
Complex Metabolomic Changes in a Combined Defect of Glycosylation and Oxidative Phosphorylation in a Patient with Pathogenic Variants in PGM1 and NDUFA13

 , , , , ,
, , , , ,  ,
,  and
and
Abstract
1. Introduction
2. Methods
2.1. Ethics
2.2. Clinical Biochemical Investigations
2.3. Genetic Testing
2.4. Cell Culture
2.5. Oxygraphy and Enzymology
2.6. 13C6-Glucose and Galactose Tracer Studies
2.7. Relative Quantification of Metabolites by LC/MS
2.8. Lipidomics
2.9. Statistics
3. Results
3.1. Clinical Presentation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proband | PGM1-CDG | Leigh Syndrome | NDUFA13 Deficiency | |
|---|---|---|---|---|
| Gene(s) affected | PGM1, NDUF13A | PGM1 | >75 genes involved in OXPHOS and OXPHOS assembly, stability and activity. Most commonly associated with complex I deficiency (>1/3 of patients) [9,11,31]. | NDUFA13 |
| Genome | Nuclear | Nuclear | Nuclear, mitochondrial | Nuclear |
| Frequency | First case with combined PGM1 and NDUFA13 deficiency. | Not known. More than 70 patients reported to date [15,32]. | 1:40,000 [9,10], varies between different affected genes. | Not known, 13 patients 11 families reported [6,7,8]. |
| Age of onset | Presented with bifid uvula at birth. In the first six months of life, presented with global developmental delay and dystrophy. | Mostly at birth [16], one adult case reported [20]. | Variable, adult/adolescent onset reported but infrequent [9,31,33]. | 11/13 [8] patients presented within the first year of life. |
| Life expectancy | Died at the age of 16 years. | Oldest reported patient is 53 years [20] old; early death due to cardiac complications has been reported in 10% of the patients [16,21,22]. | Depends on the genetic defect; in general, severely reduced [9,31], many patients die in infancy. | 12/13 [6,7,8] patients still alive at the time of this publication. Average age 7.8 years (±5.4 years). |
| Prenatal/perinatal complications | Not reported. | Frequent [16], mostly associated with dysmorphic facial features resulting in breathing and feeding difficulties. | Infrequent [9] | Not reported. |
| Progression | Yes | Usually, stable. | Yes, fast, death at early age [9]. | 11/13 slow-moderate progression [6,7,8]. |
| Congenital malformations | Bifid uvula | Bifid uvula, PRS and cleft palate [16]. | Infrequent, varies between different affected genes [9]. | Skeletal deformities and congenital glaucoma in one individual [8]. |
| Growth delay | Severe | Frequent | About half of the patients [9]. | 9/13 patients [6,7,8]. |
| Brain findings | Leigh like MRI pattern. Elevated lactate on MRS. | Majority unremarkable [16]. Thin pituitary reported in one patient [34]. | Bilateral symmetrical lesions within the brainstem and basal ganglia (“Leigh like MRI pattern”) cerebellar atrophy, thalamus and spinal cord can also be affected, elevated lactate on MRS [9,31]. | 11/13 [6,7,8], progressive cerebellar atrophy with hypersignal of the cerebellum and dentate nucleus; atrophic optic nerves; elevated lactate on MRS. |
