
Cytokine Profile Analysis During Sialodacryoadenitis Virus and Mouse Hepatitis Virus JHM Strain Infection in Primary Mixed Microglia and Astrocyte Culture—Preliminary Research

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Mixed Microglia and Astrocyte Culture
2.2. Viruses
2.3. Immune Response Profiling
2.4. ELISA
2.5. Confocal Microscopy Analysis of SDAV and MHV-JHM Infection
2.6. ROS Detection
2.7. Flow Cytometry
2.8. Statistical Analysis
3. Results
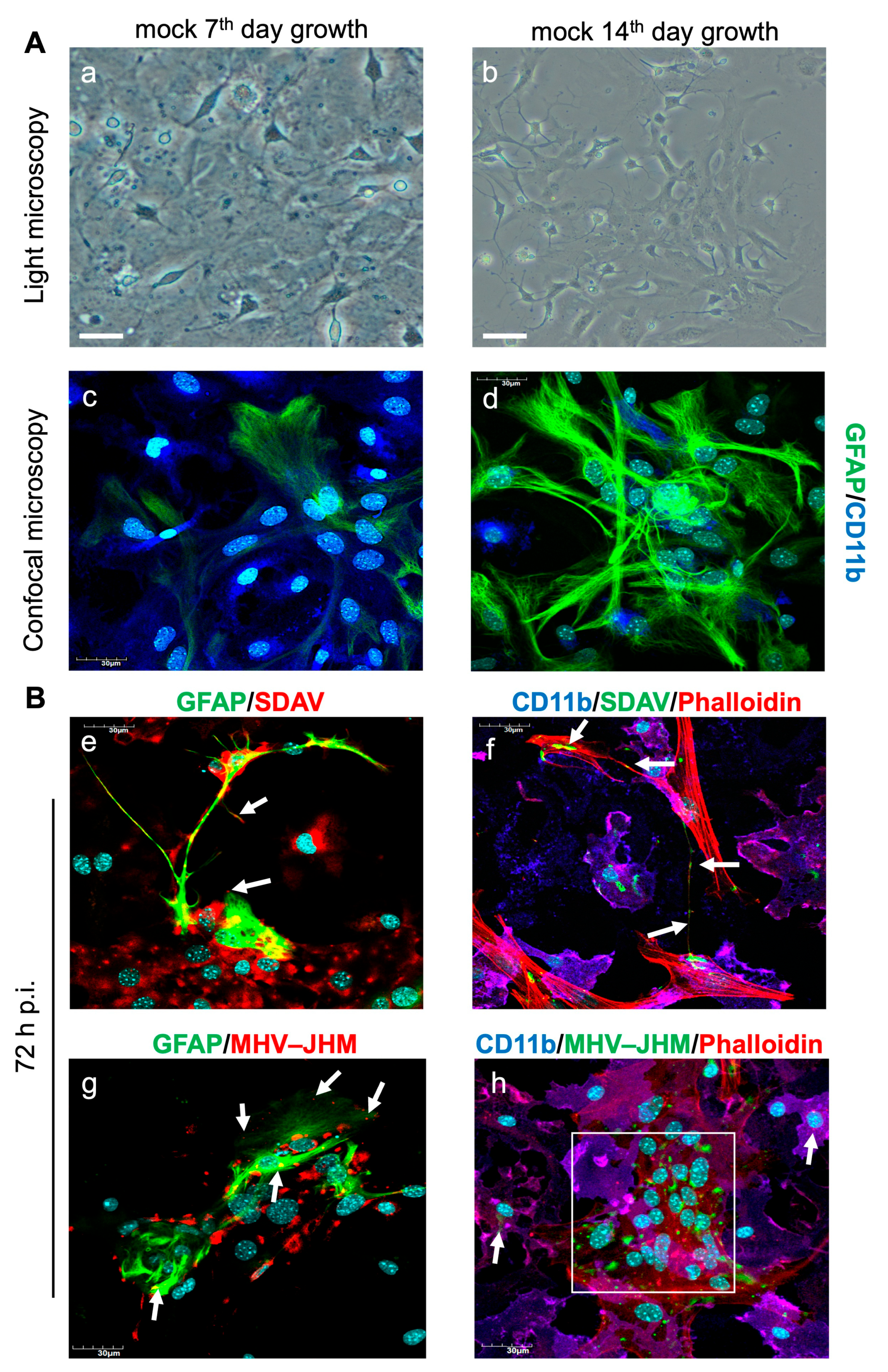
3.1. SDAV Infection of Primary Mixed Microglia and Astrocyte Cells
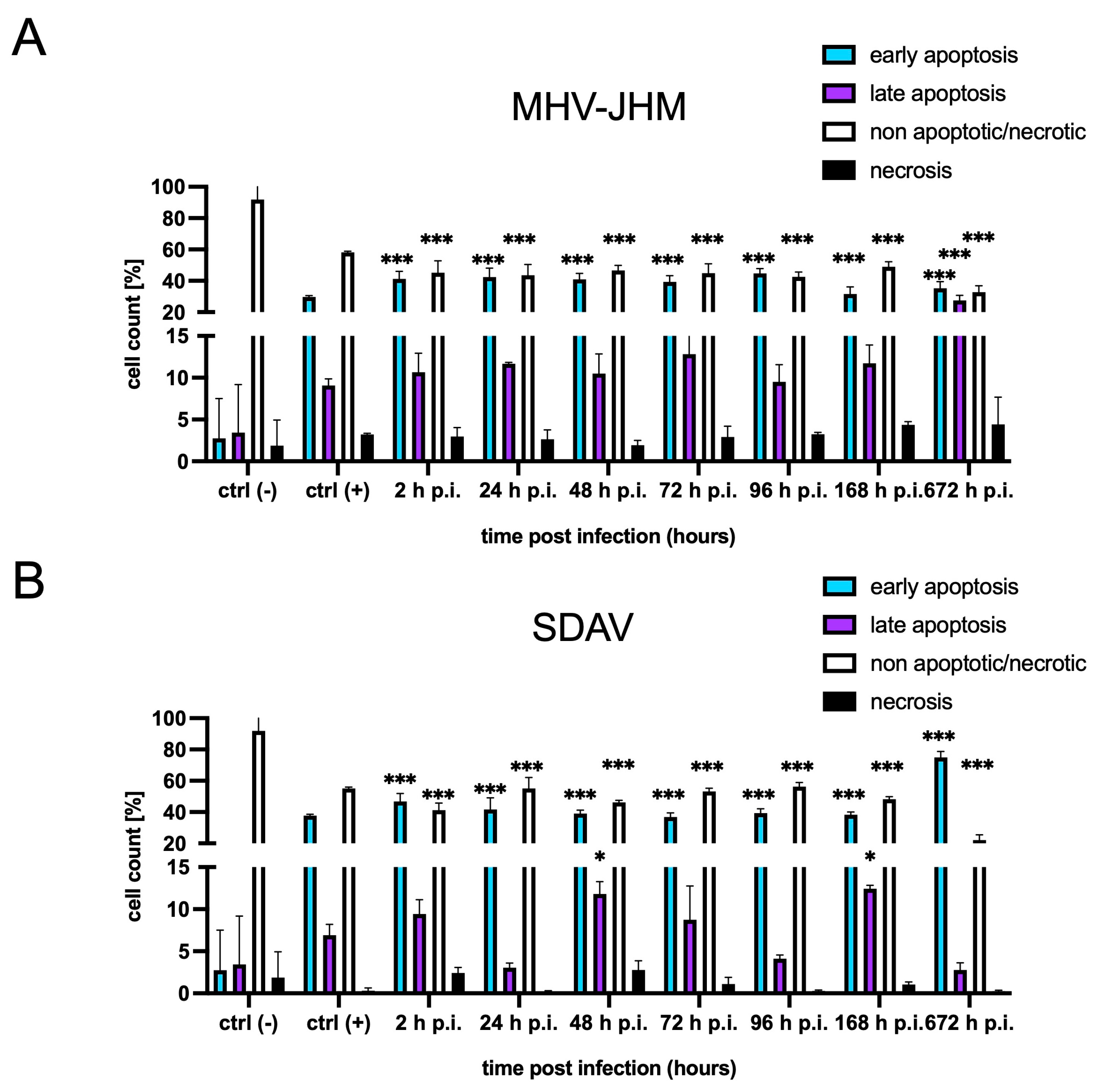
3.2. FACS Analysis of Cell Death
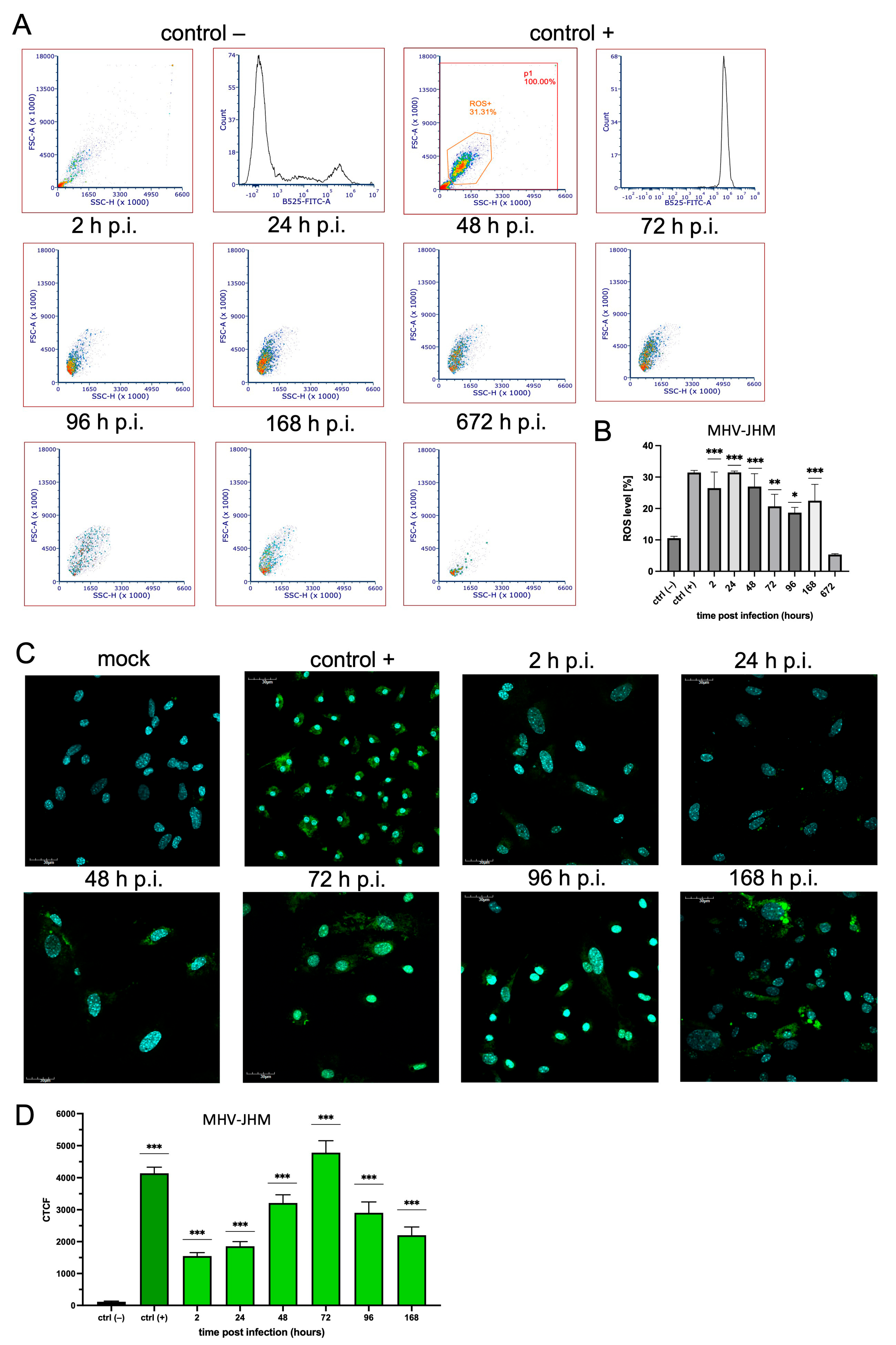
3.3. ROS Expression
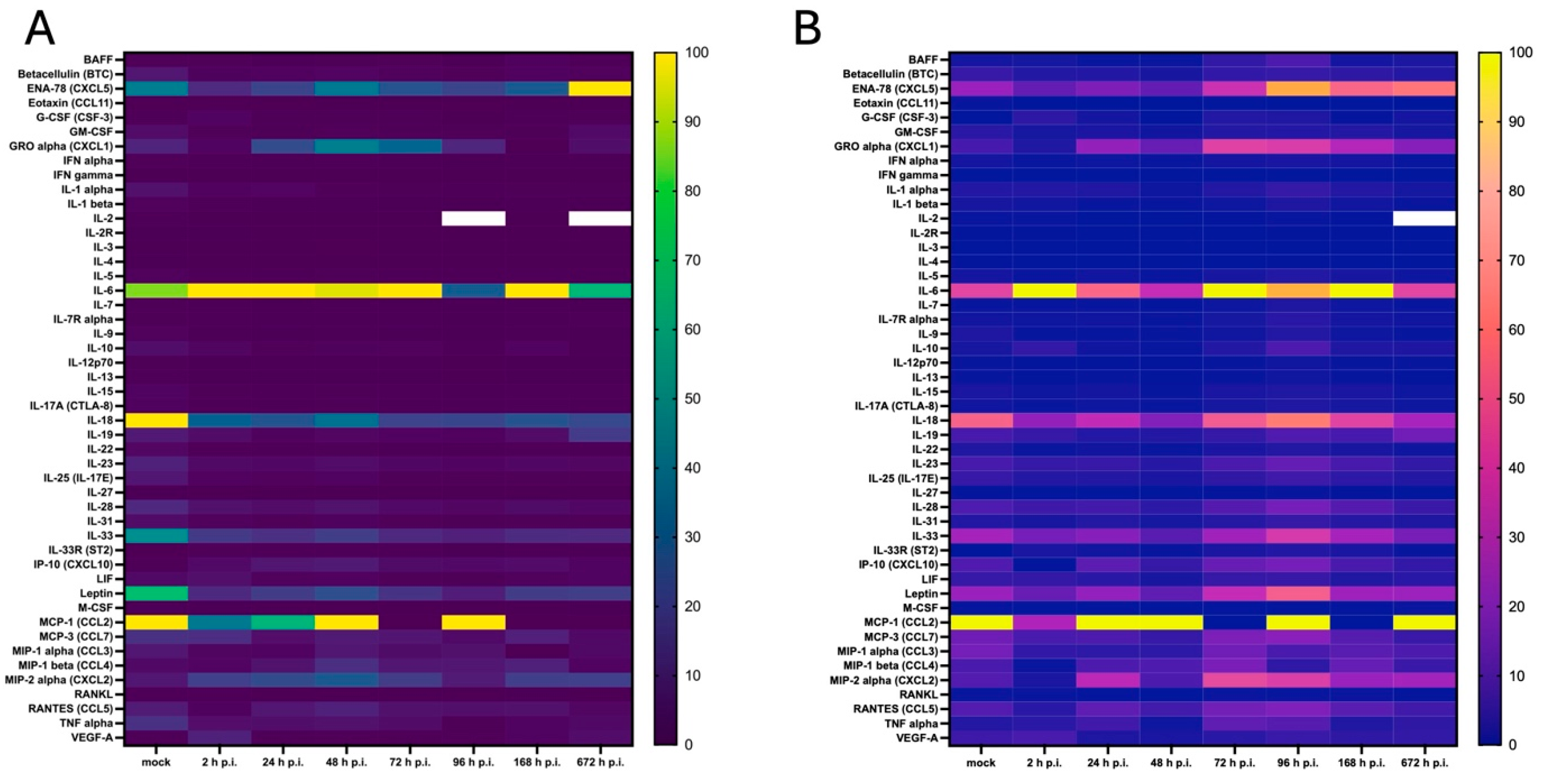
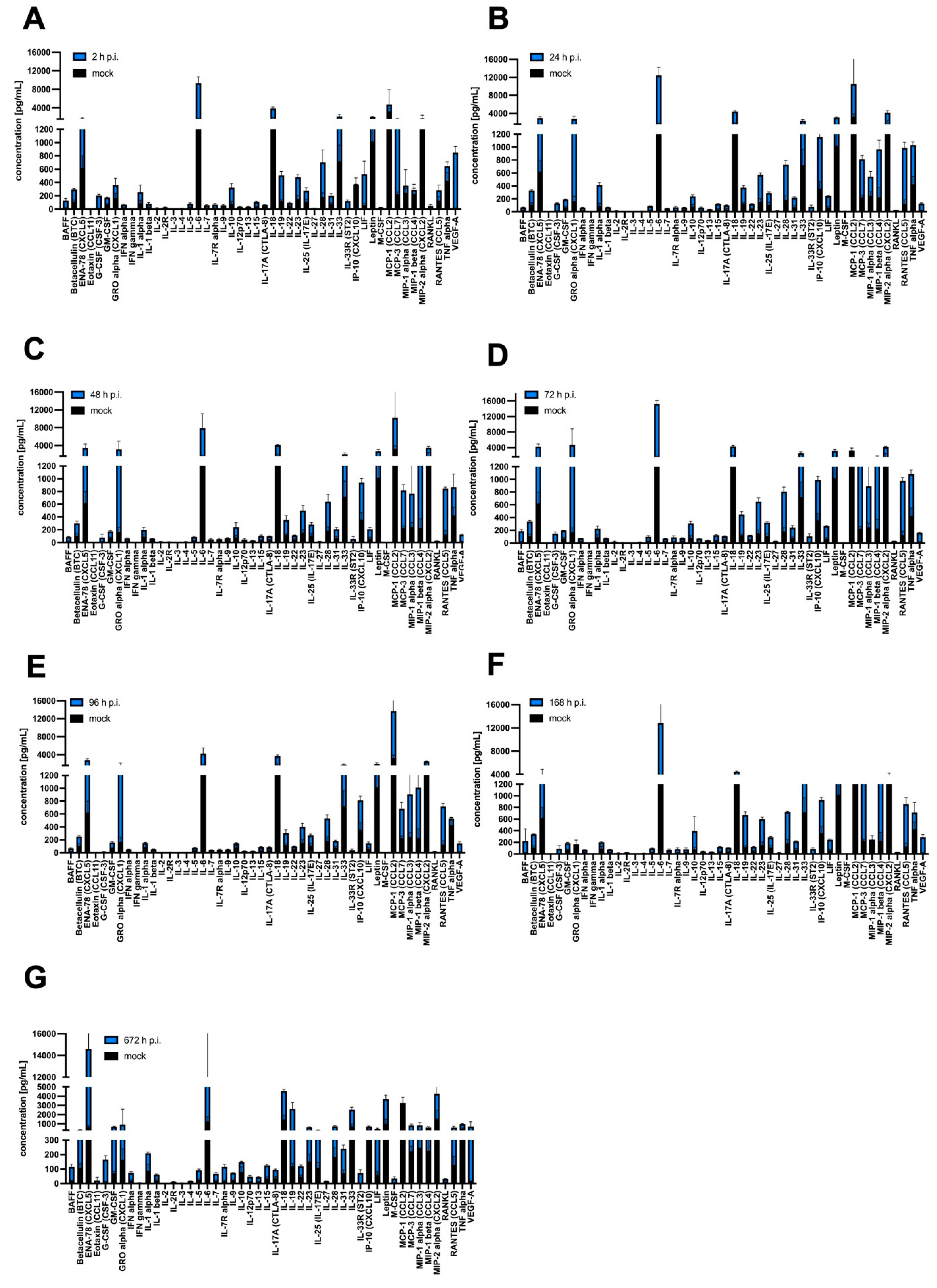
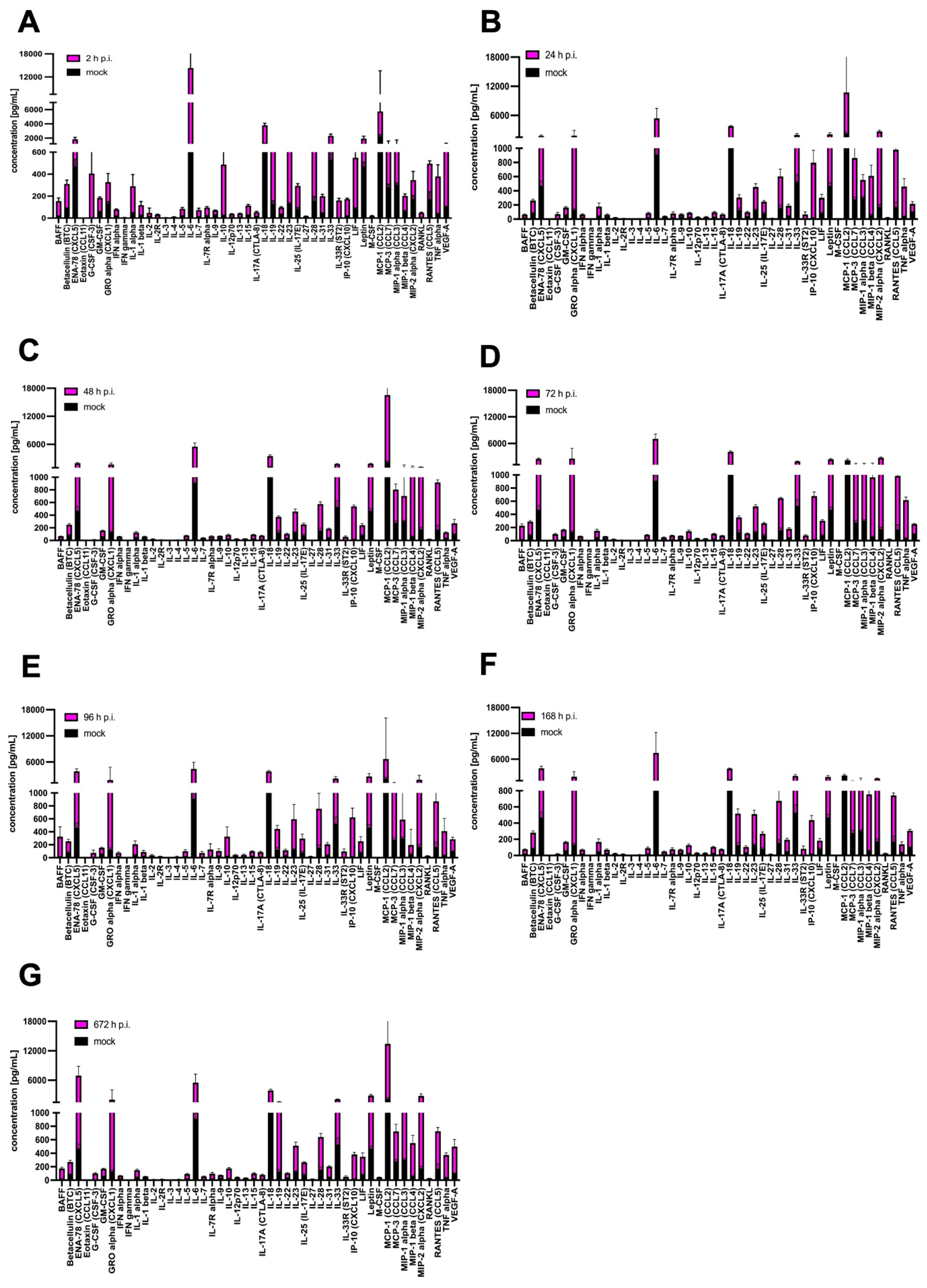
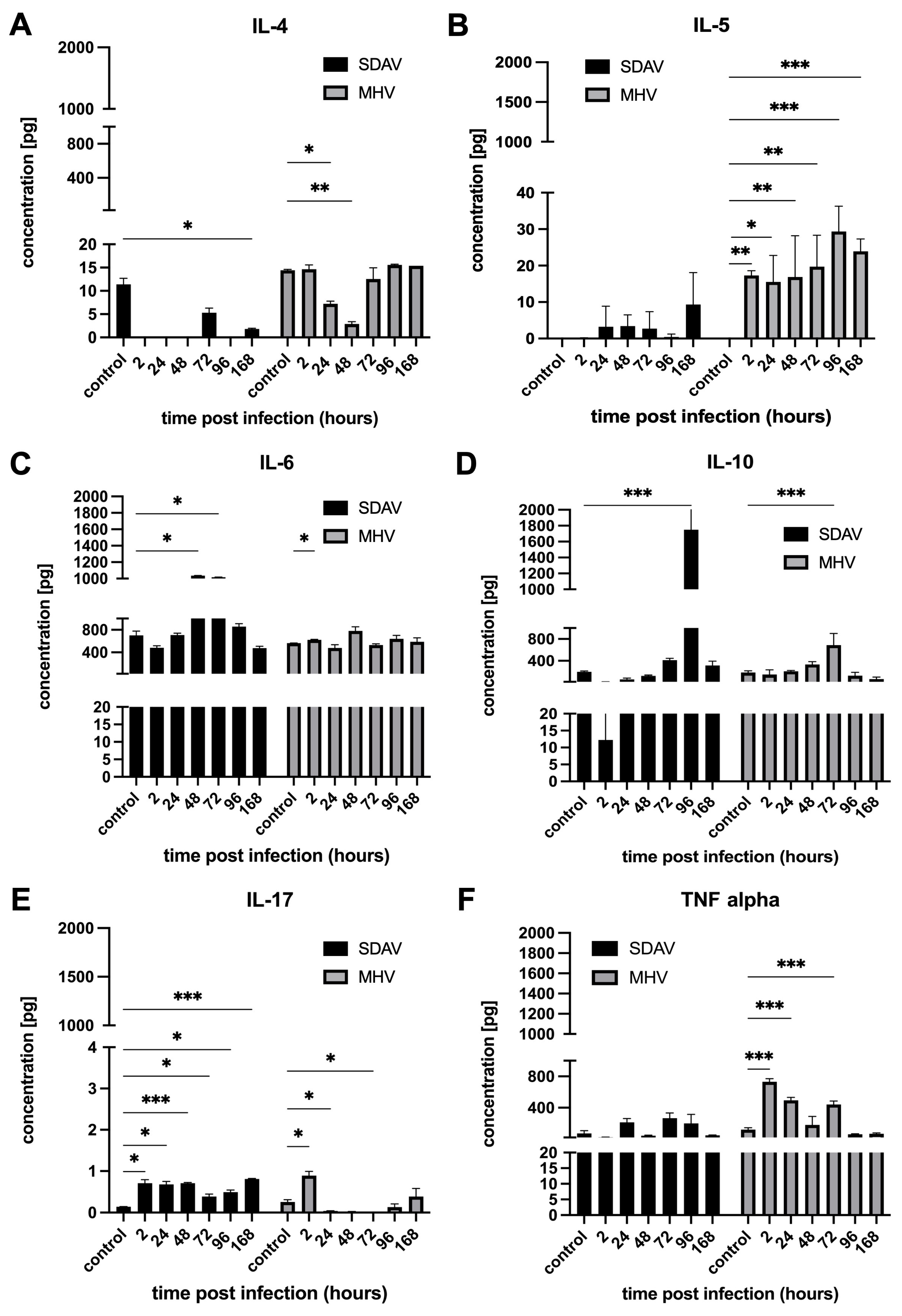
3.4. Cytokine and Chemokine Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major Findings, Mechanisms and Recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Krahel, W.D.; Bartak, M.; Cymerys, J. Acute and Long-Term SARS-CoV-2 Infection and Neurodegeneration Processes—Circulus Vitiosus. Acta Virol. 2024, 68, 12765. [Google Scholar] [CrossRef]
- Mielcarska, M.B.; Rouse, B.T. Viruses and the Brain—A Relationship Prone to Trouble. Viruses 2025, 17, 203. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-Term Effects of COVID-19: A Systematic Review and Meta-Analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef]
- Hu, C.; Chen, C.; Dong, X.-P. Impact of COVID-19 Pandemic on Patients With Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 664965. [Google Scholar] [CrossRef]
- Que, Y.; Hu, C.; Wan, K.; Hu, P.; Wang, R.; Luo, J.; Li, T.; Ping, R.; Hu, Q.; Sun, Y.; et al. Cytokine Release Syndrome in COVID-19: A Major Mechanism of Morbidity and Mortality. Int. Rev. Immunol. 2022, 41, 217–230. [Google Scholar] [CrossRef]
- De Marchi, F.; Gallo, C.; Sarnelli, M.F.; De Marchi, I.; Saraceno, M.; Cantello, R.; Mazzini, L. Accelerated Early Progression of Amyotrophic Lateral Sclerosis over the COVID-19 Pandemic. Brain Sci. 2021, 11, 1291. [Google Scholar] [CrossRef]
- Musson, L.S.; Collins, A.; Opie-Martin, S.; Bredin, A.; Hobson, E.V.; Barkhouse, E.; Coulson, M.C.; Stavroulakis, T.; Gould, R.L.; Al-Chalabi, A.; et al. Impact of the Covid-19 Pandemic on Amyotrophic Lateral Sclerosis Care in the UK. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 91–99. [Google Scholar] [CrossRef]
- Sfera, A.; Rahman, L.; Zapata-Martín del Campo, C.M.; Kozlakidis, Z. Long COVID as a Tauopathy: Of “Brain Fog” and “Fusogen Storms”. Int. J. Mol. Sci. 2023, 24, 12648. [Google Scholar] [CrossRef] [PubMed]
- Jorgačevski, J.; Potokar, M. Immune Functions of Astrocytes in Viral Neuroinfections. Int. J. Mol. Sci. 2023, 24, 3514. [Google Scholar] [CrossRef]
