
Quitting Your Day Job in Response to Stress: Cell Survival and Cell Death Require Secondary Cytoplasmic Roles of Cyclin C and Med13


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Mediator Kinase Module
2.1. Structure and Function of the MKM
2.1.1. MKM Structure
2.1.2. Function of the MKM in Yeast
2.1.3. Function of the MKM in Mammalian Cells
2.2. Regulation of the MKM
2.2.1. Dynamic MKM Promoter Recruitment and Expulsion
2.2.2. MKM Disassembly Following Cell Death Cues Triggered by ROS
2.2.3. MKM Disassembly Following Cell Survival Cues Triggered by Nitrogen Starvation
3. Roles of Cyclin C in the Cytoplasm
3.1. The Role of Cyclin C Stress-Induced Mitochondrial Hyperfission
3.1.1. Two Classes of Mitochondrial Fission
3.1.2. Cyclin C Is Required for Stress-Induced and Mitochondrial Hyperfission in Mammals
3.1.3. Cyclin C Is Required for Stress-Induced Mitochondrial Hyperfission in Yeast
3.2. Cytoplasmic Cyclin C Promotes Regulated Cell Death
3.2.1. Mitochondrial Hyperfission and Intrinsic Regulated Cell Death (iRCD)
3.2.2. Cyclin C and Cell Death in Mammals

3.2.3. Cyclin C and Cell Death in Yeast
4. Roles of Med13 in the Cytoplasm
4.1. Role of Med13 in P-Body Assembly Following Starvation in Yeast
4.1.1. P-Body Assembly Following Stress
4.1.2. Med13’s Role in P-Body Assembly in Yeast
4.2. Med13’s Role in Cargo Hitchhiking Autophagy in Yeast
4.2.1. Outline of Autophagy Pathways
4.2.2. Med13 Is Degraded by Cargo Hitchhiking Autophagy in Yeast
4.2.3. Snx4 Promotes CHA in Yeast
4.2.4. CHA Uses Phagophores Built by the Atg17 Scaffold Complex
4.2.5. Ksp1 Is the Selective Autophagy Receptor Proteins for Med13 in CHA
4.2.6. The Nucleoporin Gle1 Is Required for CHA of Med13
5. Diseases Associated with the MKM
5.1. Tumor-Suppressive Roles of Cyclin C–CDK8/19
5.1.1. Notch Signaling
5.1.2. JAK-STAT Signaling
5.2. Oncogene Roles of Cyclin C–CDK8/19
5.3. Role of Cyclin C in TDP-43-Mediated Cell Death
5.3.1. Role of TDP-43 in Maintaining Homeostasis
5.3.2. Aberrant Cytoplasmic Roles of TDP-43
5.3.3. Cyclin C and TDP-43
5.4. Diseases Associated with MED13 Biology
6. Conclusions
7. Future Prospectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | Adenosine Monophosphate-activated Protein Kinase |
| ALS | Amyloid Lateral Sclerosis |
| AIM | Atg8 Interacting Motif |
| BAR | Bin–Amphiphysin–Rvs |
| BMC | Biomolecular Condensate |
| cGAS | Second messenger cyclic GMP–AMP |
| ChIP | Chromatin Immunoprecipitation |
| CWI | Cell Wall Integrity pathway |
| cMED | Core Mediator Complex |
| HAD | Holoenzyme-associating Domain |
| IMM | Inner Mitochondrial Membrane |
| IDR | Intrinsically Disordered Region |
| iRCD | Intrinsic Regulated Cell Death |
| LLPS | Liquid–liquid Phase Separation |
| MKM | Mediator Kinase Module |
| OMM | Outer Mitochondrial Membrane |
| MOMP | Mitochondrial Outer Membrane permeabilization |
| MEFs | Mouse Embryonic Fibroblasts |
| NAC | N-acetyl-cysteine |
| mPTP | Mitochondrial Permeability Transition Pore |
| mRNPs | Non-translating mRNA-protein complexes |
| NPC | Nuclear Pore Complex |
| PI(3)P | Phosphatidylinositol-3-phosphate |
| PX | Phox Homology |
| PrLD | Prion-Like Domains |
| P-Body | Processing Body |
| RCD | Regulated Cell Death |
| RNPs | Ribonucleoproteins |
| RNAPII | RNA Polymerase II |
| RRM | RNA Recognition Motif |
| STING | Cyclic GMP–AMP receptor stimulator of interferon genes |
| SRGs | Stress Response Genes |
| T-ALL | T-Cell Acute Lymphoblastic Leukemia |
| TFs | Transcription Factors |
| TIFs | Translation Initiation Factors |
| UPS | Ubiquitin–Proteasome System |
References
- Koyuncu, S.; Loureiro, R.; Lee, H.J.; Wagle, P.; Krueger, M.; Vilchez, D. Rewiring of the ubiquitinated proteome determines ageing in C. elegans. Nature 2021, 596, 285–290. [Google Scholar] [CrossRef]
- Kevei, É.; Hoppe, T. Ubiquitin sets the timer: Impacts on aging and longevity. Nat. Struct. Mol. Biol. 2014, 21, 290–292. [Google Scholar] [CrossRef]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. It Is All about (U)biquitin: Role of Altered Ubiquitin-Proteasome System and UCHL1 in Alzheimer Disease. Oxid. Med. Cell. Longev. 2016, 2016, 2756068. [Google Scholar] [CrossRef]
- Abdullah, M.O.; Zeng, R.X.; Margerum, C.L.; Papadopoli, D.; Monnin, C.; Punter, K.B.; Chu, C.; Al-Rofaidi, M.; Al-Tannak, N.F.; Berardi, D.; et al. Mitochondrial hyperfusion via metabolic sensing of regulatory amino acids. Cell Rep. 2022, 40, 111198. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, Y.; Shin, H.; Hwang, S.; Park, J.; Song, E.J. Ubiquitin–proteasome system as a target for anticancer treatment—An update. Arch. Pharmacal Res. 2023, 46, 573–597. [Google Scholar] [CrossRef]
- Ajmal, M.R. Protein Misfolding and Aggregation in Proteinopathies: Causes, Mechanism and Cellular Response. Diseases 2023, 11, 30. [Google Scholar] [CrossRef]
- Louros, N.; Schymkowitz, J.; Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 2023, 24, 912–933. [Google Scholar] [CrossRef] [PubMed]
- Dawes, I.W.; Perrone, G.G. Stress and ageing in yeast. FEMS Yeast Res. 2019, 20, foz085. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef]
- Cazzanelli, G.; Pereira, F.; Alves, S.; Francisco, R.; Azevedo, L.; Dias Carvalho, P.; Almeida, A.; Côrte-Real, M.; Oliveira, M.J.; Lucas, C.; et al. The Yeast Saccharomyces cerevisiae as a Model for Understanding RAS Proteins and their Role in Human Tumorigenesis. Cells 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, F.; Medeiros, R.; Lopes, I.; Campos, C.; Ferraz, M.P.; Silva, F.; Alves, L.G.; Pinto, E. A Cyclam Salt as an Antifungal Agent: Interference with Candida spp. and Cryptococcus neoformans Mechanisms of Virulence. Antibiotics 2024, 13, 222. [Google Scholar] [CrossRef]
- Stieber, H.; Junghanns, L.; Wilhelm, H.; Batliner, M.; Aldejohann, A.M.; Kurzai, O.; Martin, R. The sphingolipid inhibitor myriocin increases Candida auris susceptibility to amphotericin B. Mycoses 2024, 67, e13723. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.L.; Hao, X.; Keuenhof, K.S.; Berglund, L.L.; Fischbach, A.; Ahmadpour, D.; Chawla, S.; Gómez, P.; Höög, J.L.; Widlund, P.O.; et al. Elimination of virus-like particles reduces protein aggregation and extends replicative lifespan in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2024, 121, e2313538121. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, R. Invertebrate genetic models of amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2024, 17, 1328578. [Google Scholar] [CrossRef] [PubMed]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef]
- Cooper, K.F. Till Death Do Us Part: The Marriage of Autophagy and Apoptosis. Oxid. Med. Cell. Longev. 2018, 2018, 4701275. [Google Scholar] [CrossRef]
- Cooper, K.F. Cargo hitchhiking autophagy—A hybrid autophagy pathway utilized in yeast. Autophagy 2025, 21, 500–512. [Google Scholar] [CrossRef]
- Tsai, K.L.; Sato, S.; Tomomori-Sato, C.; Conaway, R.C.; Conaway, J.W.; Asturias, F.J. A conserved Mediator-CDK8 kinase module association regulates Mediator-RNA polymerase II interaction. Nat. Struct. Mol. Biol. 2013, 20, 611–619. [Google Scholar] [CrossRef]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Fant, C.B.; Taatjes, D.J. Regulatory functions of the Mediator kinases CDK8 and CDK19. Transcription 2019, 10, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J.; Trnka, M.J.; Pellarin, R.; Greenberg, C.H.; Bushnell, D.A.; Davis, R.; Burlingame, A.L.; Sali, A.; Kornberg, R.D. Molecular architecture of the yeast Mediator complex. eLife 2015, 4, e08719. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chao, T.C.; Tsai, K.L. Structures and compositional dynamics of Mediator in transcription regulation. Curr. Opin. Struct. Biol. 2024, 88, 102892. [Google Scholar] [CrossRef]