| Other neurological presentation | Dystonic movement disorder, epilepsy and ataxia. | Present in <40% of patients, mostly related to intellectual disability [19], seizures reported in only 3 patients [16,17,18,34]. | Common [14], ataxia, abnormal spasticity and seizures. | 13/13 [6,7,8] patients, seizures, intellectual disability, bradykinesia, spasticity, axial hypotonia and truncal ataxia. |
| Developmental delay | Severe | Often [16] | The majority of the patients [9]. | 13/13 [6,7,8], ranging from mild to severe. |
| Regression | Present | Infrequent [16] | Frequent [9] | Yes |
| Intellectual disability | Severe | Present in <40%, mostly due to complications of hyperinsulinemic hypoglycemia [19]. | Frequent [9] | 8/13 [6,7,8] patients, mild to severe. |
| Eye presentation | Absent | Infrequent, 5 reported patients [16], nystagmus and strabismus. | Varies between different affected genes [9], nystagmus and optic atrophy. | 11/13 patients [8], nystagmus, optic atrophy and congenital glaucoma. |
| Hearing loss | Early bilateral hearing loss. | Infrequent, 1 reported patient [16,26]. | Varies between different affected genes [9]. | One patient with sensorineural loss [6,8]. |
| Cardiovascular | Not reported. | Approximately half of the reported patients present with cardiac abnormalities [15,16,32]. | Uncommon [9], in some patients ventricular septal defects and hypertrophic cardiomyopathy [35]. | Reported in 1 patient, hypertrophic cardiomyopathy [7]. |
| Musculoskeletal | Atrophy, truncal hypotonia. | Mare than 70% of the patients [16], rhabdomyolysis, hypotonia and elevated CK. | Frequent [9], including dystonia, hypotonia. | 10/13 [6,7,8] including hypertonia, hypotonia, truncal hypotonia and decreased muscle tone. |
| Gastrointestinal | Not reported. | GI problems uncommon [16], mostly associated with feeding difficulties due to facial dysmorphism. | Varies between different affected genes [9], dysphagia, feeding difficulties and vomiting. | 1/13 [6] Gastroesophageal reflux. |
| Liver | Mildly elevated liver transaminases. | Common [16], elevated liver enzymes, liver steatosis | Depends on the genetic defect, not frequently affected [9]. | Not reported. |
| Endocrine | Elevated TSH. | Common [16], hypothyroidism, hypogonadotropic hypogonadism, delayed puberty and hyperinsulinemia. | Common [36], depending on the genetic defect. | Not reported. |
| Complex I activity | All complexes including CI were reduced in the muscle. | Not reduced in fibroblasts [18]. | Depends on the genetic defect. | 6/13 patients tested [6,7,8], isolated CI deficiency (muscle or fibroblasts). |
| Muscle biopsy | Muscle biopsy negative for ragged fibers, positive for atrophy of type II fibers. | Not usually performed, increase of internal nuclei or fiber size variation and/or accumulation of fat or glycogen was reported [12,16]. | Frequently performed, muscle biopsy can show ragged fibers [33]. | 1 patient with showed lipid droplets in type I fibers with no ragged-red or cytochrome c oxidase negative fibers [7]. |
