
Dopamine Receptor D3 Induces Transient, mTORC1-Dependent Autophagy That Becomes Persistent, AMPK-Mediated, and Neuroprotective in Experimental Models of Huntington’s Disease

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Culture and Stable DRD2/DRD3 Transfection
2.3. Transient Q23/Q74 Transfection
2.4. Western Blot
2.5. Immunofluorescence
2.6. Quantitative and Densitometric Analysis in Immunofluorescence Material
2.7. Motor Behavior
2.8. Statistics
3. Results
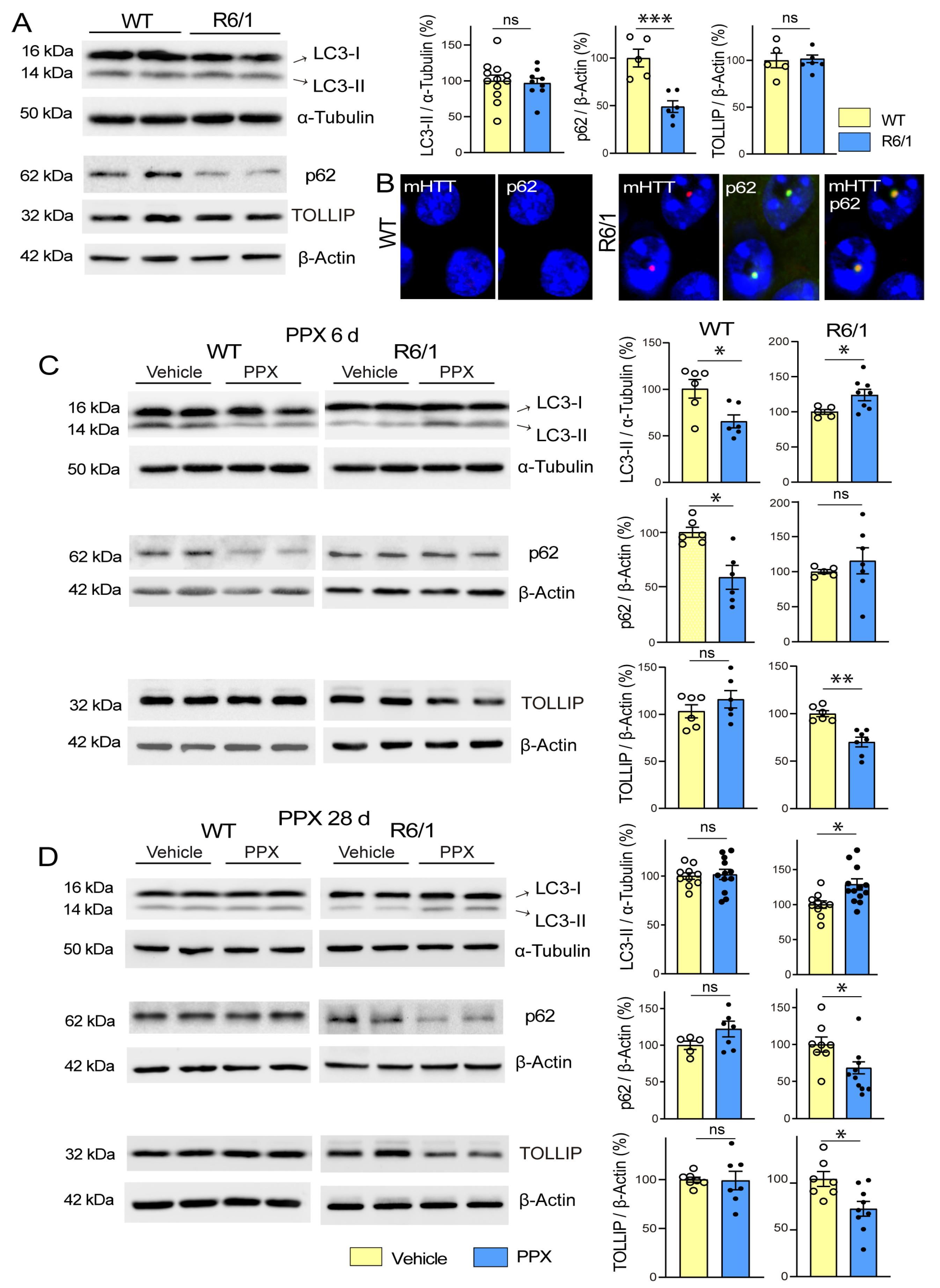
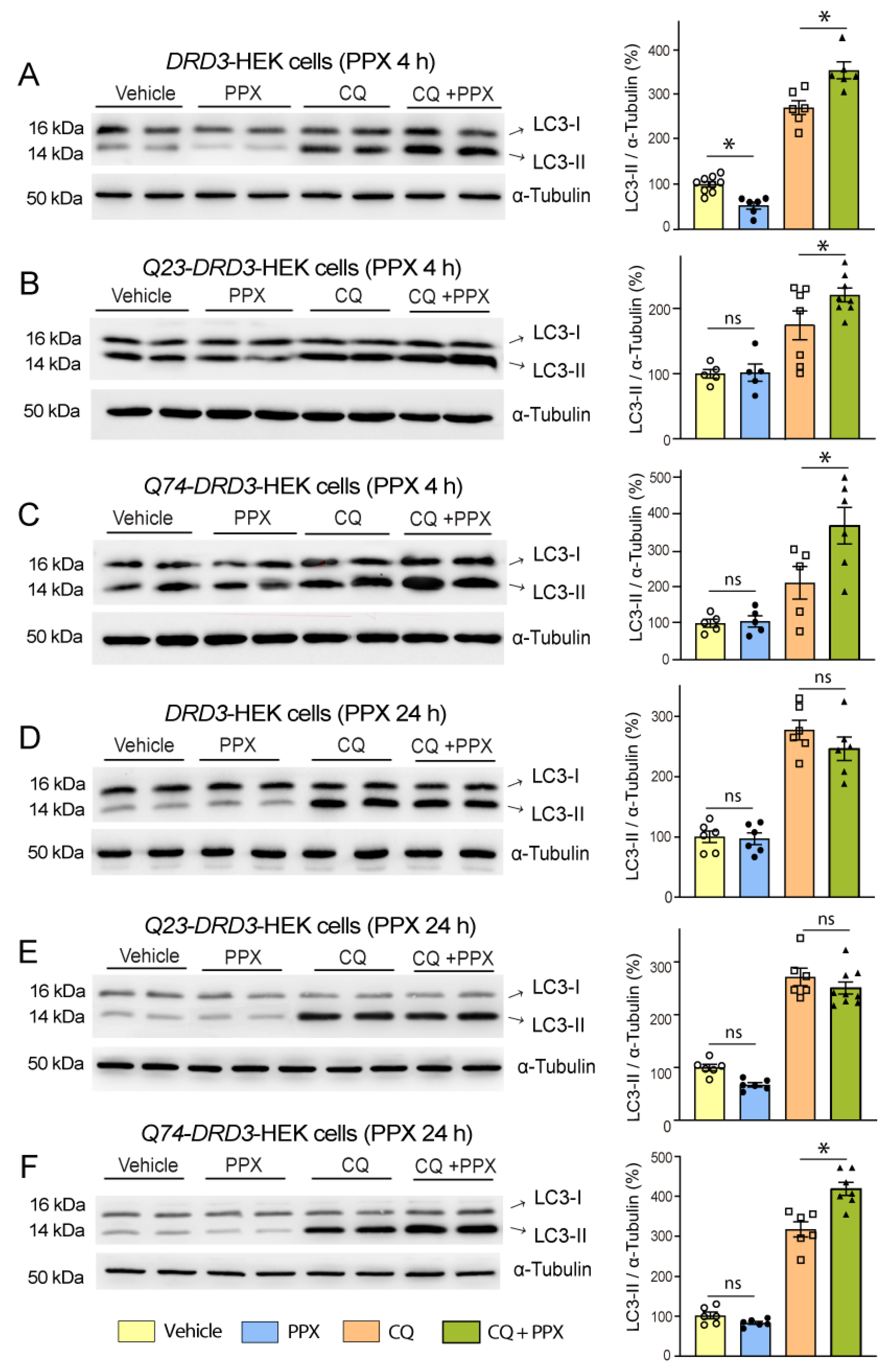
3.1. DRD3-Induced Autophagy Is Transient in WT Mice and Q23-Expressing Cells and Persistent in R6/1 Mice and Q74-Expressing Cells
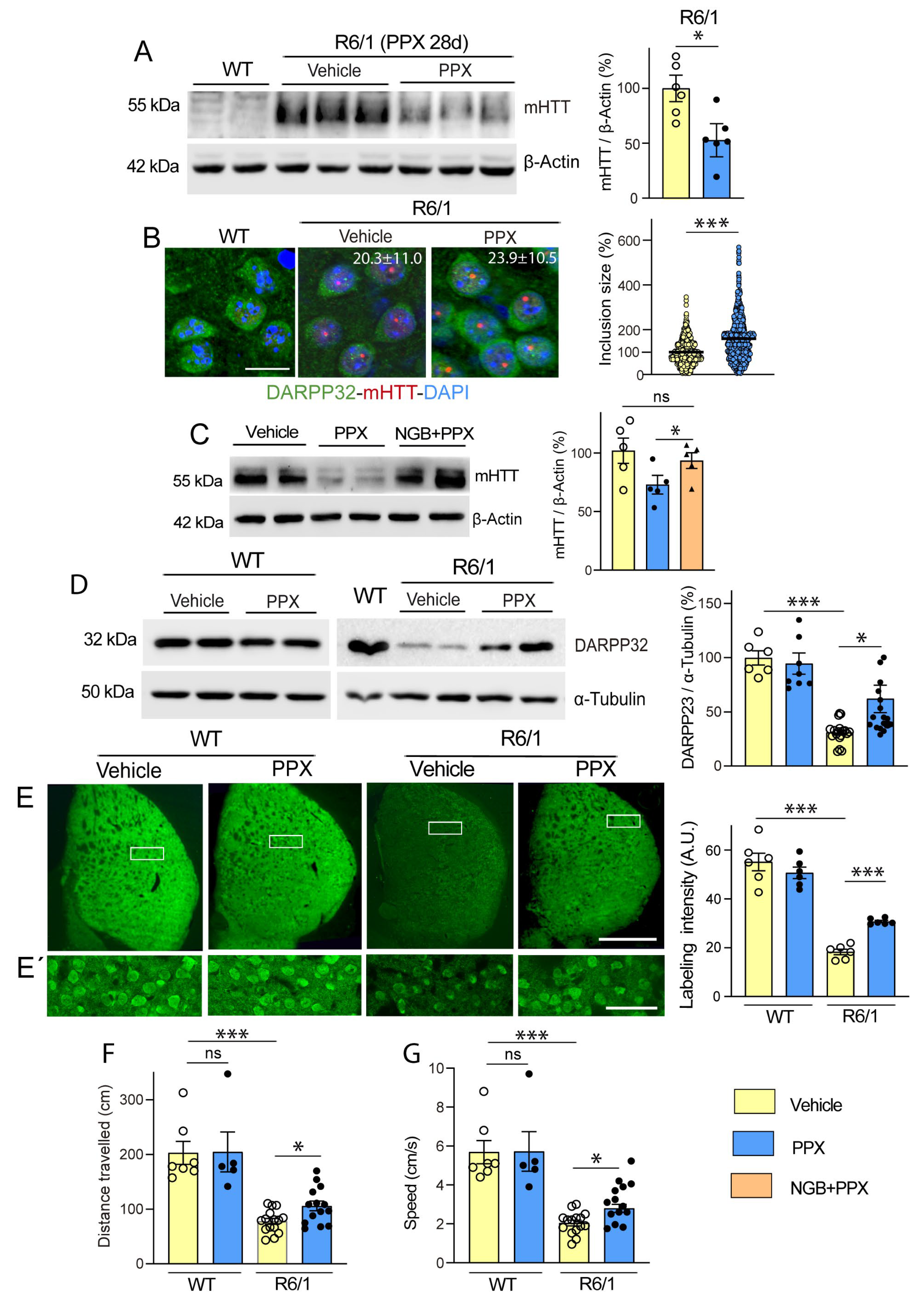
3.2. DRD3-Induced Autophagy Promotes mHTT Clearance and Neuroprotection in the Striatum of R6/1 Mice and Q74-DRD3-HEK Cells
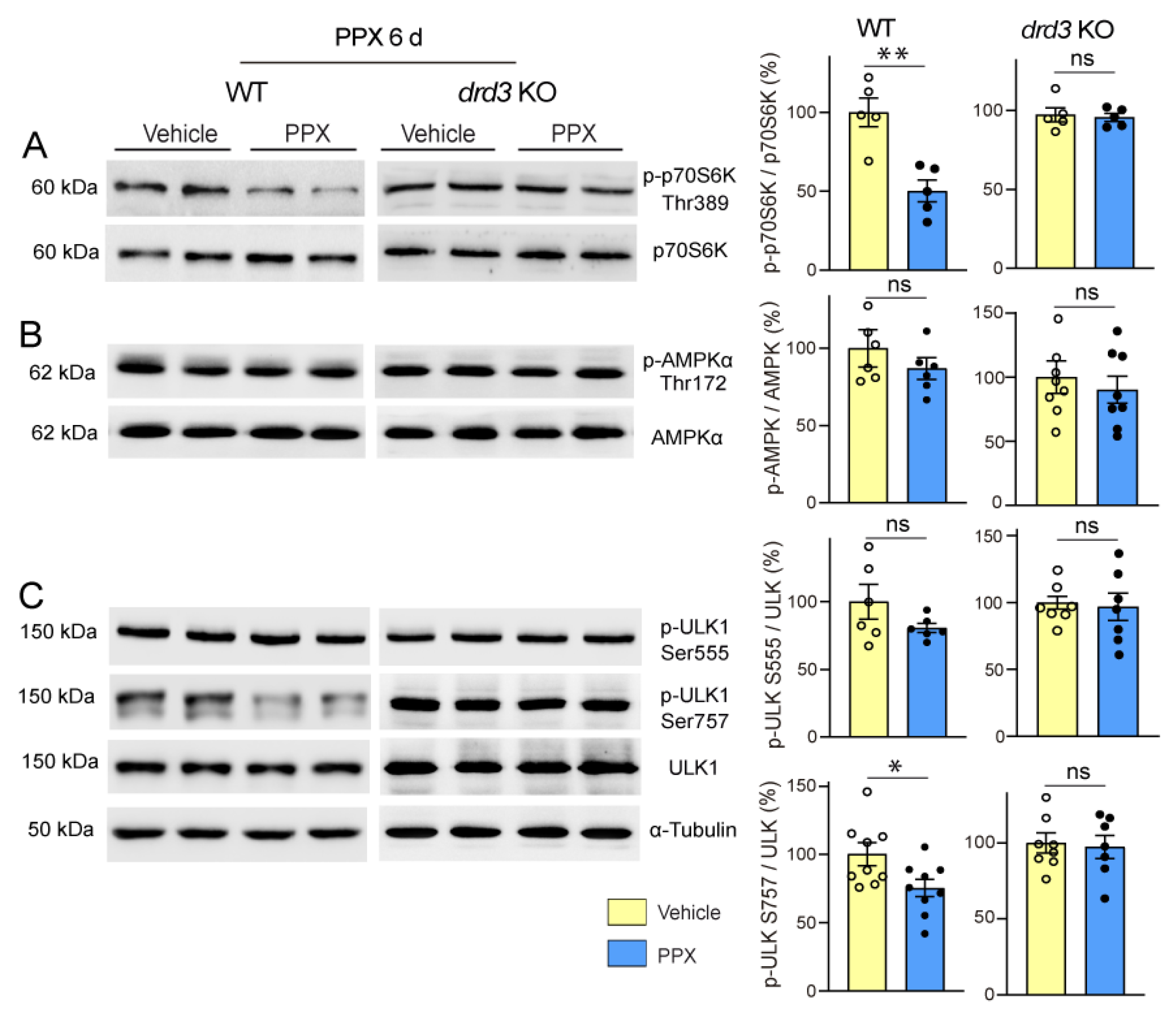
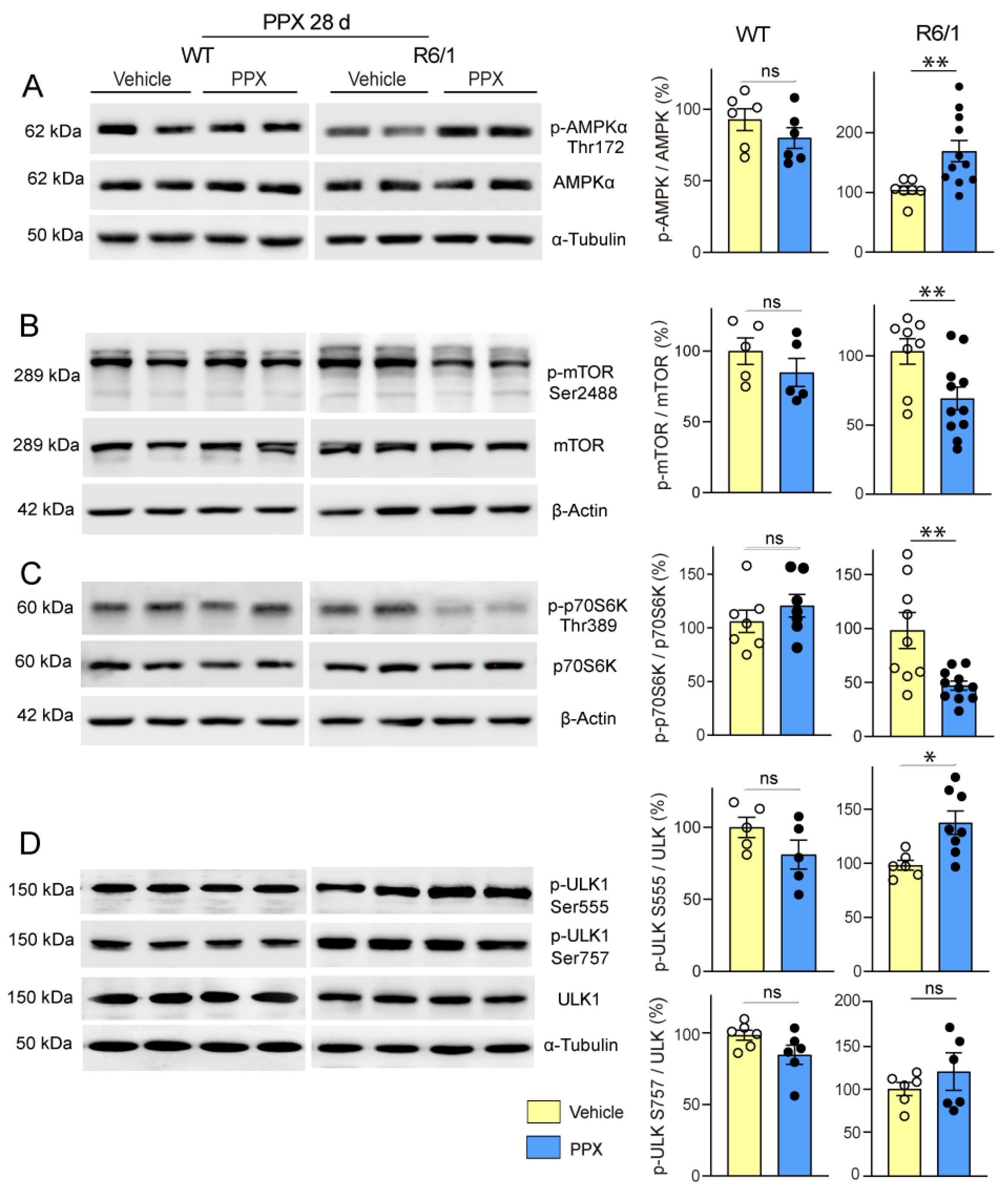
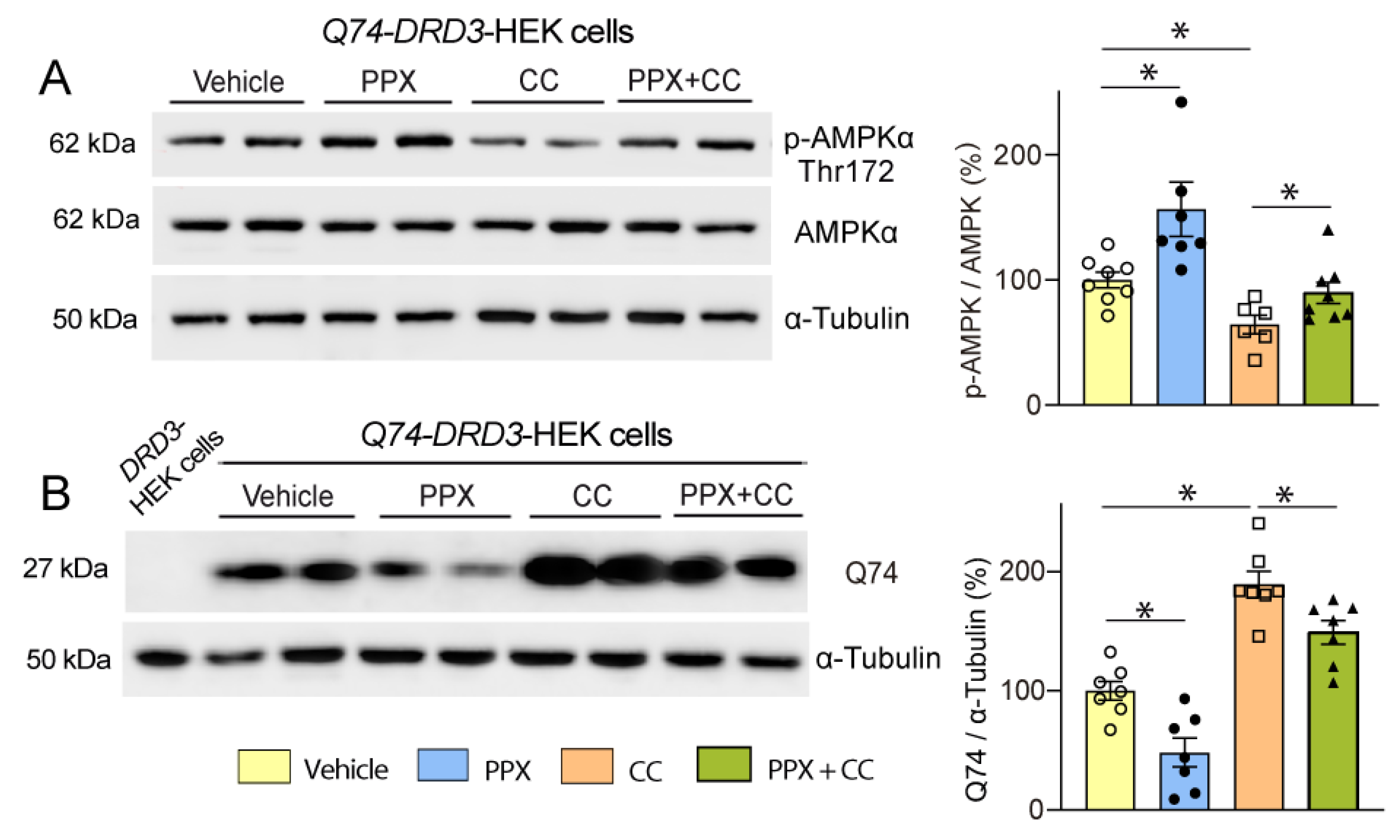
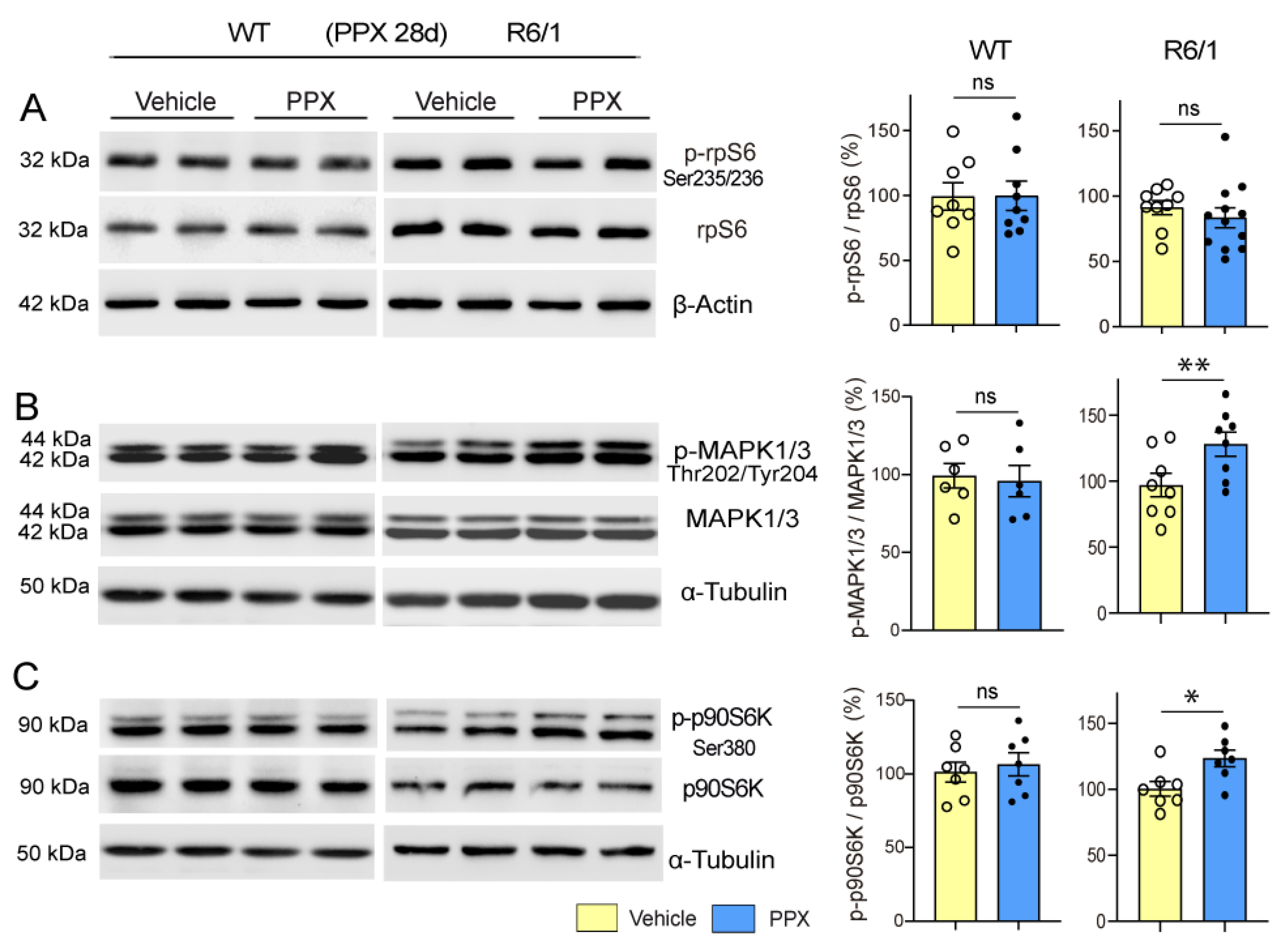
3.3. DRD3-Induced Autophagy Involves Different Signaling Pathways in Healthy and polyQ-HTT-Challenged Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H. Therapeutic strategies for targeting neurodegenerative protein misfolding Chem disorders. Curr. Opin. Chem. Biol. 2018, 44, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Klionsky, D.J. The emerging roles of autophagy in Human diseases. Biomedicines 2021, 9, 1651. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neuronal cells causes degenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Djajadikerta, A.; Keshri, S.; Pavel, M.; Prestil, R.; Ryan, L.; Rubinsztein, D.C. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, Q.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 2010, 12, 781–790. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffer, T.; Scapozza, L.; Brunner, T.; Simon, H.