
Targeted Redox Regulation α-Ketoglutarate Dehydrogenase Complex for the Treatment of Human Diseases


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. KGDHc Is a Potent mtO2•−/mtH2O2 Source
3. Dynamic Redox Control of KGDHc May Be a New Therapeutic Approach to Prevent Oxidative Distress
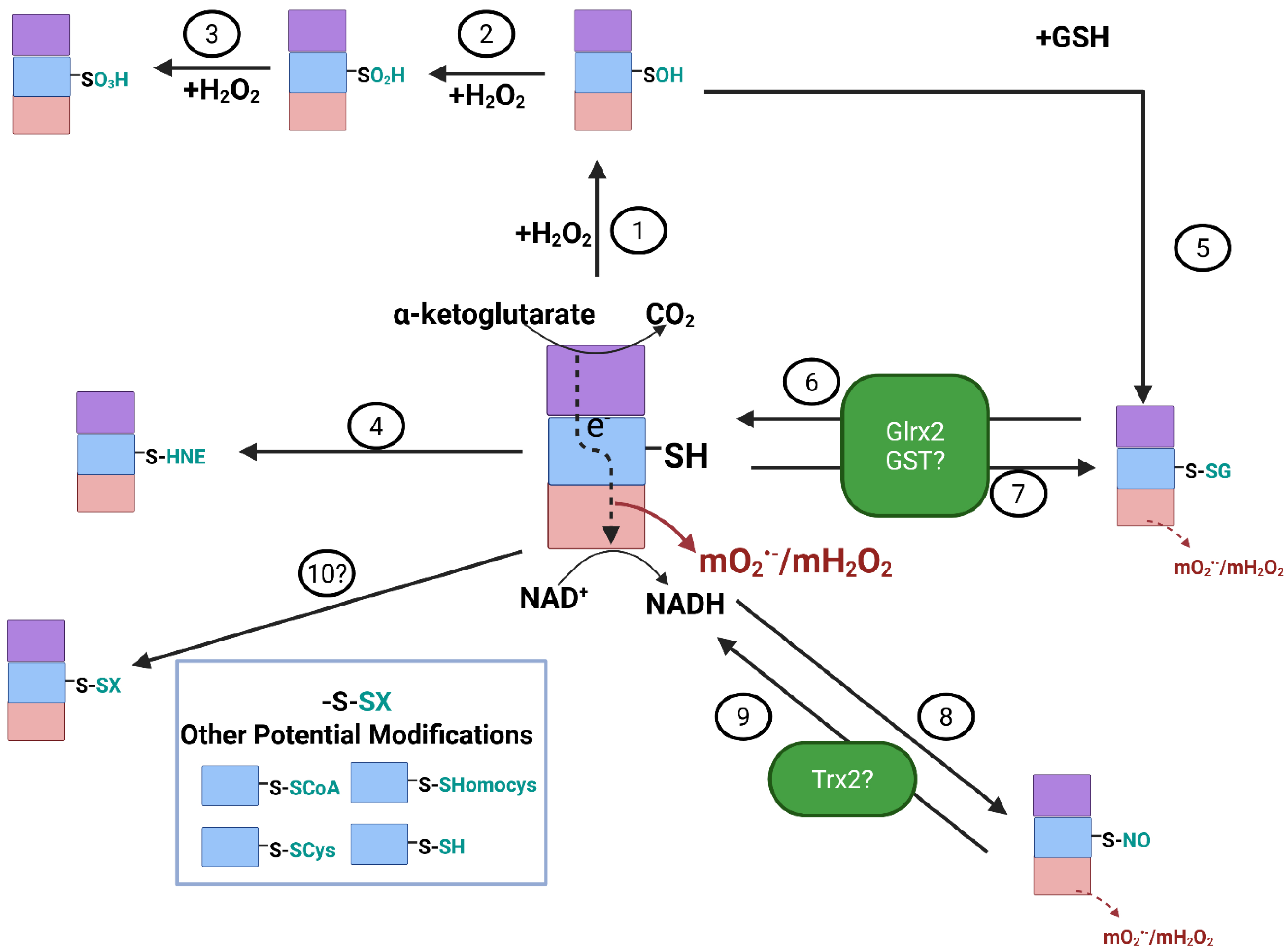
3.1. Reversible S-Glutathionylation and S-Nitrosation Regulate mO2•−/mH2O2 Production by KGDHc
3.2. The Redox Sensing Properties of KGDHc Could Be a Therapeutic Target for the Treatment of NAFLD
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Chen, X.; Zhang, M.; Yin, J.; Yang, Z.; Gao, X.; Zhang, S.; Yang, M. Molecular architecture of the mammalian 2-oxoglutarate dehydrogenase complex. Nat. Commun. 2024, 15, 8407. [Google Scholar] [CrossRef]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. alpha-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef]
- Gibson, G.E.; Park, L.C.; Sheu, K.F.; Blass, J.P.; Calingasan, N.Y. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem. Int. 2000, 36, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. An update on the role of mitochondrial alpha-ketoglutarate dehydrogenase in oxidative stress. Mol. Cell Neurosci. 2013, 55, 13–16. [Google Scholar] [CrossRef]
- Yan, L.J.; Wang, Y. Roles of Dihydrolipoamide Dehydrogenase in Health and Disease. Antioxid. Redox Signal 2023, 39, 794–806. [Google Scholar] [CrossRef]
- Heublein, M.; Burguillos, M.A.; Vogtle, F.N.; Teixeira, P.F.; Imhof, A.; Meisinger, C.; Ott, M. The novel component Kgd4 recruits the E3 subunit to the mitochondrial alpha-ketoglutarate dehydrogenase. Mol. Biol. Cell 2014, 25, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. The emerging importance of the alpha-keto acid dehydrogenase complexes in serving as intracellular and intercellular signaling platforms for the regulation of metabolism. Redox Biol. 2024, 72, 103155. [Google Scholar] [CrossRef]
- Hansen, G.E.; Gibson, G.E. The alpha-Ketoglutarate Dehydrogenase Complex as a Hub of Plasticity in Neurodegeneration and Regeneration. Int. J. Mol. Sci. 2022, 23, 12403. [Google Scholar] [CrossRef] [PubMed]
- Yap, Z.Y.; Strucinska, K.; Matsuzaki, S.; Lee, S.; Si, Y.; Humphries, K.; Tarnopolsky, M.A.; Yoon, W.H. A biallelic pathogenic variant in the OGDH gene results in a neurological disorder with features of a mitochondrial disease. J. Inherit. Metab. Dis. 2021, 44, 388–400. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Graf, A.V.; Kazantsev, A.V.; Boyko, A.I.; Aleshin, V.A.; Ksenofontov, A.L.; Bunik, V.I. Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation. Pharmaceuticals 2022, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.H.; Sandoval, H.; Nagarkar-Jaiswal, S.; Jaiswal, M.; Yamamoto, S.; Haelterman, N.A.; Putluri, N.; Putluri, V.; Sreekumar, A.; Tos, T.; et al. Loss of Nardilysin, a Mitochondrial Co-chaperone for alpha-Ketoglutarate Dehydrogenase, Promotes mTORC1 Activation and Neurodegeneration. Neuron 2017, 93, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Zayed, M.A.; Massry, S.G. Impaired activity of alpha-ketoglutarate dehydrogenase of heart mitochondria in chronic renal failure: Role of secondary hyperparathyroidism. Nephron 1991, 59, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Bertero, E.; Nickel, A.; Kohlhaas, M.; Gibson, G.E.; Heggermont, W.; Heymans, S.; Maack, C. Selective NADH communication from alpha-ketoglutarate dehydrogenase to mitochondrial transhydrogenase prevents reactive oxygen species formation under reducing conditions in the heart. Basic. Res. Cardiol. 2020, 115, 53. [Google Scholar] [CrossRef]