| Biochemical laboratory findings | Elevated CSF and plasma lactate. | Common [16], low FSH, LH, IGFB and thyroid hormones; low glucose, coagulation factor abnormalities, elevated liver enzymes and elevated CK. Lactate not commonly elevated. | Elevated CSF and plasma lactate. Elevated amino acids. Elevated urinary organic acids. Abnormal plasma carnitine panel. | Elevated plasma lactate 7 patients, elevated plasma alanine 3 patients and elevated CSF lactate in 2 patients [8]. |
| Serum Transferrin | Mixed CDG type. | Mixed CDG type, or in rare cases, CDG type II [16]. | Not routinely performed. | Not performed. |
3.2. Mitochondrial Functional Assays
3.3. Tracer Metabolomics in Proband’s Fibroblasts
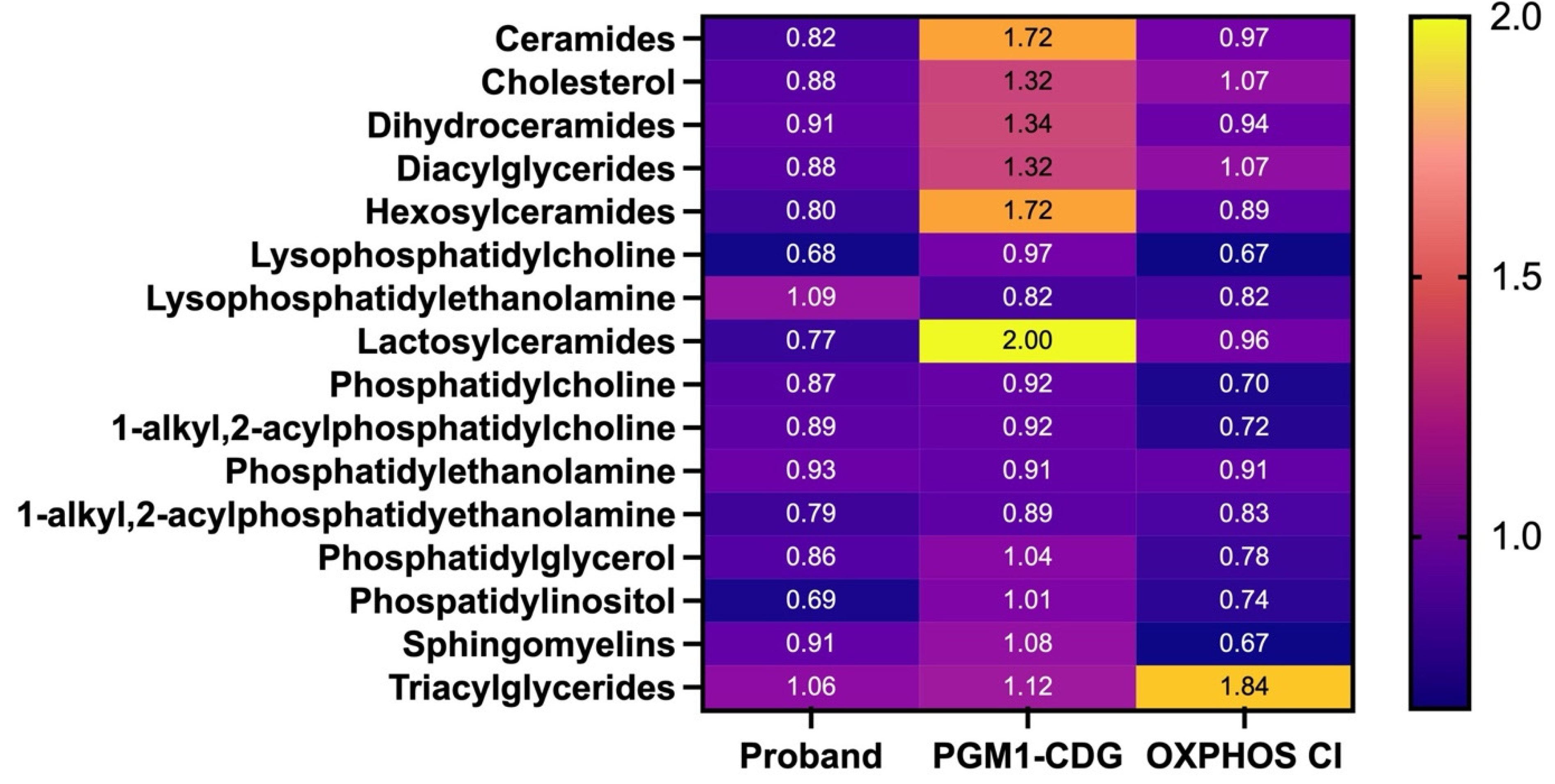
3.4. Lipidomics in Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waters, D.; Adeloye, D.; Woolham, D.; Wastnedge, E.; Patel, S.; Rudan, I. Global birth prevalence and mortality from inborn errors of metabolism: A systematic analysis of the evidence. J. Glob. Health 2018, 8, 021102. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J. An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Filosto, M.; Tsujino, S.; Lamperti, C.; Shanske, S.; Coquet, M.; Desnuelle, C.; DiMauro, S. Muscle Glycogenosis and Mitochondrial Hepatopathy in an Infant With Mutations in Both the Myophosphorylase and Deoxyguanosine Kinase Genes. Arch. Neurol. 2003, 60, 1445. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Rapsomaniki, M.; Disabella, E.; Sestito, S.; Pascale, M.G.; Moricca, M.T.; Bonapace, G.; Arbustini, E.; Strisciuglio, P. Co-existence of Phenylketonuria and Fabry disease on a 3 year-old boy: Case report. BMC Pediatr. 2010, 10, 32. [Google Scholar] [CrossRef]
- Popek, M.; Walter, M.; Fernando, M.; Lindner, M.; Schwab, K.O.; Sass, J.O. Two inborn errors of metabolism in a newborn: Glutaric aciduria type I combined with isobutyrylglycinuria. Clin. Chim. Acta 2010, 411, 2087–2091. [Google Scholar] [CrossRef]
- Angebault, C.; Charif, M.; Guegen, N.; Piro-Megy, C.; Mousson de camaret, B.; Procaccio, V.; Guichet, P.O.; Hebrard, M.; Manes, G.; Leboucq, N.; et al. Mutation in NDUFA13/GRIM19 leads to early onset hypotonia, dyskinesia and sensorial deficiencies, and mitochondrial complex I instability. Hum. Mol. Genet. 2015, 24, 3948–3955. [Google Scholar] [CrossRef]
- González-Quintana, A.; García-Consuegra, I.; Belanger-Quintana, A.; Serrano-Lorenzo, P.; Lucia, A.; Blázquez, A.; Docampo, J.; Ugalde, C.; Morán, M.; Arenas, J.; et al. Novel ndufa13 mutations associated with oxphos deficiency and leigh syndrome: A second family report. Genes 2020, 11, 855. [Google Scholar] [CrossRef]
- Kaiyrzhanov, R.; Thompson, K.; Efthymiou, S.; Mukushev, A.; Zharylkassyn, A.; Prasad, C.; Karimiani, E.G.; Alvi, J.R.; Niyazov, D.; Alahmad, A.; et al. Biallelic NDUFA13 variants lead to a neurodevelopmental phenotype with gradual neurological impairment. Brain Commun. 2025, 7, fcae453. [Google Scholar] [CrossRef]