-S. Calpain-mediated cleavage of Atg switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2014, 26, 549–555. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Scahill, R.I.; Owen, G.; Durr, A.; Leavitt, B.R.; Ross, R.A.; Borowsky, B.; Landwehrmeyer, B.; Frost, C.; Johnson, H.; et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. 2013, 12, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Hoffner, G.; Souès, S.; Djian, P. Aggregation of expanded huntingtin in the brains of patients with Huntington disease. Prion 2007, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef]
- Ehrlich, M.E. Huntington’s disease and the striatal medium spiny neuron: Cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics 2012, 9, 270–284. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Hobbs, N.Z.; Barnes, J.; Frost, C.; Henley, S.M.D.; Wild, E.J.; Macdonald, K.; Barker, R.A.; Scahill, R.I.; Fox, N.C.; Tabrizi, S.J. Onset and progression of pathologic atrophy in Huntington disease: A longitudinal MR imaging study. Am. J. Neuroradiol. 2010, 31, 1036–1041. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Damask, S.P.; Wang, J.; Haroutanian, H.; Davis, K.L.; Watson, S.J. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology 1996, 15, 17–29. [Google Scholar] [CrossRef]
- Araki, K.Y.; Sims, J.R.; Bhide, P.G. Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and postnatal development. Brain Res. 2007, 1156, 31–45. [Google Scholar] [CrossRef]
- Barroso-Chinea, P.; Luis-Ravelo, D.; Fumagallo-Reading, F.; Castro-Hernandez, J.; Salas-Hernandez, J.; Rodriguez-Nuñez, J.; Febles-Casquero, A.; Cruz-Muros, I.; Afonso-Oranmas, D.; Abreu-Gonzalez, P.; et al. DRD3 (dopamine receptor D3) but not DRD2 activates autophagy through MTORC1 inhibition preserving protein synthesis. Autophagy 2020, 16, 1279–1295. [Google Scholar] [CrossRef]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef] [PubMed]
