
The Role of Integrin β1D Mislocalization in the Pathophysiology of Calpain 3-Related Limb–Girdle Muscular Dystrophy

 , ,
, ,  , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Human Skeletal Muscle Culture
2.2. Immunofluorescence of Muscle Sections
2.3. Immunofluorescence of Cell Cultures
2.4. Wound-Healing (WH) Assay in CD56− Cells
2.5. Confocal Microscopy Analysis and Statistics
- -
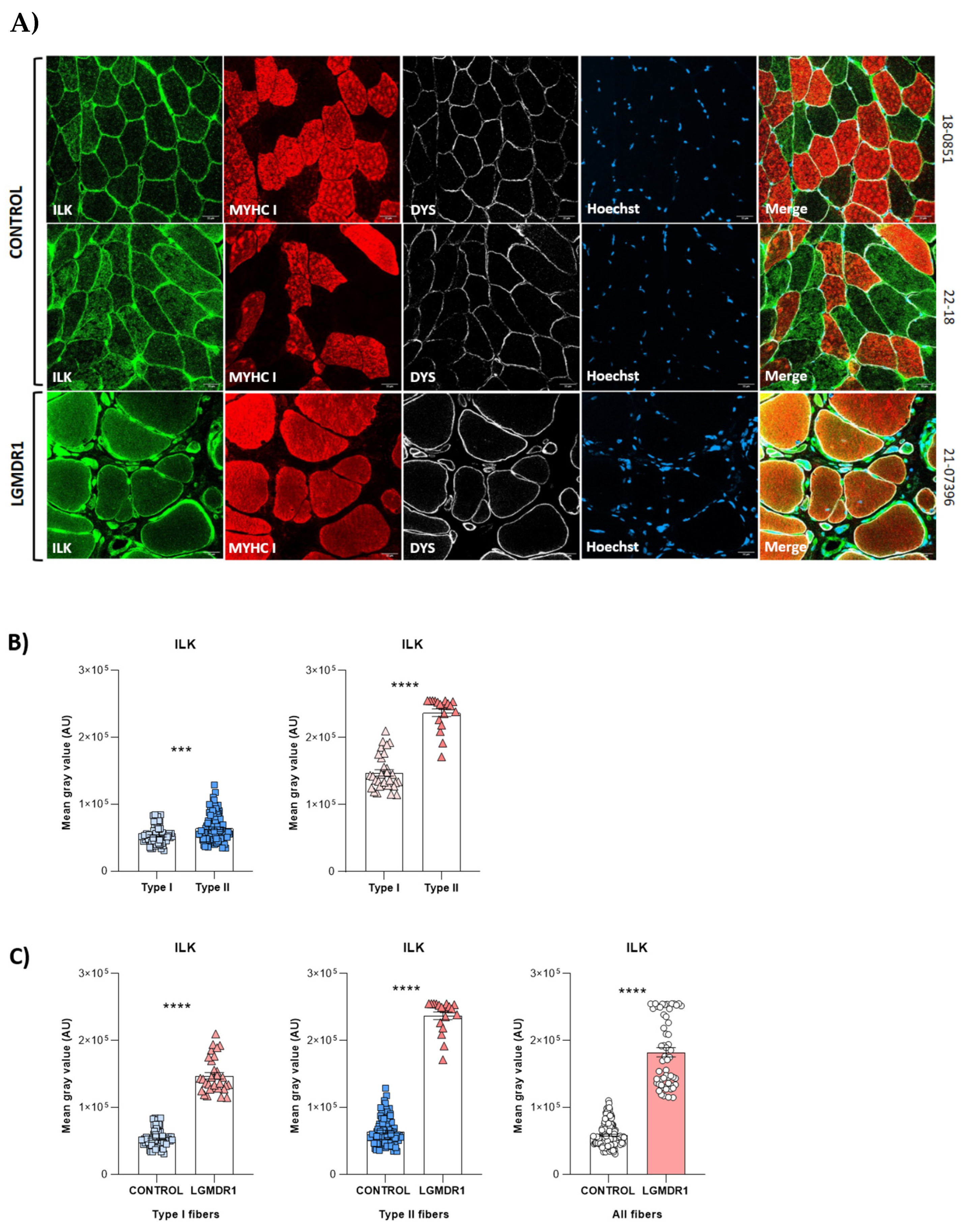
- Protein quantification of costamere proteins in the sarcoplasm of muscle fibers: Fiber segmentation and quantification of the fluorescence intensity (average fluorescence intensity within the fiber) corresponding with a direct proportion of protein expression was performed. Different amounts of the fibers were analyzed for each protein: 150–240 fibers per control/patient group for ITGβ1D analysis; 65–200 fibers per control/patient group for TLN1; 40–80 fibers per control/patient group for vinculin (VCL); 140–400 fibers per control/patient group for focal adhesion kinase (FAK); and 55–160 fibers per group for integrin-linked kinase (ILK). The statistical significance was assessed using an unpaired t-test after the elimination of outliers by the Rout method (Q = 1%).