- Rahman, S. Leigh syndrome. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2023; Volume 194, pp. 43–63. [Google Scholar]
- Schubert Baldo, M.; Vilarinho, L. Molecular basis of Leigh syndrome: A current look. Orphanet J. Rare Dis. 2020, 15, 31. [Google Scholar] [CrossRef]
- Rahman, S. Complex I deficiency remains the most frequent cause of Leigh syndrome spectrum. Brain Commun. 2025, 7, fcae470. [Google Scholar] [CrossRef]
- Tegtmeyer, L.C.; Rust, S.; van Scherpenzeel, M.; Ng, B.G.; Losfeld, M.-E.; Timal, S.; Raymond, K.; He, P.; Ichikawa, M.; Veltman, J.; et al. Multiple Phenotypes in Phosphoglucomutase 1 Deficiency. N. Engl. J. Med. 2014, 370, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, T.; Vissing, J.; Petit, F.; Piraud, M.; Orngreen, M.C.; Andersen, G.; Claeys, K.G.; Wary, C.; Hogrel, J.-Y.; Laforêt, P. Muscle Glycogenosis Due to Phosphoglucomutase 1 Deficiency. N. Engl. J. Med. 2009, 361, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Sugie, H.; Kobayashi, J.; Sugie, Y.; Ichimura, M.; Miyamoto, R.; Ito, T.; Shimizu, K.; Igarashi, Y. Infantile muscle glycogen storage disease. Neurology 1988, 38, 602. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, S.; Johnsen, C.; Schulze, A.; Lail, G.; Guilder, L.; Schwartz, K.; Schultz, M.; Mercimek-Andrews, S.; Boyer, S.; Morava, E. Novel insights into the phenotype and long-term D-gal treatment in PGM1-CDG: A case series. Ther. Adv. Rare Dis. 2023, 4, 26330040221150269. [Google Scholar] [CrossRef]
- Altassan, R.; Radenkovic, S.; Edmondson, A.C.; Barone, R.; Brasil, S.; Cechova, A.; Coman, D.; Donoghue, S.; Falkenstein, K.; Ferreira, V.; et al. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management. J. Inherit. Metab. Dis. 2021, 44, 148–163. [Google Scholar] [CrossRef]
- Wong, S.Y.-W.; Beamer, L.J.; Gadomski, T.; Honzik, T.; Mohamed, M.; Wortmann, S.B.; Brocke Holmefjord, K.S.; Mork, M.; Bowling, F.; Sykut-Cegielska, J.; et al. Defining the Phenotype and Assessing Severity in Phosphoglucomutase-1 Deficiency. J. Pediatr. 2016, 175, 130–136.e8. [Google Scholar] [CrossRef]
- Radenkovic, S.; Bird, M.J.; Emmerzaal, T.L.; Wong, S.Y.; Felgueira, C.; Stiers, K.M.; Sabbagh, L.; Himmelreich, N.; Poschet, G.; Windmolders, P.; et al. The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am. J. Hum. Genet. 2019, 104, 835–846. [Google Scholar] [CrossRef]
- Radenkovic, S.; Witters, P.; Morava, E. Central nervous involvement is common in PGM1-CDG. Mol. Genet. Metab. 2018, 125, 200–204. [Google Scholar] [CrossRef]
- Voermans, N.C.; Preisler, N.; Madsen, K.L.; Janssen, M.C.H.; Kusters, B.; Abu Bakar, N.; Conte, F.; Lamberti, V.M.L.; Nusman, F.; van Engelen, B.G.; et al. PGM1 deficiency: Substrate use during exercise and effect of treatment with galactose. Neuromuscul. Disord. 2017, 27, 370–376. [Google Scholar] [CrossRef]