- Luis-Ravelo, D.; Estévez-Silva, H.; Barroso-Chinea, P.; Afonso-Oramas, D.; Salas-Hernandez, J.; Rodriguez-Nuñez, J.; Acevedo-Arocena, A.; Marcellino, D.; Gonzalez-Hernandez, T. Pramipexole reduces soluble mutant huntingtin and protects striatal neurons through dopamine D3 receptors in a genetic model of Huntington’s disease. Exp. Neurol. 2018, 299, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Bolivar, V.J.; Manley, K.; Messer, A. Early exploratory behavior abnormalities in R6/1 Huntington’s disease transgenic mice. Brain Res. 2004, 1005, 29–35. [Google Scholar] [CrossRef]
- Wilson, S.M.; Wurst, M.G.; Whatley, M.F.; Daniels, R.N. Classics in Chemical Neuroscience: Pramipexole. ACS Chem. Neurosci. 2020, 11, 2506–2512. [Google Scholar] [CrossRef]
- Li, T.; Zou, S.; Zhang, Z.; Liu, M.; Liang, Z. Efficacy of pramipexole on quality of life in patients with Parkinson’s disease: A systematic review and meta-analysis. BMC Neurol. 2022, 22, 320. [Google Scholar] [CrossRef]
- Collins, G.T.; Newman, A.H.; Grundt, P.; Rice, K.C.; Husbands, S.; Chauvignac, C.; Chen, J.; Wang, S.; Woods, J.H. Yawning and hypothermia in rats: Effects of dopamine D3 and D2 agonists and antagonists. Psychopharmacology 2007, 193, 159–170. [Google Scholar] [CrossRef]
- Castro-Hernández, J.; Afonso-Oramas, D.; Cruz-Muros, I.; Salas-Hernandez, J.; Barroso-Chinea, P.; Moratalla, R.; Millan, J.M.; Gonzalez-Hernandez, T. Prolonged treatment with pramipexole promotes physical interaction of striatal dopamine D3 autoreceptors with dopamine transporters to reduce dopamine uptake. Neurobiol. Dis. 2015, 74, 325–335. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and humans. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Atwook, B.K.; Lopez, J.; Wager-Miller, J.; Mackie, K.; Straiker, A. Expression of G protein coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genom. 2011, 12, 14. [Google Scholar] [CrossRef]