- Chao, T.C.; Chen, S.F.; Kim, H.J.; Tang, H.C.; Tseng, H.C.; Xu, A.; Palao, L., 3rd; Khadka, S.; Li, T.; Huang, M.F.; et al. Structural basis of the human transcriptional Mediator regulated by its dissociable kinase module. Mol. Cell 2024, 84, 3932–3949.e3910. [Google Scholar] [CrossRef]
- Knuesel, M.T.; Meyer, K.D.; Donner, A.J.; Espinosa, J.M.; Taatjes, D.J. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell. Biol. 2009, 29, 650–661. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Donner, A.J.; Espinosa, J.M. CDK8: A positive regulator of transcription. Transcription 2010, 1, 4–12. [Google Scholar] [CrossRef]
- Echalier, A.; Endicott, J.A.; Noble, M.E. Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim. Biophys. Acta 2010, 1804, 511–519. [Google Scholar] [CrossRef]
- Schneider, E.V.; Bottcher, J.; Blaesse, M.; Neumann, L.; Huber, R.; Maskos, K. The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder. J. Mol. Biol. 2011, 412, 251–266. [Google Scholar] [CrossRef]
- Li, Y.C.; Chao, T.C.; Kim, H.J.; Cholko, T.; Chen, S.F.; Li, G.; Snyder, L.; Nakanishi, K.; Chang, C.E.; Murakami, K.; et al. Structure and noncanonical Cdk8 activation mechanism within an Argonaute-containing Mediator kinase module. Sci. Adv. 2021, 7, eabd4484. [Google Scholar] [CrossRef]
- Kuchin, S.; Yeghiayan, P.; Carlson, M. Cyclin-dependent protein kinase and cyclin homologs SSN3 and SSN8 contribute to transcriptional control in yeast. Proc. Natl. Acad. Sci. USA 1995, 92, 4006–4010. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.F.; Mallory, M.J.; Smith, J.B.; Strich, R. Stress and developmental regulation of the yeast C-type cyclin Ume3p (Srb11p/Ssn8p). EMBO J. 1997, 16, 4665–4675. [Google Scholar] [CrossRef]
- Cooper, K.F.; Mallory, M.J.; Strich, R. Oxidative stress-induced destruction of the yeast C-type cyclin Ume3p requires phosphatidylinositol-specific phospholipase C and the 26S proteasome. Mol. Cell. Biol. 1999, 19, 3338–3348. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.F.; Strich, R. Saccharomyces cerevisiae C-type cyclin Ume3p/Srb11p is required for efficient induction and execution of meiotic development. Eukaryot. Cell 2002, 1, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Holstege, F.C.; Jennings, E.G.; Wyrick, J.J.; Lee, T.I.; Hengartner, C.J.; Green, M.R.; Golub, T.R.; Lander, E.S.; Young, R.A. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 1998, 95, 717–728. [Google Scholar] [CrossRef]
- Wahi, M.; Johnson, A.D. Identification of genes required for alpha 2 repression in Saccharomyces cerevisiae. Genetics 1995, 140, 79–90. [Google Scholar] [CrossRef]
- Bjorklund, S.; Gustafsson, C.M. The yeast Mediator complex and its regulation. Trends Biochem. Sci. 2005, 30, 240–244. [Google Scholar] [CrossRef]
- van de Peppel, J.; Kettelarij, N.; van Bakel, H.; Kockelkorn, T.T.; van Leenen, D.; Holstege, F.C. Mediator expression profiling epistasis reveals a signal transduction pathway with antagonistic submodules and highly specific downstream targets. Mol. Cell 2005, 19, 511–522. [Google Scholar] [CrossRef]
- Surosky, R.T.; Strich, R.; Esposito, R.E. The yeast UME5 gene regulates the stability of meiotic mRNAs in response to glucose. Mol. Cell. Biol. 1994, 14, 3446–3458. [Google Scholar] [CrossRef]
- Strich, R.; Slater, M.R.; Esposito, R.E. Identification of negative regulatory genes that govern the expression of early meiotic genes in yeast. Proc. Natl. Acad. Sci. USA 1989, 86, 10018–10022. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.; Kobor, M.S.; Kuriakose, N.; Greenblatt, J.; Sadowski, I. GAL4 is regulated by the RNA polymerase II holoenzyme-associated cyclin-dependent protein kinase SRB10/CDK8. Mol. Cell 1999, 3, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.D.; Hanley, S.E.; Doyle, S.J.; Beluch, K.; Strich, R.; Cooper, K.F. Cyclin C-Cdk8 Kinase Phosphorylation of Rim15 Prevents the Aberrant Activation of Stress Response Genes. Front. Cell Dev. Biol. 2022, 10, 867257. [Google Scholar] [CrossRef]
- Hanley, S.E.; Willis, S.D.; Cooper, K.F. Snx4-assisted vacuolar targeting of transcription factors defines a new autophagy pathway for controlling ATG expression. Autophagy 2021, 17, 3547–3565. [Google Scholar] [CrossRef]
- Jeronimo, C.; Robert, F. The Mediator Complex: At the Nexus of RNA Polymerase II Transcription. Trends Cell Biol. 2017, 27, 765–783. [Google Scholar] [CrossRef]
- Chi, Y.; Huddleston, M.J.; Zhang, X.; Young, R.A.; Annan, R.S.; Carr, S.A.; Deshaies, R.J. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev. 2001, 15, 1078–1092. [Google Scholar] [CrossRef]
- Nelson, C.; Goto, S.; Lund, K.; Hung, W.; Sadowski, I. Srb10/Cdk8 regulates yeast filamentous growth by phosphorylating the transcription factor Ste12. Nature 2003, 421, 187–190. [Google Scholar] [CrossRef]
- Law, M.J.; Ciccaglione, K. Fine-Tuning of Histone H3 Lys4 Methylation During Pseudohyphal Differentiation by the CDK Submodule of RNA Polymerase II. Genetics 2015, 199, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Friedson, B.; Willis, S.D.; Shcherbik, N.; Campbell, A.N.; Cooper, K.F. The CDK8 kinase module: A novel player in the transcription of translation initiation and ribosomal genes. Mol. Biol. Cell 2025, 36, ar2. [Google Scholar] [CrossRef]
- Shore, D.; Albert, B. Ribosome biogenesis and the cellular energy economy. Curr. Biol. 2022, 32, R611–R617. [Google Scholar] [CrossRef]
- Hirai, H.; Ohta, K. Comparative Research: Regulatory Mechanisms of Ribosomal Gene Transcription in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Biomolecules 2023, 13, 288. [Google Scholar] [CrossRef] [PubMed]
- Stieg, D.C.; Cooper, K.F.; Strich, R. The extent of cyclin C promoter occupancy directs changes in stress-dependent transcription. J. Biol. Chem. 2020, 295, 16280–16291. [Google Scholar] [CrossRef]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dolken, L.; Strobl, B.; Muller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef]
- Steinparzer, I.; Sedlyarov, V.; Rubin, J.D.; Eislmayr, K.; Galbraith, M.D.; Levandowski, C.B.; Vcelkova, T.; Sneezum, L.; Wascher, F.; Amman, F.; et al. Transcriptional Responses to IFN-γ Require Mediator Kinase-Dependent Pause Release and Mechanistically Distinct CDK8 and CDK19 Functions. Mol. Cell 2019, 76, 485–499.e488. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef]
- Morris, E.J.; Ji, J.-Y.; Yang, F.; Di Stefano, L.; Herr, A.; Moon, N.-S.; Kwon, E.-J.; Haigis, K.M.; Näär, A.M.; Dyson, N.J. E2F1 represses β-catenin transcription and is antagonized by both pRB and CDK8. Nature 2008, 455, 552–556. [Google Scholar] [CrossRef]
- Zhao, X.; Feng, D.; Wang, Q.; Abdulla, A.; Xie, X.J.; Zhou, J.; Sun, Y.; Yang, E.S.; Liu, L.P.; Vaitheesvaran, B.; et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J. Clin. Investig. 2012, 122, 2417–2427. [Google Scholar] [CrossRef]
- Vincent, O.; Kuchin, S.; Hong, S.P.; Townley, R.; Vyas, V.K.; Carlson, M. Interaction of the Srb10 kinase with Sip4, a transcriptional activator of gluconeogenic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 5790–5796. [Google Scholar] [CrossRef] [PubMed]