- Maguire, D.; Talwar, D.; Shiels, P.G.; McMillan, D. The role of thiamine dependent enzymes in obesity and obesity related chronic disease states: A systematic review. Clin. Nutr. ESPEN 2018, 25, 8–17. [Google Scholar] [CrossRef]
- Atlante, S.; Visintin, A.; Marini, E.; Savoia, M.; Dianzani, C.; Giorgis, M.; Surun, D.; Maione, F.; Schnutgen, F.; Farsetti, A.; et al. alpha-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell Death Dis. 2018, 9, 756. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- Massey, V.; Muller, F.; Feldberg, R.; Schuman, M.; Sullivan, P.A.; Howell, L.G.; Mayhew, S.G.; Matthews, R.G.; Foust, G.P. The reactivity of flavoproteins with sulfite. Possible relevance to the problem of oxygen reactivity. J. Biol. Chem. 1969, 244, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Massey, V.; Strickland, S.; Mayhew, S.G.; Howell, L.G.; Engel, P.C.; Matthews, R.G.; Schuman, M.; Sullivan, P.A. The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem. Biophys. Res. Commun. 1969, 36, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Nemeria, N.S.; Torocsik, B.; Tretter, L.; Nilsson, M.; Jordan, F.; Adam-Vizi, V. Formation of reactive oxygen species by human and bacterial pyruvate and 2-oxoglutarate dehydrogenase multienzyme complexes reconstituted from recombinant components. Free Radic. Biol. Med. 2015, 89, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Craig Ayre, D.; Christian, S.L. Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biol. 2016, 8, 285–297. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Gardiner, D.; O’Brien, M. 2-Oxoglutarate dehydrogenase is a more significant source of O2·−/H2O2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radic. Biol. Med. 2016, 97, 501–512. [Google Scholar] [CrossRef]
- Zundorf, G.; Kahlert, S.; Bunik, V.I.; Reiser, G. alpha-Ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience 2009, 158, 610–616. [Google Scholar] [CrossRef]
- Horvath, G.; Svab, G.; Komlodi, T.; Ravasz, D.; Kacso, G.; Doczi, J.; Chinopoulos, C.; Ambrus, A.; Tretter, L. Reverse and Forward Electron Flow-Induced H2O2 Formation Is Decreased in alpha-Ketoglutarate Dehydrogenase (alpha-KGDH) Subunit (E2 or E3) Heterozygote Knock Out Animals. Antioxidants 2022, 11, 1487. [Google Scholar] [CrossRef]
- Szabo, E.; Nagy, B.; Czajlik, A.; Komlodi, T.; Ozohanics, O.; Tretter, L.; Ambrus, A. Mitochondrial Alpha-Keto Acid Dehydrogenase Complexes: Recent Developments on Structure and Function in Health and Disease. Subcell. Biochem. 2024, 104, 295–381. [Google Scholar] [CrossRef]
- Grayson, C.; Faerman, B.; Koufos, O.; Mailloux, R.J. Fatty acid oxidation drives mitochondrial hydrogen peroxide production by alpha-ketoglutarate dehydrogenase. J. Biol. Chem. 2024, 300, 107159. [Google Scholar] [CrossRef]