- -
- Blood vessel quantification in skeletal muscle: The blood vessels of 450–890 fibers (after fiber segmentation) were analyzed per group.
- -
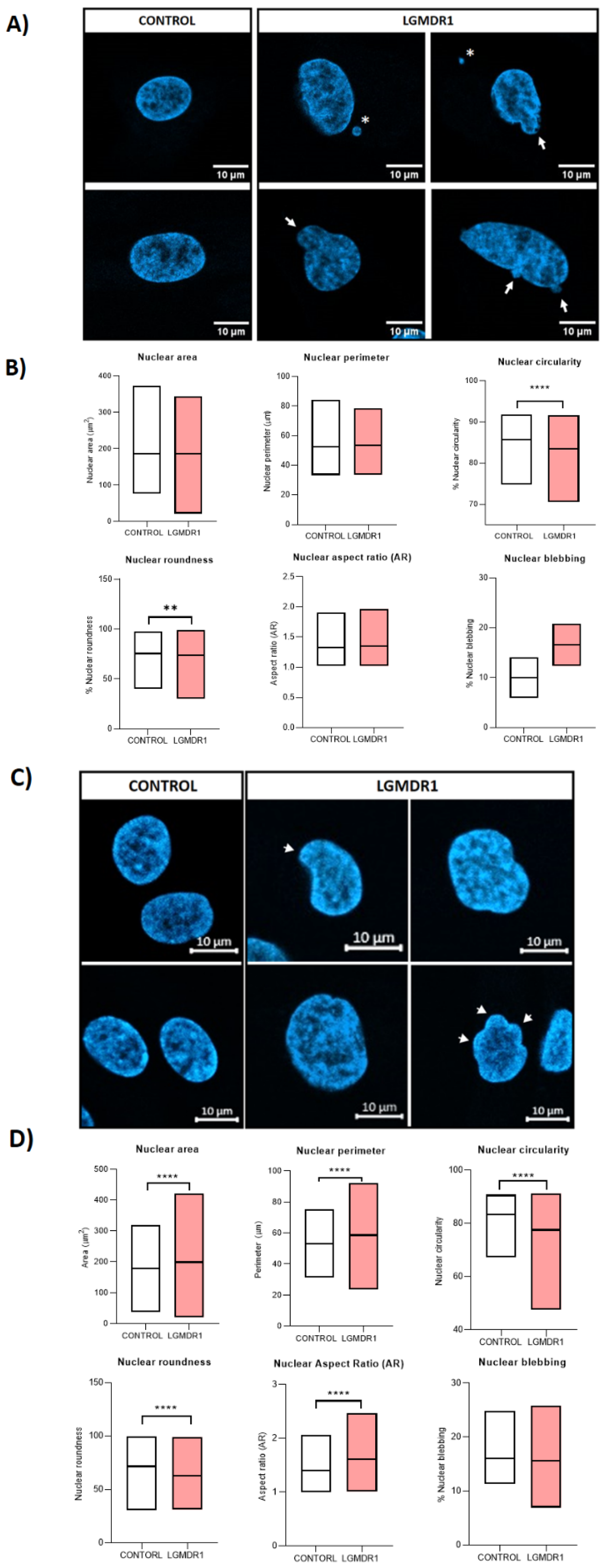
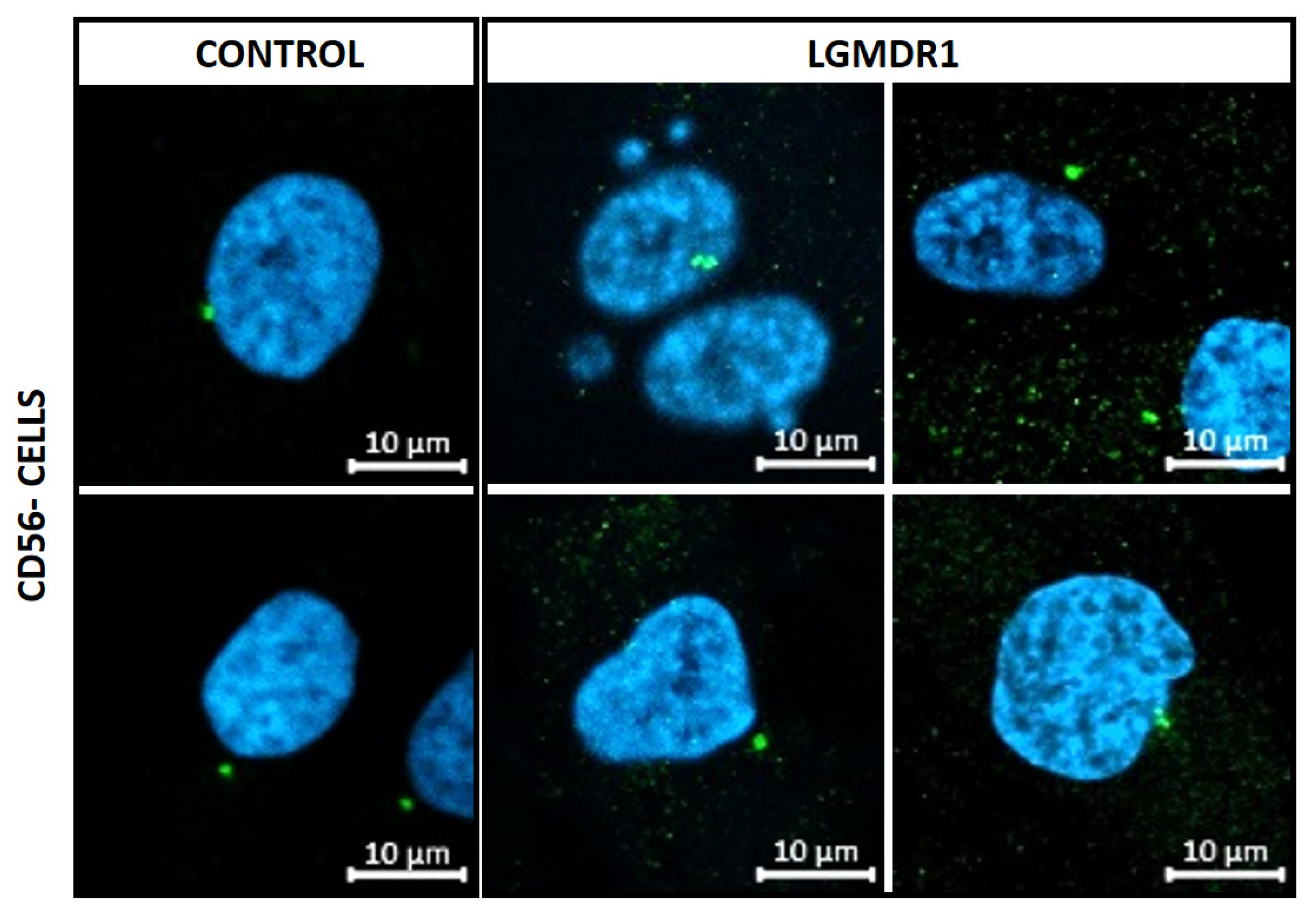
- Nuclear morphology analysis in cells and skeletal muscle tissue sections: Nucleus segmentation and shape descriptor (circularity, roundness, aspect ratio, area and perimeter) analyses were performed. Overall, 800–1000 nuclei were analyzed per group for CD56− cells, 700–1000 nuclei per group for myoblasts, 180–200 nuclei for myotubes and 500–700 nuclei for skeletal muscle sections. Only laminin-delimitated myonuclei were considered for the analysis of skeletal muscle nuclei. Statistical significance was assessed using an unpaired t-test after the elimination of outliers via the Rout method (Q = 1%).