- Wenk, D.; Khan, S.; Ignatchenko, V.; Hübner, H.; Gmeiner, P.; Weikert, D.; Pischetsrieder, M.; Kislinger, T. Phosphoproteomic Analysis of Dopamine D2 Receptor Signaling Reveals Interplay of G-Protein- and β-Arrestin-Mediated Effects. J. Proteome Res. 2023, 22, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Diaz, J.; Sokoloff, P.; Griffon, N. Interactions of GIPC with dopamine D2, D3 but not D4 receptors define a novel mode of regulation of G protein-coupled receptors. Mol. Biol. Cell 2004, 15, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Narain, Y.; Wyttenbach, A.; Rankin, J.; Furlong, R.A.; Rubinsztein, D.C. A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet. 1999, 36, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, B.J.K. The Mouse Brain in Stereotaxic Coordinates, Compact, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Leggio, G.M.; Bucolo, C.; Platania, C.B.; Salomone, S.; Drago, F. Current drug treatments targeting dopamine D3 receptor. Pharmacol. Ther. 2016, 165, 164–177. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 455, 77–88. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Klionsky, D.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A. Guidelines for the use and interpretation of assays for monitoring autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef]
- Oguro, A.; Kubota, H.; Shimizu, M.; Ishiura, S.; Atomi, Y. Protective role of the ubiquitin binding protein Tollip against the toxicity of polyglutamine-expansion proteins. Neurosci. Lett. 2011, 503, 234–239. [Google Scholar] [CrossRef]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef]
- Nagaoka, U.; Kim, K.; Jana, N.R.; Doi, H.; Maruyama, M.; Mitsui, K.; Oyama, F.; Nukina, N. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 2004, 91, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Seidel, K.; Heinsen, H.; Vonsattel, J.P.; den Dunnen, W.F.; Korf, H.W. Huntington’s disease (HD): The neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016, 26, 726–740. [Google Scholar] [CrossRef]