- Elmlund, H.; Baraznenok, V.; Lindahl, M.; Samuelsen, C.O.; Koeck, P.J.; Holmberg, S.; Hebert, H.; Gustafsson, C.M. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc. Natl. Acad. Sci. USA 2006, 103, 15788–15793. [Google Scholar] [CrossRef]
- Osman, S.; Mohammad, E.; Lidschreiber, M.; Stuetzer, A.; Bazso, F.L.; Maier, K.C.; Urlaub, H.; Cramer, P. The Cdk8 kinase module regulates interaction of the mediator complex with RNA polymerase II. J. Biol. Chem. 2021, 296, 100734. [Google Scholar] [CrossRef]
- Petrenko, N.; Jin, Y.; Wong, K.H.; Struhl, K. Mediator Undergoes a Compositional Change during Transcriptional Activation. Mol. Cell 2016, 64, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Larimore, E.A.; Fissel, B.M.; Swanger, J.; Taatjes, D.J.; Clurman, B.E. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 2013, 27, 151–156. [Google Scholar] [CrossRef]
- Mo, X.; Kowenz-Leutz, E.; Xu, H.; Leutz, A. Ras induces mediator complex exchange on C/EBP beta. Mol. Cell 2004, 13, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Pavri, R.; Lewis, B.; Kim, T.K.; Dilworth, F.J.; Erdjument-Bromage, H.; Tempst, P.; de Murcia, G.; Evans, R.; Chambon, P.; Reinberg, D. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol. Cell 2005, 18, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Lambert, E.; Puwakdandawa, K.; Tao, Y.F.; Robert, F. From structure to molecular condensates: Emerging mechanisms for Mediator function. FEBS J. 2023, 290, 286–309. [Google Scholar] [CrossRef]
- Akoulitchev, S.; Chuikov, S.; Reinberg, D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 2000, 407, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 2004, 16, 509–520. [Google Scholar] [CrossRef]
- Postlmayr, A.; Dumeau, C.E.; Wutz, A. Cdk8 is required for establishment of H3K27me3 and gene repression by Xist and mouse development. Development 2020, 147, dev175141. [Google Scholar] [CrossRef]
- Anandhakumar, J.; Moustafa, Y.W.; Chowdhary, S.; Kainth, A.S.; Gross, D.S. Evidence for Multiple Mediator Complexes in Yeast Independently Recruited by Activated Heat Shock Factor. Mol. Cell. Biol. 2016, 36, 1943–1960. [Google Scholar] [CrossRef]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Nayak, S.; Iwasa, J.; Taatjes, D.J. The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2022, 23, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Luyties, O.; Taatjes, D.J. The Mediator kinase module: An interface between cell signaling and transcription. Trends Biochem. Sci. 2022, 47, 314–327. [Google Scholar] [CrossRef]
- Gonzalez, D.; Hamidi, N.; Del Sol, R.; Benschop, J.J.; Nancy, T.; Li, C.; Francis, L.; Tzouros, M.; Krijgsveld, J.; Holstege, F.C.; et al. Suppression of Mediator is regulated by Cdk8-dependent Grr1 turnover of the Med3 coactivator. Proc. Natl. Acad. Sci. USA 2014, 111, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T.; et al. The molecular architecture of the nuclear pore complex. Nature 2007, 450, 695–701. [Google Scholar] [CrossRef]
- Adler, A.S.; McCleland, M.L.; Truong, T.; Lau, S.; Modrusan, Z.; Soukup, T.M.; Roose-Girma, M.; Blackwood, E.M.; Firestein, R. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res. 2012, 72, 2129–2139. [Google Scholar] [CrossRef]
- Chen, M.; Liang, J.; Ji, H.; Yang, Z.; Altilia, S.; Hu, B.; Schronce, A.; McDermott, M.S.J.; Schools, G.P.; Lim, C.U.; et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc. Natl. Acad. Sci. USA 2017, 114, 10208–10213. [Google Scholar] [CrossRef]
- Hirst, K.; Fisher, F.; McAndrew, P.C.; Goding, C.R. The transcription factor, the Cdk, its cyclin and their regulator: Directing the transcriptional response to a nutritional signal. EMBO J. 1994, 13, 5410–5420. [Google Scholar] [CrossRef]
- Lenssen, E.; Azzouz, N.; Michel, A.; Landrieux, E.; Collart, M.A. The Ccr4-not complex regulates Skn7 through Srb10 kinase. Eukaryot. Cell 2007, 6, 2251–2259. [Google Scholar] [CrossRef]
- Freitas, K.A.; Belk, J.A.; Sotillo, E.; Quinn, P.J.; Ramello, M.C.; Malipatlolla, M.; Daniel, B.; Sandor, K.; Klysz, D.; Bjelajac, J.; et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science 2022, 378, eabn5647. [Google Scholar] [CrossRef]
- Chen, M.; Li, J.; Zhang, L.; Wang, L.; Cheng, C.; Ji, H.; Altilia, S.; Ding, X.; Cai, G.; Altomare, D.; et al. CDK8 and CDK19: Positive regulators of signal-induced transcription and negative regulators of Mediator complex proteins. Nucleic Acids Res. 2023, 51, 7288–7313. [Google Scholar] [CrossRef] [PubMed]
- Stieg, D.C.; Chang, K.T.; Cooper, K.F.; Strich, R. Cyclin C Regulated Oxidative Stress Responsive Transcriptome in Mus musculus Embryonic Fibroblasts. G3 2019, 9, 1901–1908. [Google Scholar] [CrossRef]
- Nowak, S.J.; Corces, V.G. Phosphorylation of histone H3: A balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004, 20, 214–220. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef]
- Kim, T.W.; Kwon, Y.J.; Kim, J.M.; Song, Y.H.; Kim, S.N.; Kim, Y.J. MED16 and MED23 of Mediator are coactivators of lipopolysaccharide- and heat-shock-induced transcriptional activators. Proc. Natl. Acad. Sci. USA 2004, 101, 12153–12158. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Cozzolino, K.A.; Sanford, L.; Hunter, S.; Molison, K.; Erickson, B.; Courvan, M.C.S.; Jones, T.; Ajit, D.; Galbraith, M.D.; Espinosa, J.M.; et al. Mediator kinase inhibition suppresses hyperactive interferon signaling in Down syndrome. eLife 2025, 13, RP100197. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef]
- Jeronimo, C.; Langelier, M.F.; Bataille, A.R.; Pascal, J.M.; Pugh, B.F.; Robert, F. Tail and Kinase Modules Differently Regulate Core Mediator Recruitment and Function In Vivo. Mol. Cell 2016, 64, 455–466. [Google Scholar] [CrossRef]
- Barette, C.; Jariel-Encontre, I.; Piechaczyk, M.; Piette, J. Human cyclin C protein is stabilized by its associated kinase cdk8, independently of its catalytic activity. Oncogene 2001, 20, 551–562. [Google Scholar] [CrossRef]
- Cooper, K.F.; Scarnati, M.S.; Krasley, E.; Mallory, M.J.; Jin, C.; Law, M.J.; Strich, R. Oxidative-stress-induced nuclear to cytoplasmic relocalization is required for Not4-dependent cyclin C destruction. J. Cell Sci. 2012, 125, 1015–1026. [Google Scholar] [CrossRef]
- Wang, K.; Yan, R.; Cooper, K.F.; Strich, R. Cyclin C mediates stress-induced mitochondrial fission and apoptosis. Mol. Biol. Cell 2015, 26, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Ponce, J.M.; Coen, G.; Spitler, K.M.; Dragisic, N.; Martins, I.; Hinton, A.; Mungai, M.; Tadinada, S.M.; Zhang, H.; Oudit, G.Y.; et al. Stress-Induced Cyclin C Translocation Regulates Cardiac Mitochondrial Dynamics. J. Am. Heart Assoc. 2020, 9, e014366. [Google Scholar] [CrossRef] [PubMed]
- Truman, A.W.; Millson, S.H.; Nuttall, J.M.; King, V.; Mollapour, M.; Prodromou, C.; Pearl, L.H.; Piper, P.W. Expressed in the yeast Saccharomyces cerevisiae, human ERK5 is a client of the Hsp90 chaperone that complements loss of the Slt2p (Mpk1p) cell integrity stress-activated protein kinase. Eukaryot. Cell 2006, 5, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef]
- Krasley, E.; Cooper, K.F.; Mallory, M.J.; Dunbrack, R.; Strich, R. Regulation of the oxidative stress response through Slt2p-dependent destruction of cyclin C in Saccharomyces cerevisiae. Genetics 2006, 172, 1477–1486. [Google Scholar] [CrossRef]