- Oldford, C.; Kuksal, N.; Gill, R.; Young, A.; Mailloux, R.J. Estimation of the hydrogen peroxide producing capacities of liver and cardiac mitochondria isolated from C57BL/6N and C57BL/6J mice. Free Radic. Biol. Med. 2019, 135, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Slade, L.; Chalker, J.; Kuksal, N.; Young, A.; Gardiner, D.; Mailloux, R.J. Examination of the superoxide/hydrogen peroxide forming and quenching potential of mouse liver mitochondria. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Oldford, C.; Mailloux, R.J. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol. 2020, 28, 101339. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I. 2-Oxo acid dehydrogenase complexes in redox regulation. Eur. J. Biochem. 2003, 270, 1036–1042. [Google Scholar] [CrossRef]
- Chalifoux, O.; Faerman, B.; Mailloux, R.J. Mitochondrial hydrogen peroxide production by pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase in oxidative eustress and oxidative distress. J. Biol. Chem. 2023, 299, 105399. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef]
- Bunik, V.I.; Denton, T.T.; Xu, H.; Thompson, C.M.; Cooper, A.J.; Gibson, G.E. Phosphonate analogues of alpha-ketoglutarate inhibit the activity of the alpha-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry 2005, 44, 10552–10561. [Google Scholar] [CrossRef] [PubMed]
- Artiukhov, A.V.; Kazantsev, A.V.; Lukashev, N.V.; Bellinzoni, M.; Bunik, V.I. Selective Inhibition of 2-Oxoglutarate and 2-Oxoadipate Dehydrogenases by the Phosphonate Analogs of Their 2-Oxo Acid Substrates. Front. Chem. 2020, 8, 596187. [Google Scholar] [CrossRef]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site I(Q) of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef]
- Chalifoux, O.; Sterman, S.; Faerman, B.; Li, M.; Trezza, S.; Michalak, M.; Agellon, L.B.; Mailloux, R.J. MitoSNO inhibits mitochondrial hydrogen peroxide (mtH2O2) generation by alpha-ketoglutarate dehydrogenase (KGDH). J. Biol. Chem. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Grayson, C.; Chalifoux, O.; Russo, M.S.T.; Avizonis, D.Z.; Sterman, S.; Faerman, B.; Koufos, O.; Agellon, L.B.; Mailloux, R.J. Ablating the glutaredoxin-2 (Glrx2) gene protects male mice against non-alcoholic fatty liver disease (NAFLD) by limiting oxidative distress. Free Radic. Biol. Med. 2024, 224, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Kabysheva, M.S.; Storozhevykh, T.P.; Pinelis, V.G.; Bunik, V.I. Synthetic regulators of the 2-oxoglutarate oxidative decarboxylation alleviate the glutamate excitotoxicity in cerebellar granule neurons. Biochem. Pharmacol. 2009, 77, 1531–1540. [Google Scholar] [CrossRef]
- Trofimova, L.; Lovat, M.; Groznaya, A.; Efimova, E.; Dunaeva, T.; Maslova, M.; Graf, A.; Bunik, V. Behavioral impact of the regulation of the brain 2-oxoglutarate dehydrogenase complex by synthetic phosphonate analog of 2-oxoglutarate: Implications into the role of the complex in neurodegenerative diseases. Int. J. Alzheimers Dis. 2010, 2010, 749061. [Google Scholar] [CrossRef]
- Chen, H.; Denton, T.T.; Xu, H.; Calingasan, N.; Beal, M.F.; Gibson, G.E. Reductions in the mitochondrial enzyme alpha-ketoglutarate dehydrogenase complex in neurodegenerative disease-beneficial or detrimental? J. Neurochem. 2016, 139, 823–838. [Google Scholar] [CrossRef]