- Girault, J.A.; Nairn, A.C. DARPP-32 40 years later. Adv. Pharmacol. 2021, 90, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, P.; De Cristofaro, T.; Affaitati, A.; Pizzulo, G.M.; Feliciello, A.; Criscuolo, C.; De Michele, G.; Filla, A.; Avvedimento, E.V.; Varrone, S. DNA damage induced by polyglutamine-expanded proteins. Hum. Mol. Genet. 2003, 12, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [CrossRef]
- Sutton, L.P.; Caron, M.G. Essential role of D1R in the regulation of mTOR complex1 signaling induced by cocaine. Neuropharmacology 2015, 99, 610–619. [Google Scholar] [CrossRef]
- Xu, S.; Kang, U.G. Region-specific activation of the AMPK system by cocaine: The role of D1 and D2 receptors. Pharmacol. Biochem. Behav. 2016, 146–147, 28–34. [Google Scholar] [CrossRef]
- Melick, C.H.; Lama-Sherpa, T.D.; Curukovic, A.; Jewell, J.L. G-Protein Coupled Receptor Signaling and Mammalian Target of Rapamycin Complex 1 Regulation. Mol. Pharmacol. 2022, 101, 181–190. [Google Scholar] [CrossRef]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardy, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.; Kim, J.; Shaw, R.J.; Guam, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef]
- Copp, J.; Manning, G.; Hunter, T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): Phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009, 69, 1821–1827. [Google Scholar] [CrossRef]
- Pearson, R.B.; Dennis, P.B.; Han, J.W.; Williamson, N.A.; Kozma, S.C.; Wettenhall, R.E.; Thomas, G. The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 1995, 14, 5279–5287. [Google Scholar] [CrossRef]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Gubas, A.; Dikic, I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022, 289, 75–89. [Google Scholar] [CrossRef]