- Jin, C.; Parshin, A.V.; Daly, I.; Strich, R.; Cooper, K.F. The cell wall sensors Mtl1, Wsc1, and Mid2 are required for stress-induced nuclear to cytoplasmic translocation of cyclin C and programmed cell death in yeast. Oxid. Med. Cell. Longev. 2013, 2013, 320823. [Google Scholar] [CrossRef]
- Jin, C.; Strich, R.; Cooper, K.F. Slt2p phosphorylation induces cyclin C nuclear-to-cytoplasmic translocation in response to oxidative stress. Mol. Biol. Cell 2014, 25, 1396–1407. [Google Scholar] [CrossRef]
- Stieg, D.C.; Willis, S.D.; Ganesan, V.; Ong, K.L.; Scuorzo, J.; Song, M.; Grose, J.; Strich, R.; Cooper, K.F. A complex molecular switch directs stress-induced cyclin C nuclear release through SCF(Grr1)-mediated degradation of Med13. Mol. Biol. Cell 2018, 29, 363–375. [Google Scholar] [CrossRef]
- Khakhina, S.; Cooper, K.F.; Strich, R. Med13p prevents mitochondrial fission and programmed cell death in yeast through nuclear retention of cyclin C. Mol. Biol. Cell 2014, 25, 2807–2816. [Google Scholar] [CrossRef]
- Cooper, K.F.; Khakhina, S.; Kim, S.K.; Strich, R. Stress-induced nuclear-to-cytoplasmic translocation of cyclin C promotes mitochondrial fission in yeast. Dev. Cell 2014, 28, 161–173. [Google Scholar] [CrossRef]
- Nash, P.; Tang, X.; Orlicky, S.; Chen, Q.; Gertler, F.B.; Mendenhall, M.D.; Sicheri, F.; Pawson, T.; Tyers, M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 2001, 414, 514–521. [Google Scholar] [CrossRef]
- Ang, X.L.; Wade Harper, J. SCF-mediated protein degradation and cell cycle control. Oncogene 2005, 24, 2860–2870. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Carlson, M. Regulation of snf1 protein kinase in response to environmental stress. J. Biol. Chem. 2007, 282, 16838–16845. [Google Scholar] [CrossRef]
- Willis, S.D.; Stieg, D.C.; Ong, K.L.; Shah, R.; Strich, A.K.; Grose, J.H.; Cooper, K.F. Snf1 cooperates with the CWI MAPK pathway to mediate the degradation of Med13 following oxidative stress. Microb. Cell 2018, 5, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.F.; Strich, R. Functional analysis of the Ume3p/Srb11p-RNA polymerase II holoenzyme interaction. Gene Expr. 1999, 8, 43–57. [Google Scholar] [PubMed]
- Jezek, J.; Chang, K.T.; Joshi, A.M.; Strich, R. Mitochondrial translocation of cyclin C stimulates intrinsic apoptosis through Bax recruitment. EMBO Rep. 2019, 20, e47425. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, Y.; Wu, X.; Chen, J.; Zhou, Q.; Liu, B.; Zhang, L.; Yi, C. Interplay of energy metabolism and autophagy. Autophagy 2024, 20, 4–14. [Google Scholar] [CrossRef]
- Willis, S.D.; Hanley, S.E.; Beishke, T.; Tati, P.D.; Cooper, K.F. Ubiquitin-proteasome-mediated cyclin C degradation promotes cell survival following nitrogen starvation. Mol. Biol. Cell 2020, 31, 1015–1031. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- Hanley, S.E.; Willis, S.D.; Doyle, S.J.; Strich, R.; Cooper, K.F. Ksp1 is an autophagic receptor protein for the Snx4-assisted autophagy of Ssn2/Med13. Autophagy 2024, 20, 397–415. [Google Scholar] [CrossRef]
- Hanley, S.E.; Willis, S.D.; Friedson, B.; Cooper, K.F. Med13 is required for efficient P-body recruitment and autophagic degradation of Edc3 following nitrogen starvation. Mol. Biol. Cell 2024, 35, ar142. [Google Scholar] [CrossRef]
- Torres, J.; Di Como, C.J.; Herrero, E.; De La Torre-Ruiz, M.A. Regulation of the cell integrity pathway by rapamycin-sensitive TOR function in budding yeast. J. Biol. Chem. 2002, 277, 43495–43504. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Reichert, A.S. Mitochondrial Dynamics at Different Levels: From Cristae Dynamics to Interorganellar Cross Talk. Annu. Rev. Biophys. 2024, 53, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. The Functional Impact of Mitochondrial Structure Across Subcellular Scales. Front. Physiol. 2020, 11, 541040. [Google Scholar] [CrossRef] [PubMed]
- Westrate, L.M.; Drocco, J.A.; Martin, K.R.; Hlavacek, W.S.; MacKeigan, J.P. Mitochondrial morphological features are associated with fission and fusion events. PLoS ONE 2014, 9, e95265. [Google Scholar] [CrossRef] [PubMed]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef]
- Lewis, T.L., Jr.; Kwon, S.K.; Lee, A.; Shaw, R.; Polleux, F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 2018, 9, 5008. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Zou, W.; Yang, L.; Lu, H.; Li, M.; Ji, D.; Slone, J.; Huang, T. Application of super-resolution microscopy in mitochondria-dynamic diseases. Adv. Drug Deliv. Rev. 2023, 200, 115043. [Google Scholar] [CrossRef] [PubMed]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Ganesan, V.; Willis, S.D.; Chang, K.T.; Beluch, S.; Cooper, K.F.; Strich, R. Cyclin C directly stimulates Drp1 GTP affinity to mediate stress-induced mitochondrial hyperfission. Mol. Biol. Cell 2019, 30, 302–311. [Google Scholar] [CrossRef]
- Ježek, J.; Smethurst, D.G.J.; Stieg, D.C.; Kiss, Z.A.C.; Hanley, S.E.; Ganesan, V.; Chang, K.T.; Cooper, K.F.; Strich, R. Cyclin C: The Story of a Non-Cycling Cyclin. Biology 2019, 8, 3. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Hoeppner, S.; Baumli, S.; Cramer, P. Structure of the mediator subunit cyclin C and its implications for CDK8 function. J. Mol. Biol. 2005, 350, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.T.; Karren, M.A.; Bhar, D.; Shaw, J.M. A novel motif in the yeast mitochondrial dynamin Dnm1 is essential for adaptor binding and membrane recruitment. J. Cell Biol. 2012, 199, 613–622. [Google Scholar] [CrossRef]
- Guo, Q.; Koirala, S.; Perkins, E.M.; McCaffery, J.M.; Shaw, J.M. The mitochondrial fission adaptors Caf4 and Mdv1 are not functionally equivalent. PLoS ONE 2012, 7, e53523. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Lipton, S.A. Mitochondrial dynamics in cell death and neurodegeneration. Cell. Mol. Life Sci. 2010, 67, 3435–3447. [Google Scholar] [CrossRef]
- Sheridan, C.; Martin, S.J. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 2010, 10, 640–648. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef]
- Sheridan, C.; Delivani, P.; Cullen, S.P.; Martin, S.J. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol. Cell 2008, 31, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Jenner, A.; Peña-Blanco, A.; Salvador-Gallego, R.; Ugarte-Uribe, B.; Zollo, C.; Ganief, T.; Bierlmeier, J.; Mund, M.; Lee, J.E.; Ries, J.; et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 2022, 41, e108587. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Schellenberg, B.; Wang, P.; Keeble, J.A.; Rodriguez-Enriquez, R.; Walker, S.; Owens, T.W.; Foster, F.; Tanianis-Hughes, J.; Brennan, K.; Streuli, C.H.; et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell 2013, 49, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Subburaj, Y.; Cosentino, K.; Axmann, M.; Pedrueza-Villalmanzo, E.; Hermann, E.; Bleicken, S.; Spatz, J.; García-Sáez, A.J. Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 2015, 6, 8042. [Google Scholar] [CrossRef]
- Pena-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Manon, S.; Chaudhuri, B.; Guerin, M. Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett. 1997, 415, 29–32. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, aao6047. [Google Scholar] [CrossRef]