- Liu, L.; Liu, J.; Bao, J.; Bai, Q.; Wang, G. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Sun, M.; You, H.; Hu, X.; Luo, Y.; Zhang, Z.; Song, Y.; An, J.; Lu, H. Microglia–Astrocyte Interaction in Neural Development and Neural Pathogenesis. Cells 2023, 12, 1942. [Google Scholar] [CrossRef]
- Low, R.N.; Low, R.J.; Akrami, A. A Review of Cytokine-Based Pathophysiology of Long COVID Symptoms. Front. Med. 2023, 10, 1011936. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; Von Bergwelt-Baildon, M.S. Cytokine Release Syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Funk, K.E.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation During RNA Viral Infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef]
- Galán, M.; Vigón, L.; Fuertes, D.; Murciano-Antón, M.A.; Casado-Fernández, G.; Domínguez-Mateos, S.; Mateos, E.; Ramos-Martín, F.; Planelles, V.; Torres, M.; et al. Persistent Overactive Cytotoxic Immune Response in a Spanish Cohort of Individuals With Long-COVID: Identification of Diagnostic Biomarkers. Front. Immunol. 2022, 13, 848886. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation Pathways: A General Review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Ismail, F.S.; Faustmann, T.J.; Faustmann, P.M.; Corvace, F. Microglia as Potential Key Regulators in Viral-Induced Neuroinflammation. Front. Cell. Neurosci. 2024, 18, 1426079. [Google Scholar] [CrossRef]
- Xu, P.; Yu, Y.; Wu, P. Role of Microglia in Brain Development after Viral Infection. Front. Cell Dev. Biol. 2024, 12, 1340308. [Google Scholar] [CrossRef]
- Chhatbar, C.; Prinz, M. The Roles of Microglia in Viral Encephalitis: From Sensome to Therapeutic Targeting. Cell. Mol. Immunol. 2021, 18, 250–258. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Muñoz-Carrillo, J.L.; Contreras, J.; Gutierrez-Coronado, O.; Villalobos-Gutiérrez, P.; Ramos Gracia, L.G.; Hernández-Reyes, V. Cytokine Profiling Plays a Crucial Role in Activating Immune System to Clear Infectious Pathogens; IntechOpen: London, UK, 2018; ISBN 978-1-78985-151-9. [Google Scholar]
- Bixler, S.L.; Goff, A.J. The Role of Cytokines and Chemokines in Filovirus Infection. Viruses 2015, 7, 5489–5507. [Google Scholar] [CrossRef]
- Düsedau, H.P.; Steffen, J.; Figueiredo, C.A.; Boehme, J.D.; Schultz, K.; Erck, C.; Korte, M.; Faber-Zuschratter, H.; Smalla, K.-H.; Dieterich, D.; et al. Influenza A Virus (H1N1) Infection Induces Microglial Activation and Temporal Dysbalance in Glutamatergic Synaptic Transmission. mBio 2021, 12, e01776-21. [Google Scholar] [CrossRef]