- Wang, K.; Moore, A.; Grayson, C.; Mailloux, R.J. S-nitroso-glutathione (GSNO) inhibits hydrogen peroxide production by alpha-ketoglutarate dehydrogenase: An investigation into sex and diet effects. Free Radic. Biol. Med. 2023, 204, 287–300. [Google Scholar] [CrossRef]
- Pardee, T.S.; Luther, S.; Buyse, M.; Powell, B.L.; Cortes, J. Devimistat in combination with high dose cytarabine and mitoxantrone compared with high dose cytarabine and mitoxantrone in older patients with relapsed/refractory acute myeloid leukemia: ARMADA 2000 Phase III study. Future Oncol. 2019, 15, 3197–3208. [Google Scholar] [CrossRef]
- Kumstel, S.; Schreiber, T.; Goldstein, L.; Stenzel, J.; Lindner, T.; Joksch, M.; Zhang, X.; Wendt, E.H.U.; Schonrogge, M.; Krause, B.; et al. Targeting pancreatic cancer with combinatorial treatment of CPI-613 and inhibitors of lactate metabolism. PLoS ONE 2022, 17, e0266601. [Google Scholar] [CrossRef]
- Reddy, V.B.; Boteju, L.; Boteju, A.; Shen, L.; Kassahun, K.; Reddy, N.; Sheldon, A.; Luther, S.; Hu, K. In Vitro and In Vivo Metabolism of a Novel Antimitochondrial Cancer Metabolism Agent, CPI-613, in Rat and Human. Drug Metab. Dispos. 2022, 50, 361–373. [Google Scholar] [CrossRef]
- Arnold, C.; Demuth, P.; Seiwert, N.; Wittmann, S.; Boengler, K.; Rasenberger, B.; Christmann, M.; Huber, M.; Brunner, T.; Linnebacher, M.; et al. The Mitochondrial Disruptor Devimistat (CPI-613) Synergizes with Genotoxic Anticancer Drugs in Colorectal Cancer Therapy in a Bim-Dependent Manner. Mol. Cancer Ther. 2022, 21, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P. 2-Hydroxyglutarate in Cancer Cells. Antioxid. Redox Signal 2020, 33, 903–926. [Google Scholar] [CrossRef] [PubMed]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Redox regulation of mitochondrial biogenesis. Free Radic. Biol. Med. 2012, 53, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Costa, N.J.; Dahm, C.C.; Beer, S.M.; Brown, S.E.; Filipovska, A.; Murphy, M.P. Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal 2005, 7, 999–1010. [Google Scholar] [CrossRef]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 2023, 67, 102926. [Google Scholar] [CrossRef]
- Nulton-Persson, A.C.; Szweda, L.I. Modulation of mitochondrial function by hydrogen peroxide. J. Biol. Chem. 2001, 276, 23357–23361. [Google Scholar] [CrossRef]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nat. Rev. Mol. Cell Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983 e924. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N. Oxiforms: Unique cysteine residue- and chemotype-specified chemical combinations can produce functionally-distinct proteoforms: Like how mixing primary colours creates new shades, cysteine residue- and chemotype-specified chemical combinations can produce functionally-distinct proteoforms called oxiforms. Bioessays 2023, 45, e2200248. [Google Scholar] [CrossRef]
- Cobley, J.N.; Chatzinikolaou, P.N.; Schmidt, C.A. The nonlinear cysteine redox dynamics in the i-space: A proteoform-centric theory of redox regulation. Redox Biol. 2025, 81, 103523. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef]