- Zemet, R.; Hope, K.D.; Edmondson, A.C.; Shah, R.; Patino, M.; Yesso, A.M.; Berger, J.H.; Sarafoglou, K.; Larson, A.; Lam, C.; et al. Cardiomyopathy, an uncommon phenotype of congenital disorders of glycosylation: Recommendations for baseline screening and follow-up evaluation. Mol. Genet. Metab. 2024, 142, 108513. [Google Scholar] [CrossRef]
- Fernlund, E.; Andersson, O.; Ellegård, R.; Årstrand, H.K.; Green, H.; Olsson, H.; Gunnarsson, C. The congenital disorder of glycosylation in PGM1 (PGM1-CDG) can cause severe cardiomyopathy and unexpected sudden cardiac death in childhood. Forensic Sci. Int. Genet. 2019, 43, 102111. [Google Scholar] [CrossRef] [PubMed]
- Lefeber, D.J.; Morava, E.; Jaeken, J. How to find and diagnose a CDG due to defective N-glycosylation. J. Inherit. Metab. Dis. 2011, 34, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, S.; Bleukx, S.; Engelhardt, N.; Eklund, E.; Mercimek-Andrews, S.; Edmondson, A.C.; Morava, E. Coagulation abnormalities and vascular complications are common in PGM1-CDG. Mol. Genet. Metab. 2024, 142, 108530. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.W.; Gadomski, T.; Van Scherpenzeel, M.; Honzik, T.; Hansikova, H.; Holmefjord, K.S.B.; Mork, M.; Bowling, F.; Sykut-Cegielska, J.; Koch, D.; et al. Oral D-galactose supplementation in PGM1-CDG. Genet. Med. 2017, 19, 1226–1235. [Google Scholar] [CrossRef]
- Nolting, K.; Park, J.H.; Tegtmeyer, L.C.; Zühlsdorf, A.; Grüneberg, M.; Rust, S.; Reunert, J.; Du Chesne, I.; Debus, V.; Schulze-Bahr, E.; et al. Limitations of galactose therapy in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. Rep. 2017, 13, 33–40. [Google Scholar] [CrossRef]
- Bird, M.J.; Adant, I.; Windmolders, P.; Elst, I.V.; Felgueira, C.; Altassan, R.; Gruenert, S.C.; Ghesquière, B.; Witters, P.; Cassiman, D.; et al. Oxygraphy versus enzymology for the biochemical diagnosis of primary mitochondrial disease. Metabolites 2019, 9, 220. [Google Scholar] [CrossRef]
- Adant, I.; Bird, M.; Decru, B.; Windmolders, P.; Wallays, M.; de Witte, P.; Rymen, D.; Witters, P.; Vermeersch, P.; Cassiman, D.; et al. Pyruvate and uridine rescue the metabolic profile of OXPHOS dysfunction. Mol. Metab. 2022, 63, 101537. [Google Scholar] [CrossRef]
- Centenera, M.M.; Scott, J.S.; Machiels, J.; Nassar, Z.D.; Miller, D.C.; Zinonos, I.; Dehairs, J.; Burvenich, I.J.G.; Zadra, G.; Chetta, P.M.; et al. ELOVL5 is a critical and targetable fatty acid elongase in prostate cancer. Cancer Res. 2021, 81, 1704–1718. [Google Scholar] [CrossRef]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van Den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Radenkovic, S.; Laerdahl, J.K.; Backe, P.H.; Morava, E. The role of PGM1 isoform 2 in PGM1-CDG: One step closer to genotype–phenotype correlation? J. Inherit. Metab. Dis. 2023, 46, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, N.; Chang, G.; Li, J.; Yao, R.; Shen, Y.; Wang, J.; Huang, X.; Wang, X. Clinical and molecular genetic characterization of two patients with mutations in the phosphoglucomutase 1 (PGM1) gene. J. Pediatr. Endocrinol. Metab. 2018, 31, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, J.C.; Haas, J.E.; Monnat, R.; Milstein, J.M. Hypertrophic cardiomyopathy is a component of subacute necrotizing encephalomyelopathy. J. Pediatr. 1982, 101, 706–710. [Google Scholar] [CrossRef]