- Hosseini, S.; Wilk, E.; Michaelsen-Preusse, K.; Gerhauser, I.; Baumgärtner, W.; Geffers, R.; Schughart, K.; Korte, M. Long-Term Neuroinflammation Induced by Influenza A Virus Infection and the Impact on Hippocampal Neuron Morphology and Function. J. Neurosci. 2018, 38, 3060–3080. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, Z. Immune Response in Influenza Virus Infection and Modulation of Immune Injury by Viral Neuraminidase. Virol. J. 2023, 20, 193. [Google Scholar] [CrossRef]
- Radkowski, M.; Grabarczyk, P.; Kryczka, T.; Caraballo Cortès, K.; Kubicka-Russel, D.; Janiak, M.; Osuch, S.; Perlejewski, K.; Laskus, T. Cytokine Profile and Viral Diversity in the Early Seronegative Stage of Community-Acquired Hepatitis C Virus (HCV) Infection. Sci. Rep. 2023, 13, 20045. [Google Scholar] [CrossRef]
- Casola, A.; Estes, M.K.; Crawford, S.E.; Ogra, P.L.; Ernst, P.B.; Garofalo, R.P.; Crowe, S.E. Rotavirus Infection of Cultured Intestinal Epithelial Cells Induces Secretion of CXC and CC Chemokines. Gastroenterology 1998, 114, 947–955. [Google Scholar] [CrossRef]
- Holloway, G.; Coulson, B.S. Innate Cellular Responses to Rotavirus Infection. J. Gen. Virol. 2013, 94, 1151–1160. [Google Scholar] [CrossRef]
- Malamba-Banda, C.; Mhango, C.; Benedicto-Matambo, P.; Mandolo, J.J.; Chinyama, E.; Kumwenda, O.; Barnes, K.G.; Cunliffe, N.A.; Iturriza-Gomara, M.; Jambo, K.C.; et al. Acute Rotavirus Infection Is Associated with the Induction of Circulating Memory CD4+ T Cell Subsets. Sci. Rep. 2023, 13, 9001. [Google Scholar] [CrossRef]
- Lavi, E.; Cong, L. Type I Astrocytes and Microglia Induce a Cytokine Response in an Encephalitic Murine Coronavirus Infection. Exp. Mol. Pathol. 2020, 115, 104474. [Google Scholar] [CrossRef]
- Rajayer, S.R.; Smith, S.M. Neurovirulent Cytokines Increase Neuronal Excitability in a Model of Coronavirus-Induced Neuroinflammation. Intensive Care Med. Exp. 2023, 11, 71. [Google Scholar] [CrossRef]
- Li, Y.; Fu, L.; Gonzales, D.M.; Lavi, E. Coronavirus Neurovirulence Correlates with the Ability of the Virus to Induce Proinflammatory Cytokine Signals from Astrocytes and Microglia. J. Virol. 2004, 78, 3398–3406. [Google Scholar] [CrossRef]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Corrigendum: Oligodendrocyte, Astrocyte and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 79. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local Distribution of Microglia in the Normal Adult Human Central Nervous System Differs by up to One Order of Magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Colton, C.A. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Patrycy, M.; Chodkowski, M.; Krzyzowska, M. Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration. Pathogens 2022, 11, 809. [Google Scholar] [CrossRef]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment Targets for M2 Microglia Polarization in Ischemic Stroke. Biomed. Pharmacother. 2018, 105, 518–525. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Macht, V.A. Neuro-Immune Interactions across Development: A Look at Glutamate in the Prefrontal Cortex. Neurosci. Biobehav. Rev. 2016, 71, 267–280. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Passaro, A.P.; Lebos, A.L.; Yao, Y.; Stice, S.L. Immune Response in Neurological Pathology: Emerging Role of Central and Peripheral Immune Crosstalk. Front. Immunol. 2021, 12, 676621. [Google Scholar] [CrossRef]
- Bartak, M.; Krahel, W.D.; Chodkowski, M.; Grel, H.; Walczak, J.; Pallepati, A.; Komorowski, M.; Cymerys, J. ATPase Valosin-Containing Protein (VCP) Is Involved During the Replication and Egress of Sialodacryoadenitis Virus (SDAV) in Neurons. Int. J. Mol. Sci. 2024, 25, 11633. [Google Scholar] [CrossRef]
- Bender, S.J.; Weiss, S.R. Pathogenesis of Murine Coronavirus in the Central Nervous System. J. Neuroimmune Pharmacol. 2010, 5, 336–354. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Lane, T.E.; Stohlman, S.A. Coronavirus Infection of the Central Nervous System: Host-Virus Stand-Off. Nat. Rev. Microbiol. 2006, 4, 121–132. [Google Scholar] [CrossRef]
- Nakagaki, K.; Nakagaki, K.; Taguchi, F. Receptor-Independent Spread of a Highly Neurotropic Murine Coronavirus JHMV Strain from Initially Infected Microglial Cells in Mixed Neural Cultures. J. Virol. 2005, 79, 6102–6110. [Google Scholar] [CrossRef]
- Pasick, J.; Dales, S. MHV-JHM Infections of Rodent Neuronal Cells: Replication and Trafficking of Structural Proteins and Progeny Virions. In Coronaviruses; Laude, H., Vautherot, J.-F., Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1994; Volume 342, pp. 319–325. ISBN 978-1-4613-6305-7. [Google Scholar]
- Cymerys, J.; Kowalczyk, A.; Mikołajewicz, K.; Słońska, A.; Krzyżowska, M. Nitric Oxide Influences HSV-1-Induced Neuroinflammation. Oxid. Med. Cell. Longev. 2019, 2019, e2302835. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Arch. Für Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Gaertner, D.J.; Winograd, D.F.; Compton, S.R.; Paturzo, F.X.; Smith, A.L. Development and Optimization of Plaque Assays for Rat Coronaviruses. J. Virol. Methods 1993, 43, 53–64. [Google Scholar] [CrossRef]
- Cymerys, J.; Chodkowski, M.; Słońska, A.; Krzyżowska, M.; Bańbura, M.W. Disturbances of Mitochondrial Dynamics in Cultured Neurons Infected with Human Herpesvirus Type 1 and Type 2. J. Neurovirol. 2019, 25, 765. [Google Scholar] [CrossRef]
- Boes, M.; Falter-Braun, P. Long-COVID-19: The Persisting Imprint of SARS-CoV-2 Infections on the Innate Immune System. Signal Transduct. Target. Ther. 2023, 8, 460. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Brain Mechanisms Involved in Post COVID Syndrome: A Narrative Review. Neurochem. J. 2024, 18, 397–405. [Google Scholar] [CrossRef]