- Mu, B.; Zeng, Y.; Luo, L.; Wang, K. Oxidative stress-mediated protein sulfenylation in human diseases: Past, present, and future. Redox Biol. 2024, 76, 103332. [Google Scholar] [CrossRef]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef]
- Nickel, K.; Zhu, L.; Mangalindan, R.; Snyder, J.M.; Tucker, M.; Whitson, J.; Sweetwyne, M.; Valencia, A.P.; Klug, J.; Jiang, Z.; et al. Long-term treatment with Elamipretide enhances healthy aging phenotypes in mice. Aging Pathobiol. Ther. 2022, 4, 76–83. [Google Scholar] [CrossRef]
- Mitchell, W.; Pharaoh, G.; Tyshkovskiy, A.; Campbell, M.; Marcinek, D.J.; Gladyshev, V.N. The mitochondrial-targeted peptide therapeutic elamipretide improves cardiac and skeletal muscle function during aging without detectable changes in tissue epigenetic or transcriptomic age. bioRxiv 2024. [Google Scholar] [CrossRef]
- Whitson, J.A.; Martin-Perez, M.; Zhang, T.; Gaffrey, M.J.; Merrihew, G.E.; Huang, E.; White, C.C.; Kavanagh, T.J.; Qian, W.J.; Campbell, M.D.; et al. Elamipretide (SS-31) treatment attenuates age-associated post-translational modifications of heart proteins. Geroscience 2021, 43, 2395–2412. [Google Scholar] [CrossRef]
- Zhang, H.; Alder, N.N.; Wang, W.; Szeto, H.; Marcinek, D.J.; Rabinovitch, P.S. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. Elife 2020, 9, e60827. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Rai, A.; Checker, R.; Patwardhan, R.S.; Singh, B.; Sharma, D.; Sandur, S.K. Role of protein S-Glutathionylation in cancer progression and development of resistance to anti-cancer drugs. Arch. Biochem. Biophys. 2021, 704, 108890. [Google Scholar] [CrossRef] [PubMed]
- Brzozowa-Zasada, M.; Piecuch, A.; Bajdak-Rusinek, K.; Golabek, K.; Michalski, M.; Janelt, K.; Matysiak, N. Glutaredoxin 2 Protein (Grx2) as an Independent Prognostic Factor Associated with the Survival of Colon Adenocarcinoma Patients. Int. J. Mol. Sci. 2024, 25, 1060. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, A.; Mailloux, R.J.; Adjeitey, C.N.; Harper, M.E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim. Biophys. Acta 2013, 1833, 80–89. [Google Scholar] [CrossRef]
- Humphries, K.M.; Szweda, L.I. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: Reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry 1998, 37, 15835–15841. [Google Scholar] [CrossRef]
- McLain, A.L.; Cormier, P.J.; Kinter, M.; Szweda, L.I. Glutathionylation of alpha-ketoglutarate dehydrogenase: The chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 2013, 61, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nulton-Persson, A.C.; Starke, D.W.; Mieyal, J.J.; Szweda, L.I. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry 2003, 42, 4235–4242. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.; Gardiner, D.; Kuksal, N.; Mailloux, R.J. Characterization of the impact of glutaredoxin-2 (GRX2) deficiency on superoxide/hydrogen peroxide release from cardiac and liver mitochondria. Redox Biol. 2018, 15, 216–227. [Google Scholar] [CrossRef]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef]