- Li, X.; Goobie, G.C.; Zhang, Y. Toll-interacting protein impacts on inflammation, autophagy, and vacuole trafficking in human disease. J. Mol. Med. 2021, 99, 21–31. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Shimizu, M.; Oguro-Ando, A.; Ohoto-Fujita, E.; Atomi, Y. Toll-interacting protein pathway: Degradation of an ubiquitin-binding protein. Methods Enzymol. 2014, 534, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for ineffective autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 293–12305. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hosp, F.; Frottin, F.; Ge, H.; Mann, M.; Hayer-Hartl, M.; Hartl, F.U. Soluble Oligomers of PolyQ-Expanded Huntingtin Target a Multiplicity of Key Cellular Factors. Mol. Cell 2016, 63, 951–964. [Google Scholar] [CrossRef]
- Bodner, R.A.; Outeiro, T.F.; Altmann, S.; Maxwell, M.M.; Cho, S.H.; Hyman, B.T.; McLean, P.J.; Young, A.B.; Housman, D.E.; Kazantsev, A.G. Pharmacological promotion of inclusion formation: A therapeutic approach for Huntington’s and Parkinson’s diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 4246–4251. [Google Scholar] [CrossRef]
- Hoffner, G.; Djian, P. Polyglutamine Aggregation in Huntington Disease: Does Structure Determine Toxicity? Mol. Neurobiol. 2015, 52, 1297–1314. [Google Scholar] [CrossRef]
- Bäuerlein, F.J.B.; Saha, I.; Mishra, A.; Kalemanov, M.; Martinez-Sanchez, A.; Klein, R.; Dudanova, I.; Hipp, M.S.; Hartl, F.U.; Baumeister, W.; et al. In Situ Architecture and Cellular Interactions of PolyQ Inclusions. Cell 2017, 171, 179–187.e10. [Google Scholar] [CrossRef]
- Riguet, N.; Mahul-Mellier, A.L.; Maharjan, N.; Burtscher, J.; Croisier, M.; Knott, G.; Hastings, J.; Patin, A.; Reiterer, V.; Farhan, H.; et al. Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties. Nat. Commun. 2021, 12, 6579. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, Y.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Cho, H.M.; Choi, S.Y.; Suguira, Y.; Hayasaka, T.; Setou, M.; Koh, H.; Hwang, E.M.; Park, J.Y.; Kang, S.J.; et al. (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis. 2013, 4, e919. [Google Scholar] [CrossRef]