- Prosperini, L.; Tortorella, C.; Haggiag, S.; Ruggieri, S.; Galgani, S.; Gasperini, C. Determinants of COVID-19-Related Lethality in Multiple Sclerosis: A Meta-Regression of Observational Studies. J. Neurol. 2022, 269, 2275–2285. [Google Scholar] [CrossRef]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef]
- Almulla, A.F.; Thipakorn, Y.; Zhou, B.; Vojdani, A.; Maes, M. Immune Activation and Immune-Associated Neurotoxicity in Long-COVID: A Systematic Review and Meta-Analysis of 103 Studies Comprising 58 Cytokines/Chemokines/Growth Factors. Brain. Behav. Immun. 2024, 122, 75–94. [Google Scholar] [CrossRef]
- Cron, R.Q.; Goyal, G.; Chatham, W.W. Cytokine Storm Syndrome. Annu. Rev. Med. 2023, 74, 321–337. [Google Scholar] [CrossRef]
- Gu, Y.; Zuo, X.; Zhang, S.; Ouyang, Z.; Jiang, S.; Wang, F.; Wang, G. The Mechanism behind Influenza Virus Cytokine Storm. Viruses 2021, 13, 1362. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.J.; Schulert, G.S. COVID-19 and Cytokine Storm Syndrome: Are There Lessons from Macrophage Activation Syndrome? Transl. Res. J. Lab. Clin. Med. 2021, 232, 1–12. [Google Scholar] [CrossRef]
- Percy, D.H.; Lynch, J.A.; Descôteaux, J.-P. Central Nervous System Lesions in Suckling Mice and Rats Inoculated Intranasally with Sialodacryoadenitis Virus. Vet. Pathol. 1986, 23, 42–49. [Google Scholar] [CrossRef]
- Bartak, M.; Bąska, P.; Chodkowski, M.; Tymińska, B.; Bańbura, M.W.; Cymerys, J. Neurons Cytoskeletal Architecture Remodeling during the Replication Cycle of Mouse Coronavirus MHV-JHM: A Morphological in Vitro Study. BMC Vet. Res. 2024, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Das Sarma, J. Microglia-Mediated Neuroinflammation Is an Amplifier of Virus-Induced Neuropathology. J. Neurovirol. 2014, 20, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Savarin, C.; Bergmann, C.C. Fine Tuning the Cytokine Storm by IFN and IL-10 Following Neurotropic Coronavirus Encephalomyelitis. Front. Immunol. 2018, 9, 3022. [Google Scholar] [CrossRef]
- Verma, A.K.; Zheng, J.; Mack, M.; Ginhoux, F.; Perlman, S. Differential Effects of Prostaglandin D2 Signaling on Macrophages and Microglia in Murine Coronavirus Encephalomyelitis. mBio 2021, 12, e0196921. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Sariol, A.; Meyerholz, D.K.; Perlman, S. Microglia Are Required for Protection against Lethal Coronavirus Encephalitis in Mice. J. Clin. Investig. 2018, 128, 931–943. [Google Scholar] [CrossRef]
- Mangale, V.; Syage, A.R.; Ekiz, H.A.; Skinner, D.D.; Cheng, Y.; Stone, C.L.; Brown, R.M.; O’Connell, R.M.; Green, K.N.; Lane, T.E. Microglia Influence Host Defense, Disease, and Repair Following Murine Coronavirus Infection of the Central Nervous System. Glia 2020, 68, 2345–2360. [Google Scholar] [CrossRef]