- Zha, H.; Fisk, H.A.; Yaffe, M.P.; Mahajan, N.; Herman, B.; Reed, J.C. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell. Biol. 1996, 16, 6494–6508. [Google Scholar] [CrossRef]
- Polcic, P.; Jaka, P.; Mentel, M. Yeast as a tool for studying proteins of the Bcl-2 family. Microb. Cell 2015, 2, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Lhuissier, C.; Desquiret-Dumas, V.; Girona, A.; Alban, J.; Faure, J.; Cassereau, J.; Codron, P.; Lenaers, G.; Baris, O.R.; Gueguen, N.; et al. Mitochondrial F0F1-ATP synthase governs the induction of mitochondrial fission. iScience 2024, 27, 109808. [Google Scholar] [CrossRef]
- Matsuyama, S.; Xu, Q.; Velours, J.; Reed, J.C. The Mitochondrial F0F1-ATPase Proton Pump Is Required for Function of the Proapoptotic Protein Bax in Yeast and Mammalian Cells. Mol. Cell 1998, 1, 327–336. [Google Scholar] [CrossRef]
- Sato, T.; Hanada, M.; Bodrug, S.; Irie, S.; Iwama, N.; Boise, L.H.; Thompson, C.B.; Golemis, E.; Fong, L.; Wang, H.G.; et al. Interactions among members of the Bcl-2 protein family analyzed with a yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 1994, 91, 9238–9242. [Google Scholar] [CrossRef] [PubMed]
- Légiot, A.; Céré, C.; Dupoiron, T.; Kaabouni, M.; Camougrand, N.; Manon, S. Mitochondria-Associated Membranes (MAMs) are involved in Bax mitochondrial localization and cytochrome c release. Microb. Cell 2019, 6, 257–266. [Google Scholar] [CrossRef]
- Toth, A.; Aufschnaiter, A.; Fedotovskaya, O.; Dawitz, H.; Ädelroth, P.; Büttner, S.; Ott, M. Membrane-tethering of cytochrome c accelerates regulated cell death in yeast. Cell Death Dis. 2020, 11, 722. [Google Scholar] [CrossRef]
- Sun, G. Death and survival from executioner caspase activation. Semin. Cell Dev. Biol. 2024, 156, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Herker, E.; Maldener, C.; Wissing, S.; Lachelt, S.; Herlan, M.; Fehr, M.; Lauber, K.; Sigrist, S.J.; Wesselborg, S.; et al. A caspase-related protease regulates apoptosis in yeast. Mol. Cell 2002, 9, 911–917. [Google Scholar] [CrossRef]
- Shrestha, A.; Brunette, S.; Stanford, W.L.; Megeney, L.A. The metacaspase Yca1 maintains proteostasis through multiple interactions with the ubiquitin system. Cell Discov. 2019, 5, 6. [Google Scholar] [CrossRef]
- Lee, R.E.; Brunette, S.; Puente, L.G.; Megeney, L.A. Metacaspase Yca1 is required for clearance of insoluble protein aggregates. Proc. Natl. Acad. Sci. USA 2010, 107, 13348–13353. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.K.; Sherlock, G. Yca1 metacaspase: Diverse functions determine how yeast live and let die. FEMS Yeast Res. 2023, 23, foad022. [Google Scholar] [CrossRef] [PubMed]
- Camougrand, N.; Grelaud-Coq, A.; Marza, E.; Priault, M.; Bessoule, J.J.; Manon, S. The product of the UTH1 gene, required for Bax-induced cell death in yeast, is involved in the response to rapamycin. Mol. Microbiol. 2003, 47, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Ritch, J.J.; Davidson, S.M.; Sheehan, J.J.; Austriaco, N. The Saccharomyces SUN gene, UTH1, is involved in cell wall biogenesis. FEMS Yeast Res. 2010, 10, 168–176. [Google Scholar] [CrossRef]
- Eid, R.; Boucher, E.; Gharib, N.; Khoury, C.; Arab, N.T.; Murray, A.; Young, P.G.; Mandato, C.A.; Greenwood, M.T. Identification of human ferritin, heavy polypeptide 1 (FTH1) and yeast RGI1 (YER067W) as pro-survival sequences that counteract the effects of Bax and copper in Saccharomyces cerevisiae. Exp. Cell Res. 2016, 342, 52–61. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Aguilera, A.; Austriaco, N.; Ayscough, K.; Balzan, R.; Bar-Nun, S.; Barrientos, A.; Belenky, P.; et al. Guidelines and recommendations on yeast cell death nomenclature. Microb. Cell 2018, 5, 4–31. [Google Scholar] [CrossRef]
- Strich, R.; Cooper, K.F. The dual role of cyclin C connects stress regulated gene expression to mitochondrial dynamics. Microb. Cell 2014, 1, 318–324. [Google Scholar] [CrossRef]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef]
- Wilkinson, D.; Ramsdale, M. Proteases and caspase-like activity in the yeast Saccharomyces cerevisiae. Biochem. Soc. Trans. 2011, 39, 1502–1508. [Google Scholar] [CrossRef]
- Mentel, M.; Illová, M.; Krajčovičová, V.; Kroupová, G.; Mannová, Z.; Chovančíková, P.; Polčic, P. Yeast Bax Inhibitor (Bxi1p/Ybh3p) Is Not Required for the Action of Bcl-2 Family Proteins on Cell Viability. Int. J. Mol. Sci. 2023, 24, 12011. [Google Scholar] [CrossRef]
- Buchan, J.R. Stress granule and P-body clearance: Seeking coherence in acts of disappearance. Semin. Cell Dev. Biol. 2024, 159–160, 10–26. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Nostramo, R.; Xing, S.; Zhang, B.; Herman, P.K. Insights into the Role of P-Bodies and Stress Granules in Protein Quality Control. Genetics 2019, 213, 251–265. [Google Scholar] [CrossRef]
- Ramachandran, V.; Shah, K.H.; Herman, P.K. The cAMP-dependent protein kinase signaling pathway is a key regulator of P body foci formation. Mol. Cell 2011, 43, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. RNA granules: Post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 2009, 10, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 2013, 203, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef]
- Wang, C.; Schmich, F.; Srivatsa, S.; Weidner, J.; Beerenwinkel, N.; Spang, A. Context-dependent deposition and regulation of mRNAs in P-bodies. eLife 2018, 7, e29815. [Google Scholar] [CrossRef]
- Standart, N.; Weil, D. P-Bodies: Cytosolic Droplets for Coordinated mRNA Storage. Trends Genet. 2018, 34, 612–626. [Google Scholar] [CrossRef]
- Horvathova, I.; Voigt, F.; Kotrys, A.V.; Zhan, Y.; Artus-Revel, C.G.; Eglinger, J.; Stadler, M.B.; Giorgetti, L.; Chao, J.A. The Dynamics of mRNA Turnover Revealed by Single-Molecule Imaging in Single Cells. Mol. Cell 2017, 68, 615–625 e619. [Google Scholar] [CrossRef] [PubMed]
- Hubstenberger, A.; Courel, M.; Bénard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol. Cell 2017, 68, 144–157.e145. [Google Scholar] [CrossRef]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef]
- Fromm, S.A.; Truffault, V.; Kamenz, J.; Braun, J.E.; Hoffmann, N.A.; Izaurralde, E.; Sprangers, R. The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. EMBO J. 2012, 31, 279–290. [Google Scholar] [CrossRef]
- Fromm, S.A.; Kamenz, J.; Noldeke, E.R.; Neu, A.; Zocher, G.; Sprangers, R. In vitro reconstitution of a cellular phase-transition process that involves the mRNA decapping machinery. Angew. Chem. Int. Ed. Engl. 2014, 53, 7354–7359. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Ozgur, S.; Sharma, K.; Basquin, C.; Urlaub, H.; Conti, E. Structural analysis of the yeast Dhh1–Pat1 complex reveals how Dhh1 engages Pat1, Edc3 and RNA in mutually exclusive interactions. Nucleic Acids Res. 2013, 41, 8377–8390. [Google Scholar] [CrossRef]
- Schütz, S.; Nöldeke, E.R.; Sprangers, R. A synergistic network of interactions promotes the formation of in vitro processing bodies and protects mRNA against decapping. Nucleic Acids Res. 2017, 45, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.J.; Teixeira, D.; Parker, R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 2007, 179, 437–449. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 2015, 589, 15–22. [Google Scholar] [CrossRef]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef]