- Domise, M.; Didier, S.; Marinangeli, C.; Zhao, H.; Chandakkar, P.; Buée, L.; Viollet, B.; Davies, P.; Marambaud, P.; Vingtdeux, V. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 2016, 6, 26758. [Google Scholar] [CrossRef] [PubMed]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinsons Disease. J. Parkinson Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Y.; Shi, L.; Zhong, D.; Li, Y.; Li, J.; Jin, R. AMPK: Potential Therapeutic Target for Alzheimer’s Disease. Curr. Protein Pept. Sci. 2020, 21, 66–77. [Google Scholar] [CrossRef]
- Chou, S.Y.; Lee, Y.C.; Chen, H.M.; Chiang, M.C.; Lai, H.L.; Chang, H.H.; Wu, Y.C.; Sun, C.N.; Chien, C.L.; Lin, Y.S.; et al. CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J. Neurochem. 2005, 93, 310–320. [Google Scholar] [CrossRef]
- Ju, T.C.; Chen, H.M.; Lin, J.T.; Chang, C.P.; Chang, W.C.; Kang, J.J.; Sun, C.P.; Tao, M.H.; Tu, P.H.; Chang, C.; et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J. Cell Biol. 2011, 194, 209–227. [Google Scholar] [CrossRef]
- Ma, T.C.; Buescher, J.L.; Oatis, B.; Funk, J.A.; Nash, A.J.; Carrier, R.L.; Hoyt, K.R. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 2007, 411, 98–103. [Google Scholar] [CrossRef]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Sequedo, M.D.; Parker, A.J.; Millan, J.M.; Weiss, A.; Deglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef]
- Višnjić, D.; Lalić, H.; Dembitz, V.; Smoljo, T. AICAr, a Widely Used AMPK Activator with Important AMPK-Independent Effects: A Systematic Review. Cells 2021, 10, 1095. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Del Río, C.; Tortajada-Pérez, J.; Gómez-Escribano, A.P.; Casterá, F.; Peiró, C.; Millan, J.M.; Herrero, M.J.; Vázquez-Manrique, R.P. Metformin to treat Huntington disease: A pleiotropic drug against a multi-system disorder. Mech. Ageing Dev. 2022, 204, 111670. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, S.; Lee, J.; Pak, J.; Lee, G.; Kim, Y.; Kim, J.M.; Chung, J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007, 8, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef]
- Bockaert, J.P.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.F.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luis-Ravelo, D.; Fumagallo-Reading, F.; Febles-Casquero, A.; Lopez-Fernandez, J.; Marcellino, D.J.; Gonzalez-Hernandez, T. Dopamine Receptor D3 Induces Transient, mTORC1-Dependent Autophagy That Becomes Persistent, AMPK-Mediated, and Neuroprotective in Experimental Models of Huntington’s Disease. Cells 2025, 14, 652. https://doi.org/10.3390/cells14090652
Luis-Ravelo D, Fumagallo-Reading F, Febles-Casquero A, Lopez-Fernandez J, Marcellino DJ, Gonzalez-Hernandez T. Dopamine Receptor D3 Induces Transient, mTORC1-Dependent Autophagy That Becomes Persistent, AMPK-Mediated, and Neuroprotective in Experimental Models of Huntington’s Disease. Cells. 2025; 14(9):652. https://doi.org/10.3390/cells14090652
Chicago/Turabian StyleLuis-Ravelo, Diego, Felipe Fumagallo-Reading, Alejandro Febles-Casquero, Jonathan Lopez-Fernandez, Daniel J. Marcellino, and Tomas Gonzalez-Hernandez. 2025. "Dopamine Receptor D3 Induces Transient, mTORC1-Dependent Autophagy That Becomes Persistent, AMPK-Mediated, and Neuroprotective in Experimental Models of Huntington’s Disease" Cells 14, no. 9: 652. https://doi.org/10.3390/cells14090652
APA StyleLuis-Ravelo, D., Fumagallo-Reading, F., Febles-Casquero, A., Lopez-Fernandez, J., Marcellino, D. J., & Gonzalez-Hernandez, T. (2025). Dopamine Receptor D3 Induces Transient, mTORC1-Dependent Autophagy That Becomes Persistent, AMPK-Mediated, and Neuroprotective in Experimental Models of Huntington’s Disease. Cells, 14(9), 652. https://doi.org/10.3390/cells14090652