- -
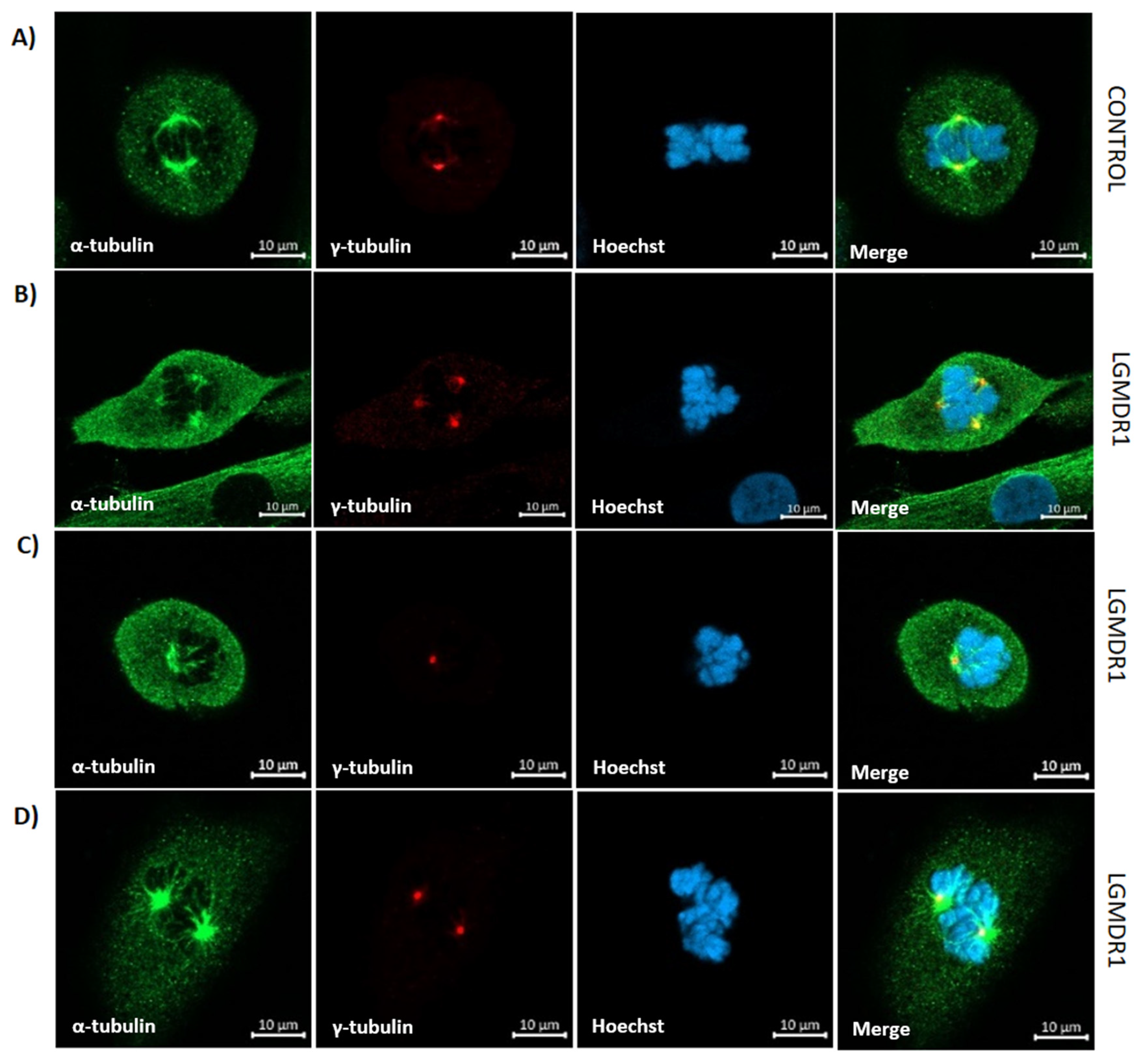
- Centrosome–nucleus distance in skeletal muscle cells: The centrosome–nucleus distance was assessed using antibodies against ϒ-tubulin. Overall, 100 CD56− cells and 150 myoblasts were analyzed per group. The statistical significance was assessed using an unpaired t-test after the elimination of outliers via the Rout method (Q = 1%).