- Sariol, A.; Mackin, S.; Allred, M.-G.; Ma, C.; Zhou, Y.; Zhang, Q.; Zou, X.; Abrahante, J.E.; Meyerholz, D.K.; Perlman, S. Microglia Depletion Exacerbates Demyelination and Impairs Remyelination in a Neurotropic Coronavirus Infection. Proc. Natl. Acad. Sci. USA 2020, 117, 24464–24474. [Google Scholar] [CrossRef]
- Syage, A.; Pachow, C.; Cheng, Y.; Mangale, V.; Green, K.N.; Lane, T.E. Microglia Influence Immune Responses and Restrict Neurologic Disease in Response to Central Nervous System Infection by a Neurotropic Murine Coronavirus. Front. Cell. Neurosci. 2023, 17, 1291255. [Google Scholar] [CrossRef]
- Liu, Y.; Cai, Y.; Zhang, X. Induction of Caspase-Dependent Apoptosis in Cultured Rat Oligodendrocytes by Murine Coronavirus Is Mediated during Cell Entry and Does Not Require Virus Replication. J. Virol. 2003, 77, 11952–11963. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X. Murine Coronavirus-Induced Oligodendrocyte Apoptosis Is Mediated through the Activation of the Fas Signaling Pathway. Virology 2007, 360, 364–375. [Google Scholar] [CrossRef]
- Jeong, G.U.; Lyu, J.; Kim, K.-D.; Chung, Y.C.; Yoon, G.Y.; Lee, S.; Hwang, I.; Shin, W.-H.; Ko, J.; Lee, J.-Y.; et al. SARS-CoV-2 Infection of Microglia Elicits Proinflammatory Activation and Apoptotic Cell Death. Microbiol. Spectr. 2022, 10, e01091-22. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Dharra, R.; Kumar Sharma, A.; Datta, S. Emerging Aspects of Cytokine Storm in COVID-19: The Role of Proinflammatory Cytokines and Therapeutic Prospects. Cytokine 2023, 169, 156287. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine Storm and Leukocyte Changes in Mild versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Gao, Z.; Zhu, Q.; Zhang, Y.; Zhao, Y.; Cai, L.; Shields, C.B.; Cai, J. Reciprocal Modulation Between Microglia and Astrocyte in Reactive Gliosis Following the CNS Injury. Mol. Neurobiol. 2013, 48, 690–701. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.A.; Wang, J.; Holmes, K.V.; Mason, R.J. Rat Coronaviruses Infect Rat Alveolar Type I Epithelial Cells and Induce Expression of CXC Chemokines. Virology 2007, 369, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.J.; Manzer, R.; Miura, T.A.; Groshong, S.D.; Ito, Y.; Travanty, E.A.; Leete, J.; Holmes, K.V.; Mason, R.J. Rat Respiratory Coronavirus Infection: Replication in Airway and Alveolar Epithelial Cells and the Innate Immune Response. J. Gen. Virol. 2009, 90, 2956–2964. [Google Scholar] [CrossRef]