- Wang, J.T.; Smith, J.; Chen, B.C.; Schmidt, H.; Rasoloson, D.; Paix, A.; Lambrus, B.G.; Calidas, D.; Betzig, E.; Seydoux, G. Regulation of RNA granule dynamics by phosphorylation of serine-rich, intrinsically disordered proteins in C. elegans. eLife 2014, 3, e04591. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019, 15, 51–61. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef]
- Jafari, M.; McCabe, M.; Cuervo, A.M. Chaperone-mediated autophagy: Mechanisms and physiological relevance. Curr. Opin. Physiol. 2022, 30, 100597. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Klionsky, D.J.; Shen, H.-M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Isoda, T.; Takeda, E.; Hosokawa, S.; Hotta-Ren, S.; Ohsumi, Y. Atg45 is an autophagy receptor for glycogen, a non-preferred cargo of bulk autophagy in yeast. iScience 2024, 27, 109810. [Google Scholar] [CrossRef]
- Takeda, E.; Isoda, T.; Hosokawa, S.; Oikawa, Y.; Hotta-Ren, S.; May, A.I.; Ohsumi, Y. Receptor-mediated cargo hitchhiking on bulk autophagy. EMBO J. 2024, 43, 3116–3140. [Google Scholar] [CrossRef] [PubMed]
- Hanley, S.E.; Cooper, K.F. Sorting Nexins in Protein Homeostasis. Cells 2020, 10, 17. [Google Scholar] [CrossRef]
- Vazquez-Sanchez, S.; Gonzalez-Lozano, M.A.; Walfenzao, A.; Li, K.W.; van Weering, J.R.T. The endosomal protein sorting nexin 4 is a synaptic protein. Sci. Rep. 2020, 10, 18239. [Google Scholar] [CrossRef]
- Poppinga, J.; Barret, N.J.; Cornelisse, L.N.; Verhage, M.; van Weering, J.R.T. Endosomal sorting protein SNX4 limits synaptic vesicle docking and release. eLife 2024, 13, RP97910. [Google Scholar] [CrossRef]
- Kim, N.Y.; Cho, M.H.; Won, S.H.; Kang, H.J.; Yoon, S.Y.; Kim, D.H. Sorting nexin-4 regulates β-amyloid production by modulating β-site-activating cleavage enzyme-1. Alzheimers Res. Ther. 2017, 9, 4. [Google Scholar] [CrossRef]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Kotani, T.; Sakai, Y.; Kirisako, H.; Kakuta, C.; Kakuta, S.; Ohsumi, Y.; Nakatogawa, H. A mechanism that ensures non-selective cytoplasm degradation by autophagy. Nat. Commun. 2023, 14, 5815. [Google Scholar] [CrossRef]
- Nemec, A.A.; Howell, L.A.; Peterson, A.K.; Murray, M.A.; Tomko, R.J., Jr. Autophagic clearance of proteasomes in yeast requires the conserved sorting nexin Snx4. J. Biol. Chem. 2017, 292, 21466–21480. [Google Scholar] [CrossRef]
- Makino, S.; Kawamata, T.; Iwasaki, S.; Ohsumi, Y. Selectivity of mRNA degradation by autophagy in yeast. Nat. Commun. 2021, 12, 2316. [Google Scholar] [CrossRef]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Shimron, F.; Peleg, Y.; Elazar, Z. Fatty acid synthase is preferentially degraded by autophagy upon nitrogen starvation in yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 1434–1439. [Google Scholar] [CrossRef]
- Liu, X.; Mao, K.; Yu, A.Y.H.; Omairi-Nasser, A.; Austin, J., 2nd; Glick, B.S.; Yip, C.K.; Klionsky, D.J. The Atg17-Atg31-Atg29 Complex Coordinates with Atg11 to Recruit the Vam7 SNARE and Mediate Autophagosome-Vacuole Fusion. Curr. Biol. 2016, 26, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Noda, N.N. Biophysical characterization of Atg11, a scaffold protein essential for selective autophagy in yeast. FEBS Open Bio 2018, 8, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Mechanisms of Selective Autophagy. Annu. Rev. Cell. Dev. Biol. 2021, 37, 143–169. [Google Scholar] [CrossRef]
- Fujioka, Y.; Alam, J.M.; Noshiro, D.; Mouri, K.; Ando, T.; Okada, Y.; May, A.I.; Knorr, R.L.; Suzuki, K.; Ohsumi, Y.; et al. Phase separation organizes the site of autophagosome formation. Nature 2020, 578, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Wang, Z.; Zhang, H. Liquid–liquid phase separation in autophagy. J. Cell Biol. 2020, 219, e202004062. [Google Scholar] [CrossRef]
- Yamamoto, H.; Fujioka, Y.; Suzuki, S.W.; Noshiro, D.; Suzuki, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ando, T.; Noda, N.N.; et al. The Intrinsically Disordered Protein Atg13 Mediates Supramolecular Assembly of Autophagy Initiation Complexes. Dev. Cell 2016, 38, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Nezis, I.P.; Tsapras, P.; Zhang, H.; Dagdas, Y.; Noda, N.N.; Nakatogawa, H.; Wirth, M.; Mouilleron, S.; McEwan, D.G.; et al. Atg8 family proteins, LIR/AIM motifs and other interaction modes. Autophagy Rep. 2023, 2, 2188523. [Google Scholar] [CrossRef]
- Laxman, S.; Tu, B.P. Multiple TORC1-associated proteins regulate nitrogen starvation-dependent cellular differentiation in Saccharomyces cerevisiae. PLoS ONE 2011, 6, e26081. [Google Scholar] [CrossRef] [PubMed]
- Umekawa, M.; Klionsky, D.J. Ksp1 kinase regulates autophagy via the target of rapamycin complex 1 (TORC1) pathway. J. Biol. Chem. 2012, 287, 16300–16310. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Bodenmiller, B.; Uotila, A.; Stahl, M.; Wanka, S.; Gerrits, B.; Aebersold, R.; Loewith, R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009, 23, 1929–1943. [Google Scholar] [CrossRef]
- Oliveira, A.P.; Ludwig, C.; Zampieri, M.; Weisser, H.; Aebersold, R.; Sauer, U. Dynamic phosphoproteomics reveals TORC1-dependent regulation of yeast nucleotide and amino acid biosynthesis. Sci. Signal 2015, 8, rs4. [Google Scholar] [CrossRef] [PubMed]
- Soulard, A.; Cremonesi, A.; Moes, S.; Schütz, F.; Jenö, P.; Hall, M.N. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol. Biol. Cell 2010, 21, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Muhlrad, D.; Parker, R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 2008, 183, 441–455. [Google Scholar] [CrossRef]
- Mitchell, S.F.; Jain, S.; She, M.; Parker, R. Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 2013, 20, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.C.; Wente, S.R. Functions of Gle1 are governed by two distinct modes of self-association. J. Biol. Chem. 2020, 295, 16813–16825. [Google Scholar] [CrossRef] [PubMed]
- Demetrick, D.J.; Matsumoto, S.; Hannon, G.J.; Okamoto, K.; Xiong, Y.; Zhang, H.; Beach, D.H. Chromosomal mapping of the genes for the human cell cycle proteins cyclin C (CCNC), cyclin E (CCNE), p21 (CDKN1) and KAP (CDKN3). Cytogenet. Cell Genet. 1995, 69, 190–192. [Google Scholar] [CrossRef]
- Offit, K.; Parsa, N.Z.; Gaidano, G.; Filippa, D.A.; Louie, D.; Pan, D.; Jhanwar, S.C.; Dalla-Favera, R.; Chaganti, R.S.K. 6q deletions define distinct clinico-pathologic subsets of non-Hodgkin’s lympoma. Blood 1993, 82, 2157–2162. [Google Scholar] [CrossRef]
- Ohata, N.; Ito, S.; Yoshida, A.; Kunisada, T.; Numoto, K.; Jitsumori, Y.; Kanzaki, H.; Ozaki, T.; Shimizu, K.; Ouchida, M. Highly frequent allelic loss of chromosome 6q16-23 in osteosarcoma: Involvement of cyclin C in osteosarcoma. Int. J. Mol. Med. 2006, 18, 1153–1158. [Google Scholar] [CrossRef]
- Clark, A.D.; Oldenbroek, M.; Boyer, T.G. Mediator kinase module and human tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 393–426. [Google Scholar] [CrossRef]
- Li, H.; Lahti, J.M.; Valentine, M.; Saito, M.; Reed, S.I.; Look, A.T.; Kidd, V.J. Molecular cloning and chromosomal localization of the human cyclin C (CCNC) and cyclin E (CCNE) genes: Deletion of the CCNC gene in human tumors. Genomics 1996, 32, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fassl, A.; Chick, J.; Inuzuka, H.; Li, X.; Mansour, M.R.; Liu, L.; Wang, H.; King, B.; Shaik, S.; et al. Cyclin C is a haploinsufficient tumour suppressor. Nat. Cell Biol. 2014, 16, 1080–1091. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of Notch signalling—Are we there yet? Nat. Rev. Drug Discov. 2014, 13, 357–378. [Google Scholar] [CrossRef] [PubMed]