- Seim, G.L.; Britt, E.C.; John, S.V.; Yeo, F.J.; Johnson, A.R.; Eisenstein, R.S.; Pagliarini, D.J.; Fan, J. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-gamma stimulation. Nat. Metab. 2019, 1, 731–742. [Google Scholar] [CrossRef]
- Seim, G.L.; John, S.V.; Arp, N.L.; Fang, Z.; Pagliarini, D.J.; Fan, J. Nitric oxide-driven modifications of lipoic arm inhibit alpha-ketoacid dehydrogenases. Nat. Chem. Biol. 2023, 19, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Hurd, T.R.; Nadtochiy, S.M.; Brookes, P.S.; Fearnley, I.M.; Lilley, K.S.; Smith, R.A.; Murphy, M.P. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): Implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 2010, 430, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Peskin, A.V.; Meotti, F.C.; Magon, N.J.; de Souza, L.F.; Salvador, A.; Winterbourn, C.C. Mechanism of glutathionylation of the active site thiols of peroxiredoxin 2. J. Biol. Chem. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid. Redox Signal 2017, 26, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, S.; Bonetto, V.; Fratelli, M.; Gianazza, E.; Eberini, I.; Massignan, T.; Salmona, M.; Chang, G.; Holmgren, A.; Ghezzi, P. Glutathionylation of human thioredoxin: A possible crosstalk between the glutathione and thioredoxin systems. Proc. Natl. Acad. Sci. USA 2002, 99, 9745–9749. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Chalker, J.; Slade, L.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 2017, 106, 302–314. [Google Scholar] [CrossRef]
- Zhou, H.L.; Premont, R.T.; Stamler, J.S. The manifold roles of protein S-nitrosylation in the life of insulin. Nat. Rev. Endocrinol. 2022, 18, 111–128. [Google Scholar] [CrossRef]
- Won, J.S.; Kim, J.; Annamalai, B.; Shunmugavel, A.; Singh, I.; Singh, A.K. Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimers Dis. 2013, 34, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019, 565, 96–100. [Google Scholar] [CrossRef]
- Jung, K.H.; Chu, K.; Ko, S.Y.; Lee, S.T.; Sinn, D.I.; Park, D.K.; Kim, J.M.; Song, E.C.; Kim, M.; Roh, J.K. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke 2006, 37, 2744–2750. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liu, Y.C.; Liu, Y.W.; Lee, Y.B.; Huang, H.C.; Chen, Y.Y.; Shih, Y.H.; Lee, Y.C.; Cheng, C.F.; Meng, T.C. Nitrite Protects Neurons Against Hypoxic Damage Through S-nitrosylation of Caspase-6. Antioxid. Redox Signal 2019, 31, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Gallage, S.; Avila, J.E.B.; Ramadori, P.; Focaccia, E.; Rahbari, M.; Ali, A.; Malek, N.P.; Anstee, Q.M.; Heikenwalder, M. A researcher’s guide to preclinical mouse NASH models. Nat. Metab. 2022, 4, 1632–1649. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, L.; Osella, A.R.; Ciccacci, F.; Giannelli, G.; Scavo, M.P. Premenopausal Syndrome and NAFLD: A New Approach Based on Gender Medicine. Biomedicines 2022, 10, 1184. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Kamzolas, I.; Harder, L.M.; Oakley, F.; Trautwein, C.; Hatting, M.; Ross, T.; Bernardo, B.; Oldenburger, A.; Hjuler, S.T.; et al. An unbiased ranking of murine dietary models based on their proximity to human metabolic dysfunction-associated steatotic liver disease (MASLD). Nat. Metab. 2024, 6, 1178–1196. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, P.A.; Tamaki, F.K.; Cardoso, A.R.; Marana, S.R.; Kowaltowski, A.J. H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol. 2015, 4, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.R.; Kakimoto, P.A.; Kowaltowski, A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: Role of very long chain acyl-CoA dehydrogenases. PLoS ONE 2013, 8, e77088. [Google Scholar] [CrossRef]