- Olivarria, G.; Lane, T.E. Evaluating the Role of Chemokines and Chemokine Receptors Involved in Coronavirus Infection. Expert Rev. Clin. Immunol. 2022, 18, 57–66. [Google Scholar] [CrossRef]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and Chemokine Receptors during COVID-19 Infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Gudowska-Sawczuk, M.; Mroczko, B. What Is Currently Known about the Role of CXCL10 in SARS-CoV-2 Infection? Int. J. Mol. Sci. 2022, 23, 3673. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Liu, C.; Su, L.; Zhang, D.; Fan, J.; Yang, Y.; Xiao, M.; Xie, J.; Xu, Y.; et al. IP-10 and MCP-1 as Biomarkers Associated with Disease Severity of COVID-19. Mol. Med. 2020, 26, 97. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, C.; Li, J.; Yuan, J.; Wei, J.; Huang, F.; Wang, F.; Li, G.; Li, Y.; Xing, L.; et al. Plasma IP-10 and MCP-3 Levels Are Highly Associated with Disease Severity and Predict the Progression of COVID-19. J. Allergy Clin. Immunol. 2020, 146, 119–127.e4. [Google Scholar] [CrossRef]
- Tegethoff, S.A.; Danziger, G.; Kühn, D.; Kimmer, C.; Adams, T.; Heintz, L.; Metz, C.; Reifenrath, K.; Angresius, R.; Mang, S.; et al. TNF-Related Apoptosis-Inducing Ligand, Interferon Gamma-Induced Protein 10, and C-Reactive Protein in Predicting the Progression of SARS-CoV-2 Infection: A Prospective Cohort Study. Int. J. Infect. Dis. 2022, 122, 178–187. [Google Scholar] [CrossRef]
- Lu, Q.; Zhu, Z.; Tan, C.; Zhou, H.; Hu, Y.; Shen, G.; Zhu, P.; Yang, G.; Xie, X. Changes of Serum IL-10, IL-1β, IL-6, MCP-1, TNF-α, IP-10 and IL-4 in COVID-19 Patients. Int. J. Clin. Pract. 2021, 75, e14462. [Google Scholar] [CrossRef]
- Madhurantakam, S.; Lee, Z.J.; Naqvi, A.; Prasad, S. Importance of IP-10 as a Biomarker of Host Immune Response: Critical Perspective as a Target for Biosensing. Curr. Res. Biotechnol. 2023, 5, 100130. [Google Scholar] [CrossRef]
- Rodríguez, A.M.; Rodríguez, J.; Giambartolomei, G.H. Microglia at the Crossroads of Pathogen-Induced Neuroinflammation. ASN Neuro 2022, 14, 17590914221104566. [Google Scholar] [CrossRef]
- Van Everbroeck, B.; Dewulf, E.; Pals, P.; Lübke, U.; Martin, J.-J.; Cras, P. The Role of Cytokines, Astrocytes, Microglia and Apoptosis in Creutzfeldt-Jakob Disease. Neurobiol. Aging 2002, 23, 59–64. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of Pro-Inflammatory Cytokines Released from Microglia in Neurodegenerative Diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The Pro-Inflammatory Cytokines in COVID-19 Pathogenesis: What Goes Wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Kujawska, M.; Mostafavi, E.; Kaushik, A. SARS-CoV-2 Getting into the Brain; Neurological Phenotype of COVID-19, and Management by Nano-Biotechnology. Neural Regen. Res. 2023, 18, 519. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive Astrocytes: The Nexus of Pathological and Clinical Hallmarks of Alzheimer’s Disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef]