- Gharaibeh, L.; Elmadany, N.; Alwosaibai, K.; Alshaer, W. Notch1 in Cancer Therapy: Possible Clinical Implications and Challenges. Mol. Pharmacol. 2020, 98, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Radtke, F. Notch signaling in solid tumors. Curr. Top. Dev. Biol. 2010, 92, 411–455. [Google Scholar] [CrossRef]
- Hyytinen, E.R.; Saadut, R.; Chen, C.; Paull, L.; Koivisto, P.A.; Vessella, R.L.; Frierson, H.F., Jr.; Dong, J.T. Defining the region(s) of deletion at 6q16-q22 in human prostate cancer. Genes. Chromosomes Cancer 2002, 34, 306–312. [Google Scholar] [CrossRef]
- Jackson, A.; Carrara, P.; Duke, V.; Sinclair, P.; Papaioannou, M.; Harrison, C.J.; Foroni, L. Deletion of 6q16-q21 in human lymphoid malignancies: A mapping and deletion analysis. Cancer Res. 2000, 60, 2775–2779. [Google Scholar]
- Sinclair, P.B.; Sorour, A.; Martineau, M.; Harrison, C.J.; Mitchell, W.A.; O’Neill, E.; Foroni, L. A fluorescence in situ hybridization map of 6q deletions in acute lymphocytic leukemia: Identification and analysis of a candidate tumor suppressor gene. Cancer Res. 2004, 64, 4089–4098. [Google Scholar] [CrossRef]
- Lobry, C.; Oh, P.; Mansour, M.R.; Look, A.T.; Aifantis, I. Notch signaling: Switching an oncogene to a tumor suppressor. Blood 2014, 123, 2451–2459. [Google Scholar] [CrossRef]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, 2304. [Google Scholar] [CrossRef] [PubMed]
- Choschzick, M.; Stergiou, C.; Gut, A.; Zoche, M.; Ross, J.S.; Moch, H. NOTCH1 and PIK3CA mutation are related to HPV-associated vulvar squamous cell carcinoma. Pathol. Res. Pract. 2023, 251, 154877. [Google Scholar] [CrossRef]
- Jezek, J.; Wang, K.; Yan, R.; Di Cristofano, A.; Cooper, K.F.; Strich, R. Synergistic repression of thyroid hyperplasia by cyclin C and Pten. J. Cell Sci. 2019, 132, jcs230029. [Google Scholar] [CrossRef]
- Wreesmann, V.B.; Ghossein, R.A.; Patel, S.G.; Harris, C.P.; Schnaser, E.A.; Shaha, A.R.; Tuttle, R.M.; Shah, J.P.; Rao, P.H.; Singh, B. Genome-wide appraisal of thyroid cancer progression. Am. J. Pathol. 2002, 161, 1549–1556. [Google Scholar] [CrossRef]
- Antico Arciuch, V.G.; Russo, M.A.; Dima, M.; Kang, K.S.; Dasrath, F.; Liao, X.H.; Refetoff, S.; Montagna, C.; Di Cristofano, A. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget 2011, 2, 1109–1126. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef]
- Ho, T.-Y.; Sung, T.-Y.; Pan, S.-L.; Huang, W.-J.; Hsu, K.-C.; Hsu, J.-Y.; Lin, T.E.; Hsu, C.-M.; Yang, C.-R. The study of a novel CDK8 inhibitor E966-0530–45418 that inhibits prostate cancer metastasis in vitro and in vivo. Biomed. Pharmacother. 2023, 162, 114667. [Google Scholar] [CrossRef]
- Roninson, I.B.; Győrffy, B.; Mack, Z.T.; Shtil, A.A.; Shtutman, M.S.; Chen, M.; Broude, E.V. Identifying Cancers Impacted by CDK8/19. Cells 2019, 8, 821. [Google Scholar] [CrossRef]
- Porter, D.C.; Farmaki, E.; Altilia, S.; Schools, G.P.; West, D.K.; Chen, M.; Chang, B.D.; Puzyrev, A.T.; Lim, C.U.; Rokow-Kittell, R.; et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. USA 2012, 109, 13799–13804. [Google Scholar] [CrossRef] [PubMed]
- Brägelmann, J.; Klümper, N.; Offermann, A.; von Mässenhausen, A.; Böhm, D.; Deng, M.; Queisser, A.; Sanders, C.; Syring, I.; Merseburger, A.S.; et al. Pan-Cancer Analysis of the Mediator Complex Transcriptome Identifies CDK19 and CDK8 as Therapeutic Targets in Advanced Prostate Cancer. Clin. Cancer Res. 2017, 23, 1829–1840. [Google Scholar] [CrossRef]
- Menzl, I.; Witalisz-Siepracka, A.; Sexl, V. CDK8-Novel Therapeutic Opportunities. Pharmaceuticals 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef]
- Datta, N.; Chakraborty, S.; Basu, M.; Ghosh, M.K. Tumor Suppressors Having Oncogenic Functions: The Double Agents. Cells 2020, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Grabher, C.; von Boehmer, H.; Look, A.T. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2006, 6, 347–359. [Google Scholar] [CrossRef]
- Dotto, G.P. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat. Rev. Cancer 2009, 9, 587–595. [Google Scholar] [CrossRef]
- Shen, L.; Shi, Q.; Wang, W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018, 7, 25. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Pantano, S.; D’Ambrogio, A.; Buratti, E.; Brindisi, A.; Marchetti, C.; Romano, M.; Baralle, F.E. Human, Drosophila, and C.elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 2005, 348, 575–588. [Google Scholar] [CrossRef]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef]
- Gottschalk, W.K.; Lutz, M.W.; He, Y.T.; Saunders, A.M.; Burns, D.K.; Roses, A.D.; Chiba-Falek, O. The Broad Impact of TOM40 on Neurodegenerative Diseases in Aging. J. Park. Dis. Alzheimers Dis. 2014, 1, 12. [Google Scholar] [CrossRef]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 2017, 7, 7709. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed]
- Lucini, C.B.; Braun, R.J. Mitochondrion-Dependent Cell Death in TDP-43 Proteinopathies. Biomedicines 2021, 9, 376. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Wang, Y.T.; Kuo, P.H.; Chiang, C.H.; Liang, J.R.; Chen, Y.R.; Wang, S.; Shen, J.C.; Yuan, H.S. The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 2013, 288, 9049–9057. [Google Scholar] [CrossRef] [PubMed]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ngo, S.T. Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis. Metabolites 2022, 12, 709. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Schwenk, B.M.; Hartmann, H.; Serdaroglu, A.; Schludi, M.H.; Hornburg, D.; Meissner, F.; Orozco, D.; Colombo, A.; Tahirovic, S.; Michaelsen, M.; et al. TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 2016, 35, 2350–2370. [Google Scholar] [CrossRef]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Bose, J.K.; Huang, C.C.; Shen, C.K. Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Kang, Y.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470.e455. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649 e618. [Google Scholar] [CrossRef]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovascular Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Bharathi, V.; Girdhar, A.; Patel, B.K. Role of CNC1 gene in TDP-43 aggregation-induced oxidative stress-mediated cell death in S. cerevisiae model of ALS. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118993. [Google Scholar] [CrossRef] [PubMed]
- Riva, N.; Domi, T.; Pozzi, L.; Lunetta, C.; Schito, P.; Spinelli, E.G.; Cabras, S.; Matteoni, E.; Consonni, M.; Bella, E.D.; et al. Update on recent advances in amyotrophic lateral sclerosis. J. Neurol. 2024, 271, 4693–4723. [Google Scholar] [CrossRef]
- Daniels, D.L.; Ford, M.J.; Schwinn, M.K.; Benink, H.A.; Galbraith, M.D.; Amunugama, R.; Jones, R.J.; Allen, D.; Okazaki, N.; Yamakawa, H.; et al. Mutual Exclusivity of MED12/MED12L, MED13/13L, and CDK8/19Paralogs Revealed within the CDK-Mediator Kinase Module. J. Proteom. Bioinform. 2013, 2013, 1–7. [Google Scholar]
- Bessenyei, B.; Balogh, I.; Mokánszki, A.; Ujfalusi, A.; Pfundt, R.; Szakszon, K. MED13L-related intellectual disability due to paternal germinal mosaicism. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006124. [Google Scholar] [CrossRef]