- Varughese, R.; Rahman, S. Endocrine Dysfunction in Primary Mitochondrial Diseases. Endocr. Rev. 2025, bnaf002. [Google Scholar] [CrossRef]
- Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V.; et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab. 2018, 27, 1007–1025.e5. [Google Scholar] [CrossRef]
- Wu, Z.; Bezwada, D.; Cai, F.; Harris, R.C.; Ko, B.; Sondhi, V.; Pan, C.; Vu, H.S.; Nguyen, P.T.; Faubert, B.; et al. Electron transport chain inhibition increases cellular dependence on purine transport and salvage. Cell Metab. 2024, 36, 1504–1520.e9. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Radenkovic, S.; Fitzpatrick-Schmidt, T.; Byeon, S.K.; Madugundu, A.K.; Saraswat, M.; Lichty, A.; Wong, S.Y.W.; McGee, S.; Kubiak, K.; Ligezka, A.; et al. Expanding the clinical and metabolic phenotype of DPM2 deficient congenital disorders of glycosylation. Mol. Genet. Metab. 2021, 132, 27–37. [Google Scholar] [CrossRef]
- Ren, C.; Liu, J.; Zhou, J.; Liang, H.; Zhu, Y.; Wang, Q.; Leng, Y.; Zhang, Z.; Yuan, Y.; Wang, Z.; et al. Lipidomic profiling of plasma samples from patients with mitochondrial disease. Biochem. Biophys. Res. Commun. 2018, 500, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Imran, A.; Qasim, M.; Zafar, S.; Kamran, S.K.S.; Razzaq, A.; Aziz, N.; et al. Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis. 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radenkovic, S.; Adant, I.; Bird, M.J.; Swinnen, J.V.; Cassiman, D.; Kozicz, T.; Gruenert, S.C.; Ghesquière, B.; Morava, E. Complex Metabolomic Changes in a Combined Defect of Glycosylation and Oxidative Phosphorylation in a Patient with Pathogenic Variants in PGM1 and NDUFA13. Cells 2025, 14, 638. https://doi.org/10.3390/cells14090638
Radenkovic S, Adant I, Bird MJ, Swinnen JV, Cassiman D, Kozicz T, Gruenert SC, Ghesquière B, Morava E. Complex Metabolomic Changes in a Combined Defect of Glycosylation and Oxidative Phosphorylation in a Patient with Pathogenic Variants in PGM1 and NDUFA13. Cells. 2025; 14(9):638. https://doi.org/10.3390/cells14090638
Chicago/Turabian StyleRadenkovic, Silvia, Isabelle Adant, Matthew J. Bird, Johannes V. Swinnen, David Cassiman, Tamas Kozicz, Sarah C. Gruenert, Bart Ghesquière, and Eva Morava. 2025. "Complex Metabolomic Changes in a Combined Defect of Glycosylation and Oxidative Phosphorylation in a Patient with Pathogenic Variants in PGM1 and NDUFA13" Cells 14, no. 9: 638. https://doi.org/10.3390/cells14090638
APA StyleRadenkovic, S., Adant, I., Bird, M. J., Swinnen, J. V., Cassiman, D., Kozicz, T., Gruenert, S. C., Ghesquière, B., & Morava, E. (2025). Complex Metabolomic Changes in a Combined Defect of Glycosylation and Oxidative Phosphorylation in a Patient with Pathogenic Variants in PGM1 and NDUFA13. Cells, 14(9), 638. https://doi.org/10.3390/cells14090638