- Karimkhanloo, H.; Keenan, S.N.; Bayliss, J.; De Nardo, W.; Miotto, P.M.; Devereux, C.J.; Nie, S.; Williamson, N.A.; Ryan, A.; Watt, M.J.; et al. Mouse strain-dependent variation in metabolic associated fatty liver disease (MAFLD): A comprehensive resource tool for pre-clinical studies. Sci. Rep. 2023, 13, 4711. [Google Scholar] [CrossRef] [PubMed]
- Hallsworth, K.; Adams, L.A. Lifestyle modification in NAFLD/NASH: Facts and figures. JHEP Rep. 2019, 1, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Di Ciaula, A.; Baj, J.; Molina-Molina, E.; Shanmugam, H.; Garruti, G.; Wang, D.Q.; Portincasa, P. Protocols for Mitochondria as the Target of Pharmacological Therapy in the Context of Nonalcoholic Fatty Liver Disease (NAFLD). Methods Mol. Biol. 2021, 2310, 201–246. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Tang, D.; Feng, J. Mitochondrial targeted therapies in MAFLD. Biochem. Biophys. Res. Commun. 2025, 753, 151498. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P.; Lopez-Sanchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969. [Google Scholar] [CrossRef]
- Zeng, X.F.; Varady, K.A.; Wang, X.D.; Targher, G.; Byrne, C.D.; Tayyem, R.; Latella, G.; Bergheim, I.; Valenzuela, R.; George, J.; et al. The role of dietary modification in the prevention and management of metabolic dysfunction-associated fatty liver disease: An international multidisciplinary expert consensus. Metabolism 2024, 161, 156028. [Google Scholar] [CrossRef]
- Burra, P.; Bizzaro, D.; Gonta, A.; Shalaby, S.; Gambato, M.; Morelli, M.C.; Trapani, S.; Floreani, A.; Marra, F.; Brunetto, M.R.; et al. Clinical impact of sexual dimorphism in non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Liver Int. 2021, 41, 1713–1733. [Google Scholar] [CrossRef]
- Amorim, R.; Simoes, I.C.M.; Teixeira, J.; Cagide, F.; Potes, Y.; Soares, P.; Carvalho, A.; Tavares, L.C.; Benfeito, S.; Pereira, S.P.; et al. Mitochondria-targeted anti-oxidant AntiOxCIN(4) improved liver steatosis in Western diet-fed mice by preventing lipid accumulation due to upregulation of fatty acid oxidation, quality control mechanism and antioxidant defense systems. Redox Biol. 2022, 55, 102400. [Google Scholar] [CrossRef]
- Mao, G.; Kraus, G.A.; Kim, I.; Spurlock, M.E.; Bailey, T.B.; Zhang, Q.; Beitz, D.C. A mitochondria-targeted vitamin E derivative decreases hepatic oxidative stress and inhibits fat deposition in mice. J. Nutr. 2010, 140, 1425–1431. [Google Scholar] [CrossRef]