- Hamada, N.; Iwamoto, I.; Nagata, K.I. MED13L and its disease-associated variants influence the dendritic development of cerebral cortical neurons in the mammalian brain. J. Neurochem. 2023, 165, 334–347. [Google Scholar] [CrossRef]
- Siavrienė, E.; Petraitytė, G.; Mikštienė, V.; Maldžienė, Ž.; Sasnauskienė, A.; Žitkutė, V.; Ambrozaitytė, L.; Rančelis, T.; Utkus, A.; Kučinskas, V.; et al. Molecular and Functional Characterisation of a Novel Intragenic 12q24.21 Deletion Resulting in MED13L Haploinsufficiency Syndrome. Medicina 2023, 59, 1225. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, R.; Pfalzer, A.; Bichell, T.J.; Terala, A.; Campbell, A.; Taatjes, D.; Ghoumid, J.; Grueter, C.; Bain, J.; Strich, R.; et al. The MED13L Foundation strategic research plan: A roadmap to the future. Ther. Adv. Rare Dis. 2024, 5, 26330040241290252. [Google Scholar] [CrossRef]
- Chang, K.T.; Jezek, J.; Campbell, A.N.; Stieg, D.C.; Kiss, Z.A.; Kemper, K.; Jiang, P.; Lee, H.O.; Kruger, W.D.; van Hasselt, P.M.; et al. Aberrant cyclin C nuclear release induces mitochondrial fragmentation and dysfunction in MED13L syndrome fibroblasts. iScience 2022, 25, 103823. [Google Scholar] [CrossRef]
- Snijders Blok, L.; Hiatt, S.M.; Bowling, K.M.; Prokop, J.W.; Engel, K.L.; Cochran, J.N.; Bebin, E.M.; Bijlsma, E.K.; Ruivenkamp, C.A.L.; Terhal, P.; et al. De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum. Genet. 2018, 137, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Polla, D.L.; Bhoj, E.J.; Verheij, J.B.G.M.; Wassink-Ruiter, J.S.K.; Reis, A.; Deshpande, C.; Gregor, A.; Hill-Karfe, K.; Silfhout, A.T.V.-v.; Pfundt, R.; et al. De novo variants in MED12 cause X-linked syndromic neurodevelopmental disorders in 18 females. Genet. Med. 2021, 23, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Plassche, S.V.; Brouwer, A.P. MED12-Related (Neuro)Developmental Disorders: A Question of Causality. Genes 2021, 12, 663. [Google Scholar] [CrossRef]
- Calpena, E.; Hervieu, A.; Kaserer, T.; Swagemakers, S.M.A.; Goos, J.A.C.; Popoola, O.; Ortiz-Ruiz, M.J.; Barbaro-Dieber, T.; Bownass, L.; Brilstra, E.H.; et al. De Novo Missense Substitutions in the Gene Encoding CDK8, a Regulator of the Mediator Complex, Cause a Syndromic Developmental Disorder. Am. J. Hum. Genet. 2019, 104, 709–720. [Google Scholar] [CrossRef]
- Uehara, T.; Abe, K.; Oginuma, M.; Ishitani, S.; Yoshihashi, H.; Okamoto, N.; Takenouchi, T.; Kosaki, K.; Ishitani, T. Pathogenesis of CDK8-associated disorder: Two patients with novel CDK8 variants and in vitro and in vivo functional analyses of the variants. Sci. Rep. 2020, 10, 17575. [Google Scholar] [CrossRef]
- Lee, M.; Cheon, K.; Chae, B.; Hwang, H.; Kim, H.K.; Chung, Y.J.; Song, J.Y.; Cho, H.H.; Kim, J.H.; Kim, M.R. Analysis of MED12 Mutation in Multiple Uterine Leiomyomas in South Korean patients. Int. J. Med. Sci. 2018, 15, 124–128. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Cheng, D.; Semmens, K.; McManus, E.; Chen, Q.; Meerzaman, D.; Wang, X.; Hafner, M.; Lewis, B.A.; Takahashi, H.; Devaiah, B.N.; et al. The nuclear transcription factor, TAF7, is a cytoplasmic regulator of protein synthesis. Sci. Adv. 2021, 7, eabi5751. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Catez, F.; Diaz, J.J. p53, a translational regulator: Contribution to its tumour-suppressor activity. Oncogene 2015, 34, 5513–5523. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Kowalski, A.; Betzer, C.; Larsen, S.T.; Gregersen, E.; Newcombe, E.A.; Bermejo, M.C.; Bendtsen, V.W.; Diemer, J.; Ernstsen, C.V.; Jain, S.; et al. Monomeric alpha-synuclein activates the plasma membrane calcium pump. EMBO J. 2023, 42, e111122. [Google Scholar] [CrossRef]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell 2022, 185, 2035–2056.e2033. [Google Scholar] [CrossRef]
- Friedson, B.; Cooper, K.F. Cdk8 Kinase Module: A Mediator of Life and Death Decisions in Times of Stress. Microorganisms 2021, 9, 2152. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Virgilio, L.; Silva-Lucero, M.D.; Flores-Morelos, D.S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.M.; Zacapala-Gomez, A.E.; Luna-Munoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef]
- Koller, H.; Perkins, L.B. Brewing and the Chemical Composition of Amine-Containing Compounds in Beer: A Review. Foods 2022, 11, 257. [Google Scholar] [CrossRef]
- Reiter, T.; Montpetit, R.; Byer, S.; Frias, I.; Leon, E.; Viano, R.; McLoughlin, M.; Halligan, T.; Hernandez, D.; Figueroa-Balderas, R.; et al. Transcriptomics Provides a Genetic Signature of Vineyard Site and Offers Insight into Vintage-Independent Inoculated Fermentation Outcomes. mSystems 2021, 6, e00033-21. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, Y.; Guo, Y.; Wu, F.; Zhang, Y.; Qi, X.; Wang, Z.; Wang, Q. Stress tolerance enhancement via SPT15 base editing in Saccharomyces cerevisiae. Biotechnol. Biofuels 2021, 14, 155. [Google Scholar] [CrossRef]
- Postaru, M.; Tucaliuc, A.; Cascaval, D.; Galaction, A.I. Cellular Stress Impact on Yeast Activity in Biotechnological Processes—A Short Overview. Microorganisms 2023, 11, 2522. [Google Scholar] [CrossRef] [PubMed]
- Minden, S.; Aniolek, M.; Noorman, H.; Takors, R. Performing in spite of starvation: How Saccharomyces cerevisiae maintains robust growth when facing famine zones in industrial bioreactors. Microb. Biotechnol. 2023, 16, 148–168. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Coppola, F.; Letizia, F.; Testa, B.; Sorrentino, E. Role of Yeasts in the Brewing Process: Tradition and Innovation. Processes 2021, 9, 839. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, S.; Pan, Y.; Li, X.; Liu, G.; Sun, H.; Zhang, R.; Zhang, C. Breeding of high-tolerance yeast by adaptive evolution and high-gravity brewing of mutant. J. Sci. Food Agric. 2024, 104, 686–697. [Google Scholar] [CrossRef]
- Hou, D.; Xu, X.; Wang, J.; Liu, C.; Niu, C.; Zheng, F.; Li, Q. Effect of environmental stresses during fermentation on brewing yeast and exploration on the novel flocculation-associated function of RIM15 gene. Bioresour. Technol. 2023, 379, 129004. [Google Scholar] [CrossRef]
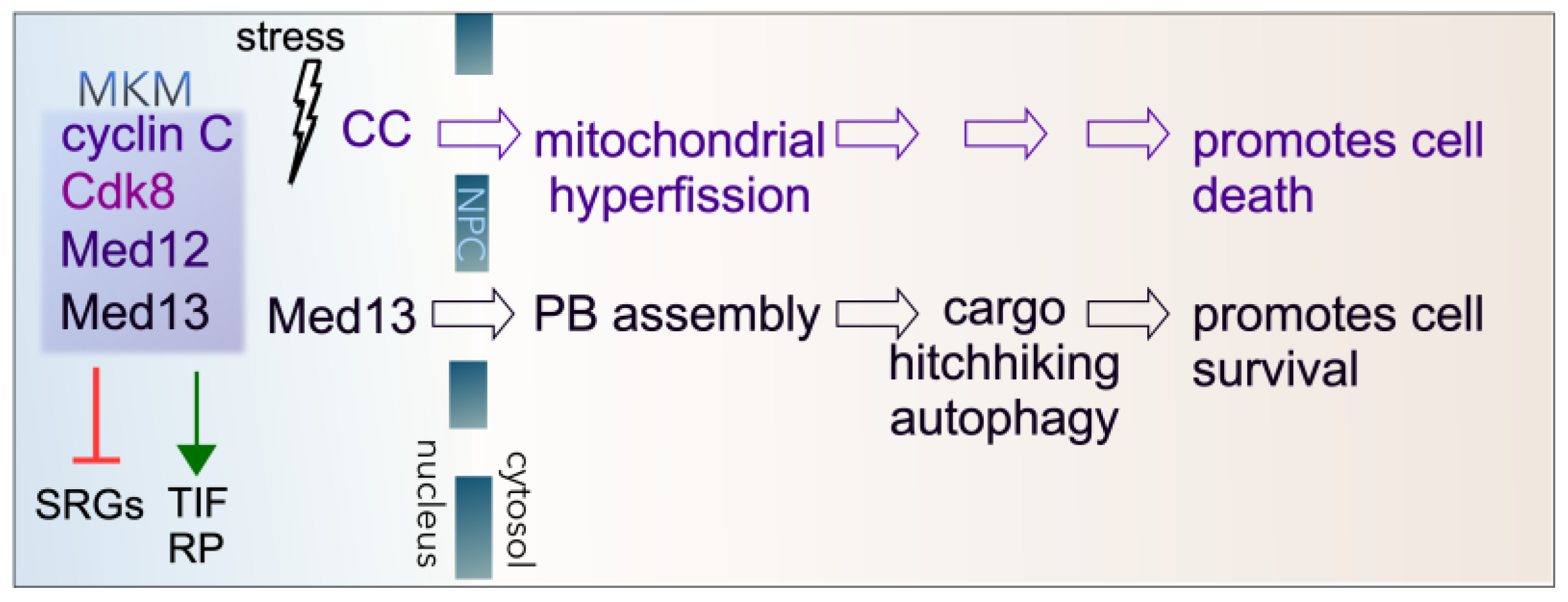

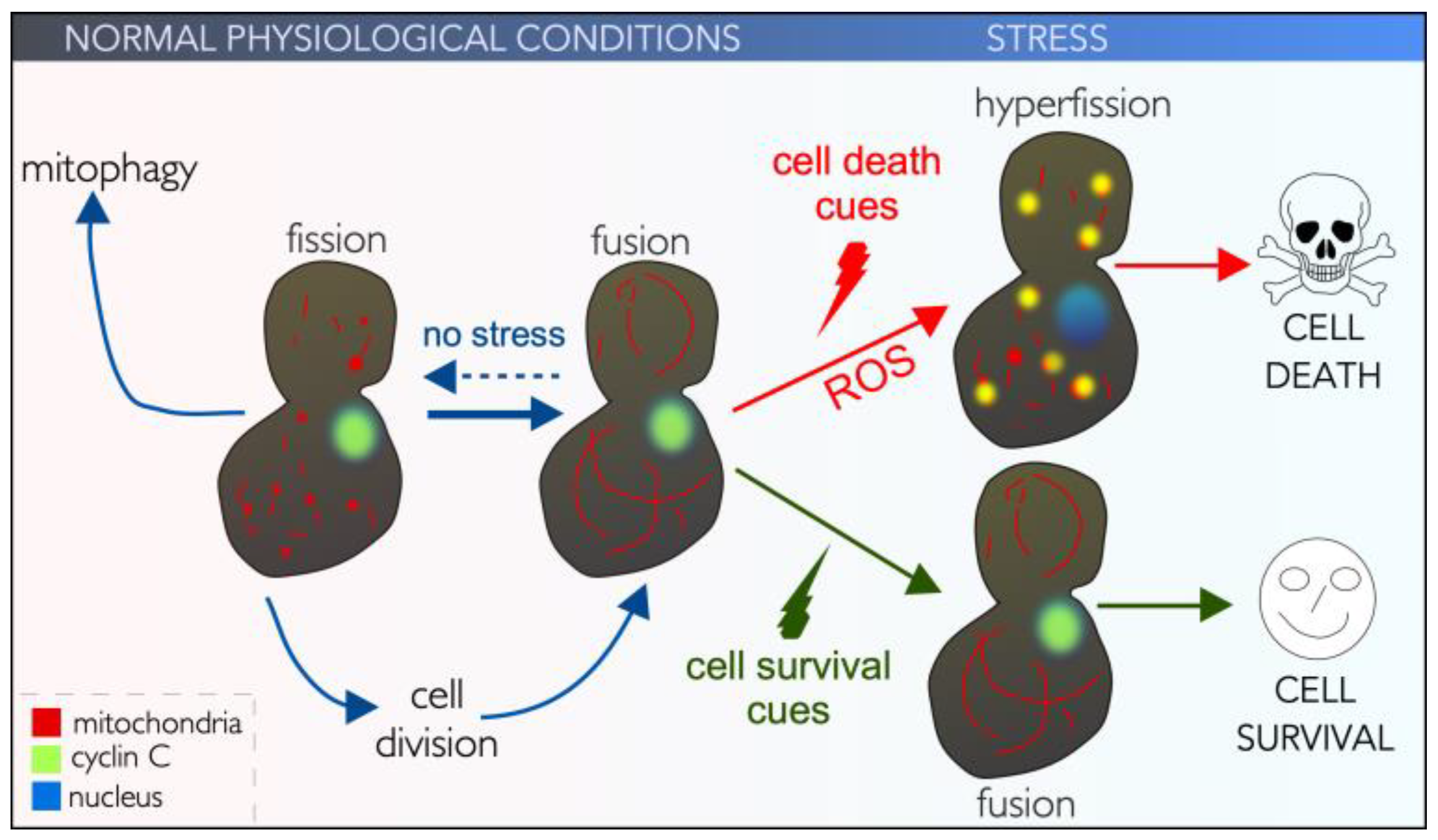
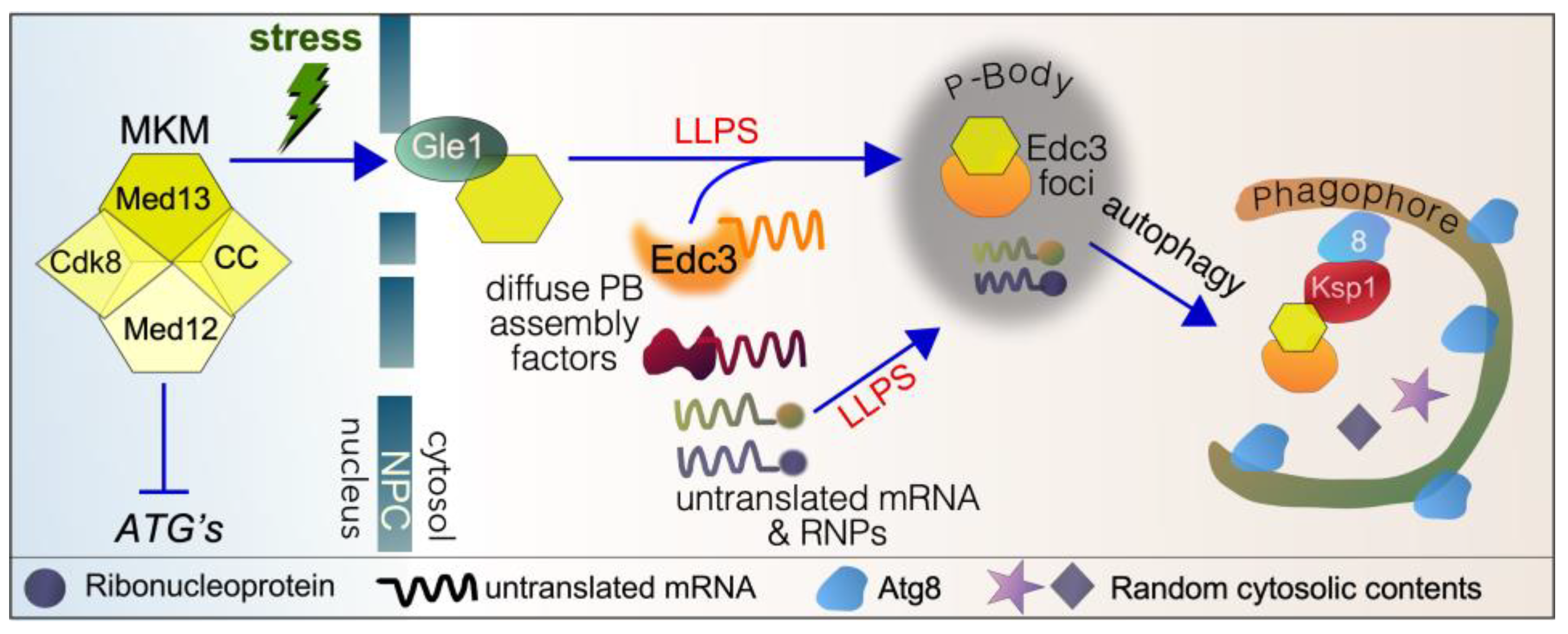
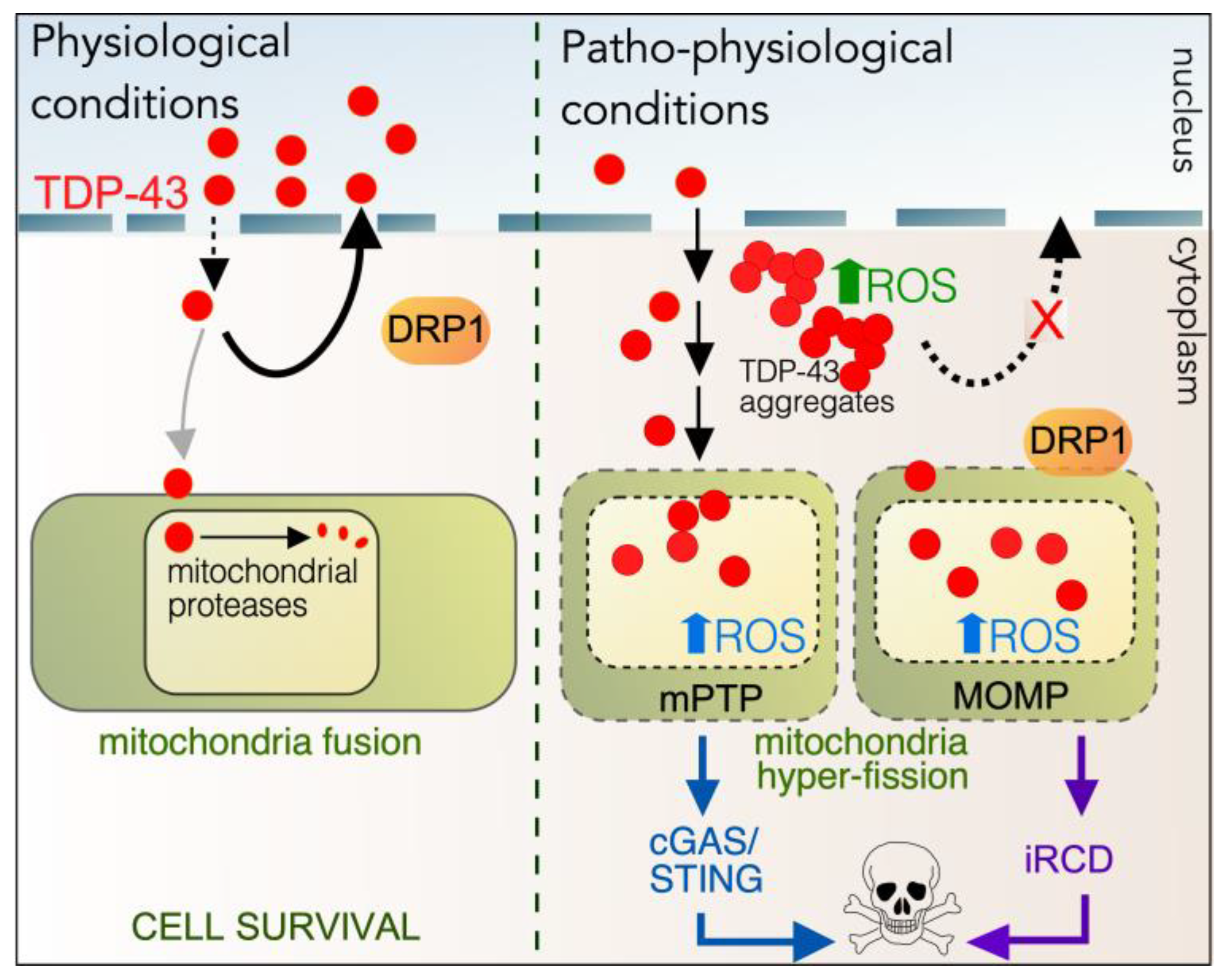
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, J.R.; Robinson, T.L.; Strich, R.; Cooper, K.F. Quitting Your Day Job in Response to Stress: Cell Survival and Cell Death Require Secondary Cytoplasmic Roles of Cyclin C and Med13. Cells 2025, 14, 636. https://doi.org/10.3390/cells14090636
Bauer JR, Robinson TL, Strich R, Cooper KF. Quitting Your Day Job in Response to Stress: Cell Survival and Cell Death Require Secondary Cytoplasmic Roles of Cyclin C and Med13. Cells. 2025; 14(9):636. https://doi.org/10.3390/cells14090636
Chicago/Turabian StyleBauer, Justin R., Tamaraty L. Robinson, Randy Strich, and Katrina F. Cooper. 2025. "Quitting Your Day Job in Response to Stress: Cell Survival and Cell Death Require Secondary Cytoplasmic Roles of Cyclin C and Med13" Cells 14, no. 9: 636. https://doi.org/10.3390/cells14090636
APA StyleBauer, J. R., Robinson, T. L., Strich, R., & Cooper, K. F. (2025). Quitting Your Day Job in Response to Stress: Cell Survival and Cell Death Require Secondary Cytoplasmic Roles of Cyclin C and Med13. Cells, 14(9), 636. https://doi.org/10.3390/cells14090636