- -
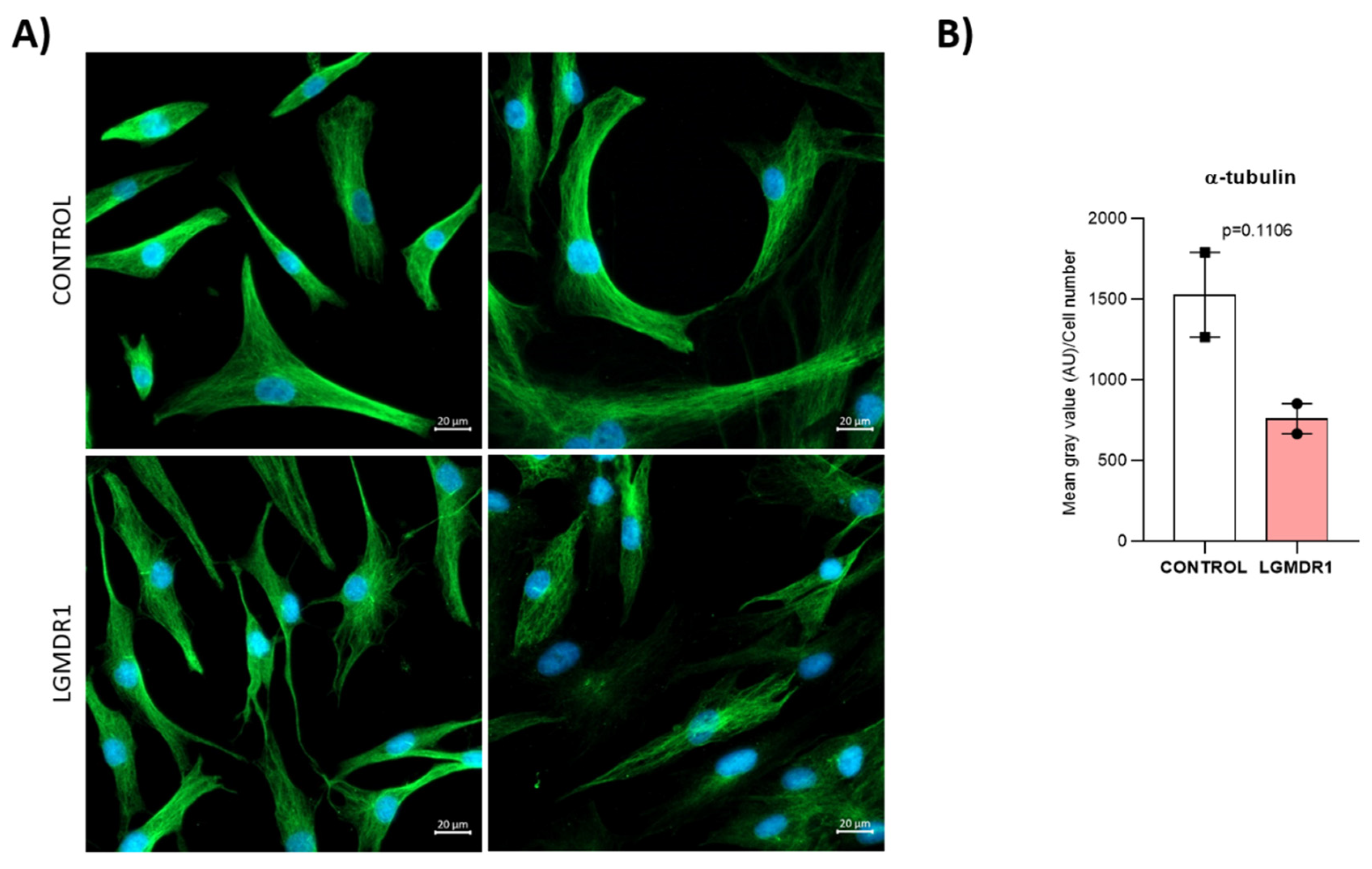
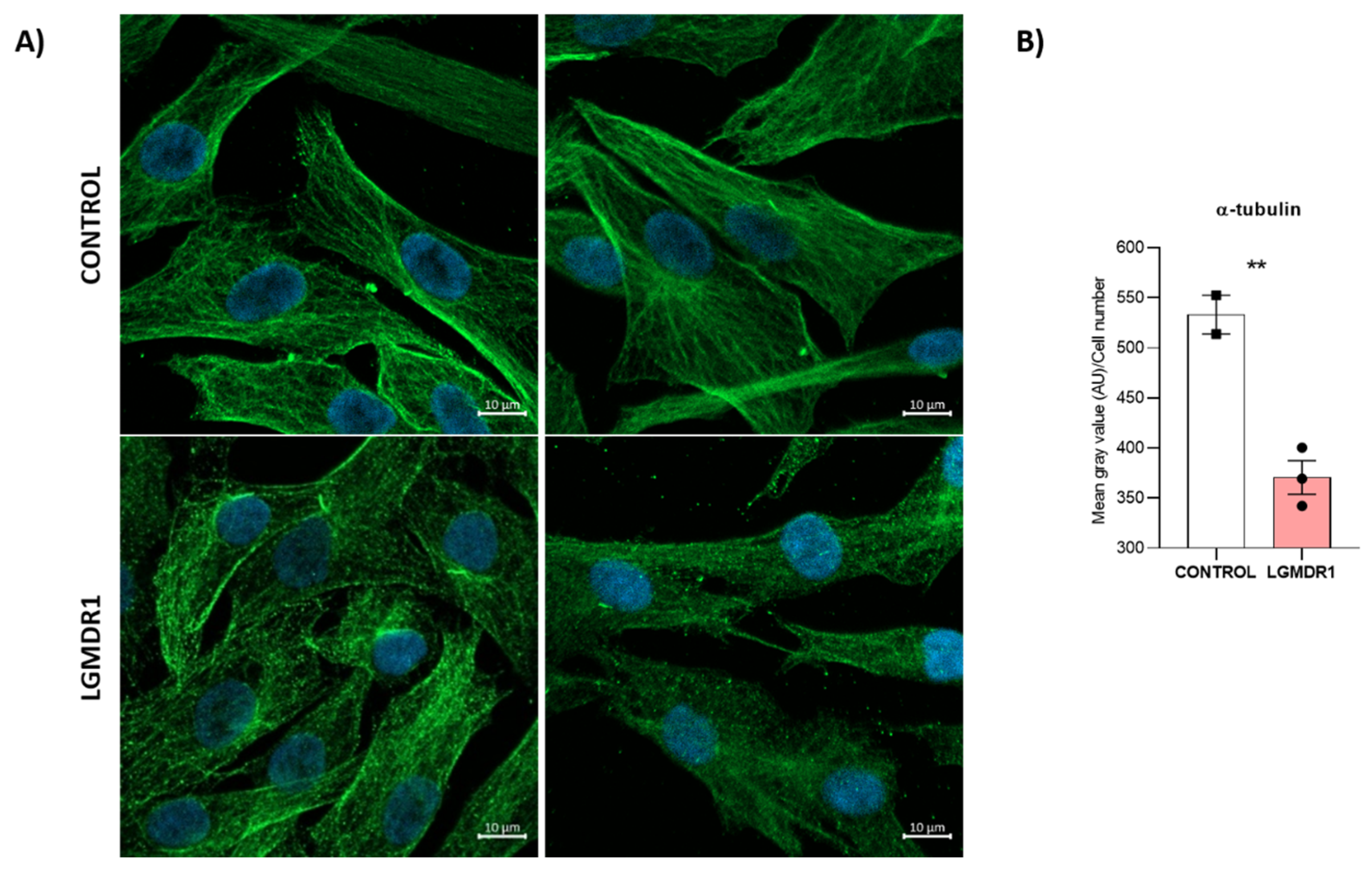
- Quantification of cytoskeletal α-tubulin in muscle cells: α-tubulin fluorescence intensity was normalized to the cell number. For CD56− cells, 250 control and 500 patient cells were assessed; for myoblasts, there were 500 control and 1200 patient cells. The statistical significance was assessed using an unpaired t-test after the elimination of outliers via the Rout method (Q = 1%).