- Rosciszewski, G.; Cadena, V.; Murta, V.; Lukin, J.; Villarreal, A.; Roger, T.; Ramos, A.J. Toll-Like Receptor 4 (TLR4) and Triggering Receptor Expressed on Myeloid Cells-2 (TREM-2) Activation Balance Astrocyte Polarization into a Proinflammatory Phenotype. Mol. Neurobiol. 2018, 55, 3875–3888. [Google Scholar] [CrossRef] [PubMed]
- Rosciszewski, G.; Cadena, V.; Auzmendi, J.; Cieri, M.B.; Lukin, J.; Rossi, A.R.; Murta, V.; Villarreal, A.; Reinés, A.; Gomes, F.C.A.; et al. Detrimental Effects of HMGB-1 Require Microglial-Astroglial Interaction: Implications for the Status Epilepticus -Induced Neuroinflammation. Front. Cell. Neurosci. 2019, 13, 380. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Target List |
|---|---|
| Cytokines: | BAFF, G-CSF (CSF-3), GM-CSF, IFN alpha, IFN gamma, IL-1 alpha, IL-1 beta, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12p70, IL-13, IL-15/IL-15R, IL-17A (CTLA-8), IL-18, IL-19, IL-22, IL-23, IL-25 (IL-17E), IL-27, IL-28, IL-31, IL-33, LIF18, M-CSF, RANKL, TNF alpha |
| Chemokines: | ENA-78 (CXCL5), Eotaxin (CCL11), GRO alpha (CXCL1), IP-10 (CXCL10), MCP-1 (CCL2), MCP-3 (CCL7), MIP-1 alpha (CCL3), MIP-1 beta (CCL4), MIP-2, RANTES (CCL5) |
| Growth factors/regulators: | Betacellulin (BTC), Leptin, VEGF-A |
| Soluble receptors: | IL-2R, IL-7R alpha, IL-33R (ST2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartak, M.; Krahel, W.D.; Gregorczyk-Zboroch, K.; Chodkowski, M.; Potârniche, A.V.; Długosz, E.; Krzyżowska, M.; Cymerys, J. Cytokine Profile Analysis During Sialodacryoadenitis Virus and Mouse Hepatitis Virus JHM Strain Infection in Primary Mixed Microglia and Astrocyte Culture—Preliminary Research. Cells 2025, 14, 637. https://doi.org/10.3390/cells14090637
Bartak M, Krahel WD, Gregorczyk-Zboroch K, Chodkowski M, Potârniche AV, Długosz E, Krzyżowska M, Cymerys J. Cytokine Profile Analysis During Sialodacryoadenitis Virus and Mouse Hepatitis Virus JHM Strain Infection in Primary Mixed Microglia and Astrocyte Culture—Preliminary Research. Cells. 2025; 14(9):637. https://doi.org/10.3390/cells14090637
Chicago/Turabian StyleBartak, Michalina, Weronika D. Krahel, Karolina Gregorczyk-Zboroch, Marcin Chodkowski, Adrian Valentin Potârniche, Ewa Długosz, Małgorzata Krzyżowska, and Joanna Cymerys. 2025. "Cytokine Profile Analysis During Sialodacryoadenitis Virus and Mouse Hepatitis Virus JHM Strain Infection in Primary Mixed Microglia and Astrocyte Culture—Preliminary Research" Cells 14, no. 9: 637. https://doi.org/10.3390/cells14090637
APA StyleBartak, M., Krahel, W. D., Gregorczyk-Zboroch, K., Chodkowski, M., Potârniche, A. V., Długosz, E., Krzyżowska, M., & Cymerys, J. (2025). Cytokine Profile Analysis During Sialodacryoadenitis Virus and Mouse Hepatitis Virus JHM Strain Infection in Primary Mixed Microglia and Astrocyte Culture—Preliminary Research. Cells, 14(9), 637. https://doi.org/10.3390/cells14090637