W. Risk factors for respiratory failure with tetraplegia after acute traumatic cervical spinal cord injury. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 9–14. [Google Scholar] [PubMed]
- Cabrera-Aldana, E.E.; Ruelas, F.; Aranda, C.; Rincon-Heredia, R.; Martínez-Cruz, A.; Reyes-Sánchez, A.; Guizar-Sahagún, G.; Tovar-y-Romo, L.B. Methylprednisolone Administration Following Spinal Cord Injury Reduces Aquaporin 4 Expression and Exacerbates Edema. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Nakamura, T.; Ikeda, T.; Takagi, K. Potent protective effects of melatonin on experimental spinal cord injury. Spine 2000, 25, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Kaptanoglu, E.; Tuncel, M.; Palaoglu, S.; Konan, A.; Demirpençe, E.; Kilinç, K. Comparison of the effects of melatonin and methylprednisolone in experimental spinal cord injury. J. Neurosurg. Spine 2000, 93, 77–84. [Google Scholar] [CrossRef]
- Li, C.; Chen, X.; Qiao, S.; Liu, X.; Liu, C.; Zhu, D.; Su, J.; Wang, Z. Melatonin lowers edema after spinal cord injury. Neural Regen. Res. 2014, 9, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Schiaveto-de-Souza, A.; da-Silva, C.A.; Defino, H.L.A.; Del Bel, E.A. Effect of melatonin on the functional recovery from experimental traumatic compression of the spinal cord. Braz. J. Med. Biol. Res. 2013, 46, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.M.; Beattie, M.S.; Bresnahan, J.C. A Sensitive and Reliable Locomotor Rating Scale for Open Field Testing in Rats. J. Neurotrauma 1995, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, A.S.; Tator, C.H. Objective clinical assessment of motor function after experimental spinal cord injury in the rat. J. Neurosurg. 1977, 47, 577–581. [Google Scholar] [CrossRef] [PubMed]
- McCoy, E.S.; Haas, B.R.; Sontheimer, H. Water permeability through aquaporin-4 is regulated by protein kinase C and becomes rate-limiting for glioma invasion. Neuroscience 2010, 168, 971–981. [Google Scholar] [CrossRef]
- Mitsuma, T.; Tani, K.; Hiroaki, Y.; Kamegawa, A.; Suzuki, H.; Hibino, H.; Kurachi, Y.; Fujiyoshi, Y. Influence of the Cytoplasmic Domains of Aquaporin-4 on Water Conduction and Array Formation. J. Mol. Biol. 2010, 402, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Gunnarson, E.; Zelenina, M.; Axehult, G.; Song, Y.; Bondar, A.; Krieger, P.; Brismar, H.; Zelenin, S.; Aperia, A. Identification of a molecular target for glutamate regulation of astrocyte water permeability. Glia 2008, 56, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Fenton, R.A.; Zeuthen, T.; MacAulay, N. Vasopressin-dependent short-term regulation of aquaporin 4 expressed in Xenopus oocytes. Neuroscience 2009, 164, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Brinton, R.D. Vasopressin-induced cytoplasmic and nuclear calcium signaling in cultured cortical astrocytes. Brain Res. 2002, 943, 117–131. [Google Scholar] [CrossRef]
- Carmosino, M.; Procino, G.; Tamma, G.; Mannucci, R.; Svelto, M.; Valenti, G. Trafficking and phosphorylation dynamics of AQP4 in histamine-treated human gastric cells. Biol. Cell 2007, 99, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Day, R.E.; Taylor, L.H.J.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and molecular mechanisms of the rapid tonicity-induced relocalization of the aquaporin 4 channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Spinal Cord | Brain | References | |||
|---|---|---|---|---|---|
| Location | Function | Location | Function | ||
| AQP1 | Unmyelinated sensory fibers in DRG, DH, and grey matter | Pain processing | Ependymal cells in the CPE | CSF production | [31,50,52,53,54,55,56,57,58,59,60,61,62,63] |
| Endothelial cells within glia limitans | Unknown | Perivascular astrocytes in white matter, and glial limitans | Cell migration, water homeostasis | ||
| Astrocytes within glia limitans, dorsal horn, central canal and white matter | Cell migration, water homeostasis | Neurons surrounding pial blood vessels | Axonal elongation | ||
| Ependymal cells within glia limitans and central canal | CSF production (*) | ||||
| AQP4 | Astrocyte end-foot processes encircling capillaries in grey and white matter | Water homeostasis, ionic homeostasis | Perivascular end-foot processes in white matter | Water and waste clearance Neuronal excitability | [50,61,62,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81] |
| Astrocyte end-foot processes enveloping myelinated axons and axonal synapses | Regulation of perisynaptic volume | ||||
| Astrocyte processes facing glia limitans and surrounding central canal | Water homeostasis (*) | Perisynaptic astrocyte end-foot processes | Synaptic function Perisynaptic volume Synapse plasticity K+ homeostasis | ||
| Fibrous astrocytes | Unknown | ||||
| Ependymal cells within glia limitans | CSF production (*), water homeostasis (*) | Subpial and subependymal astrocyte processes | Water flow | ||
| Muller cells | K+ clearance | ||||
| AQP9 | Astrocyte end-foot processes in white matter and glia limitans | Water flow (*) | Catacholinergic neurons | Energy metabolism (*) | [50,62,82,83] |
| Astrocytes in glia limitans | Water flow (*) | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halsey, A.M.; Conner, A.C.; Bill, R.M.; Logan, A.; Ahmed, Z. Aquaporins and Their Regulation after Spinal Cord Injury. Cells 2018, 7, 174. https://doi.org/10.3390/cells7100174
Halsey AM, Conner AC, Bill RM, Logan A, Ahmed Z. Aquaporins and Their Regulation after Spinal Cord Injury. Cells. 2018; 7(10):174. https://doi.org/10.3390/cells7100174
Chicago/Turabian StyleHalsey, Andrea M., Alex C. Conner, Roslyn M. Bill, Ann Logan, and Zubair Ahmed. 2018. "Aquaporins and Their Regulation after Spinal Cord Injury" Cells 7, no. 10: 174. https://doi.org/10.3390/cells7100174
APA StyleHalsey, A. M., Conner, A. C., Bill, R. M., Logan, A., & Ahmed, Z. (2018). Aquaporins and Their Regulation after Spinal Cord Injury. Cells, 7(10), 174. https://doi.org/10.3390/cells7100174