- Feillet-Coudray, C.; Fouret, G.; Ebabe Elle, R.; Rieusset, J.; Bonafos, B.; Chabi, B.; Crouzier, D.; Zarkovic, K.; Zarkovic, N.; Ramos, J.; et al. The mitochondrial-targeted antioxidant MitoQ ameliorates metabolic syndrome features in obesogenic diet-fed rats better than Apocynin or Allopurinol. Free Radic. Res. 2014, 48, 1232–1246. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ran, S.; Zhou, Y.; Shi, Q.; Yu, J.; Wang, W.; Sun, C.; Li, D.; Hu, Y.; Pan, C.; et al. Exposure to Succinate Leads to Steatosis in Non-Obese Non-Alcoholic Fatty Liver Disease by Inhibiting AMPK/PPARalpha/FGF21-Dependent Fatty Acid Oxidation. J. Agric. Food Chem. 2024, 72, 21052–21064. [Google Scholar] [CrossRef]
- Marsal-Beltran, A.; Rodriguez-Castellano, A.; Astiarraga, B.; Calvo, E.; Rada, P.; Madeira, A.; Rodriguez-Pena, M.M.; Llaurado, G.; Nunez-Roa, C.; Gomez-Santos, B.; et al. Protective effects of the succinate/SUCNR1 axis on damaged hepatocytes in NAFLD. Metabolism 2023, 145, 155630. [Google Scholar] [CrossRef] [PubMed]
- Serena, C.; Ceperuelo-Mallafre, V.; Keiran, N.; Queipo-Ortuno, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Perez-Brocal, V.; Andres-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [PubMed]
- Prime, T.A.; Blaikie, F.H.; Evans, C.; Nadtochiy, S.M.; James, A.M.; Dahm, C.C.; Vitturi, D.A.; Patel, R.P.; Hiley, C.R.; Abakumova, I.; et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2009, 106, 10764–10769. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef]
- Wilson, R.J.; Drake, J.C.; Cui, D.; Lewellen, B.M.; Fisher, C.C.; Zhang, M.; Kashatus, D.F.; Palmer, L.A.; Murphy, M.P.; Yan, Z. Mitochondrial protein S-nitrosation protects against ischemia reperfusion-induced denervation at neuromuscular junction in skeletal muscle. Free Radic. Biol. Med. 2018, 117, 180–190. [Google Scholar] [CrossRef]
- Methner, C.; Chouchani, E.T.; Buonincontri, G.; Pell, V.R.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Mitochondria selective S-nitrosation by mitochondria-targeted S-nitrosothiol protects against post-infarct heart failure in mouse hearts. Eur. J. Heart Fail. 2014, 16, 712–717. [Google Scholar] [CrossRef]
- Methner, C.; Lukowski, R.; Grube, K.; Loga, F.; Smith, R.A.; Murphy, M.P.; Hofmann, F.; Krieg, T. Protection through postconditioning or a mitochondria-targeted S-nitrosothiol is unaffected by cardiomyocyte-selective ablation of protein kinase G. Basic. Res. Cardiol. 2013, 108, 337. [Google Scholar] [CrossRef]
- Kim, M.; Stepanova, A.; Niatsetskaya, Z.; Sosunov, S.; Arndt, S.; Murphy, M.P.; Galkin, A.; Ten, V.S. Attenuation of oxidative damage by targeting mitochondrial complex I in neonatal hypoxic-ischemic brain injury. Free Radic. Biol. Med. 2018, 124, 517–524. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mailloux, R.J. Targeted Redox Regulation α-Ketoglutarate Dehydrogenase Complex for the Treatment of Human Diseases. Cells 2025, 14, 653. https://doi.org/10.3390/cells14090653
Mailloux RJ. Targeted Redox Regulation α-Ketoglutarate Dehydrogenase Complex for the Treatment of Human Diseases. Cells. 2025; 14(9):653. https://doi.org/10.3390/cells14090653
Chicago/Turabian StyleMailloux, Ryan J. 2025. "Targeted Redox Regulation α-Ketoglutarate Dehydrogenase Complex for the Treatment of Human Diseases" Cells 14, no. 9: 653. https://doi.org/10.3390/cells14090653
APA StyleMailloux, R. J. (2025). Targeted Redox Regulation α-Ketoglutarate Dehydrogenase Complex for the Treatment of Human Diseases. Cells, 14(9), 653. https://doi.org/10.3390/cells14090653