- -
- Centrosome positioning assessment in CD56− cells: The number of cells with a misoriented centrosome after a WH assay was assessed. The statistical significance was assessed using an unpaired t-test.
- -
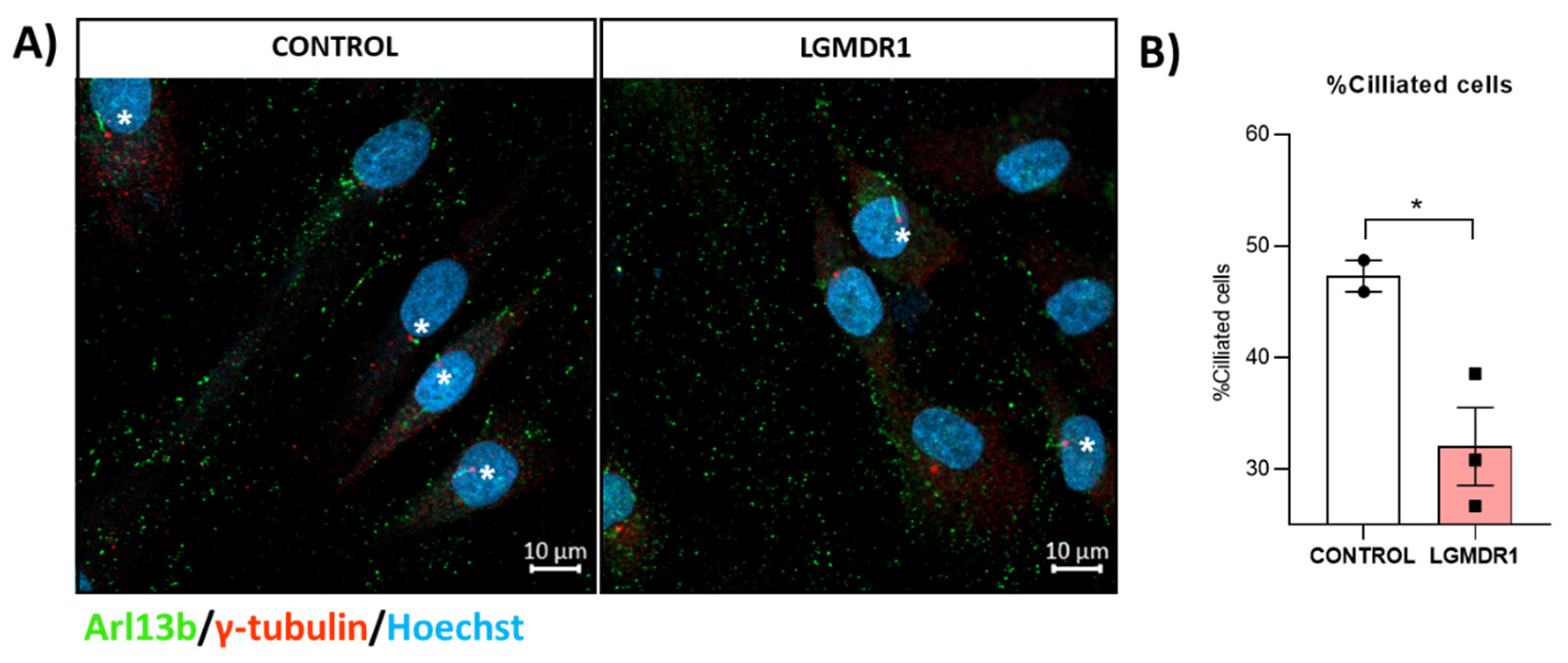
- Quantification of ciliated myoblasts: The number of ciliated cells was measured. The statistical significance was assessed using an unpaired t-test.
3. Results
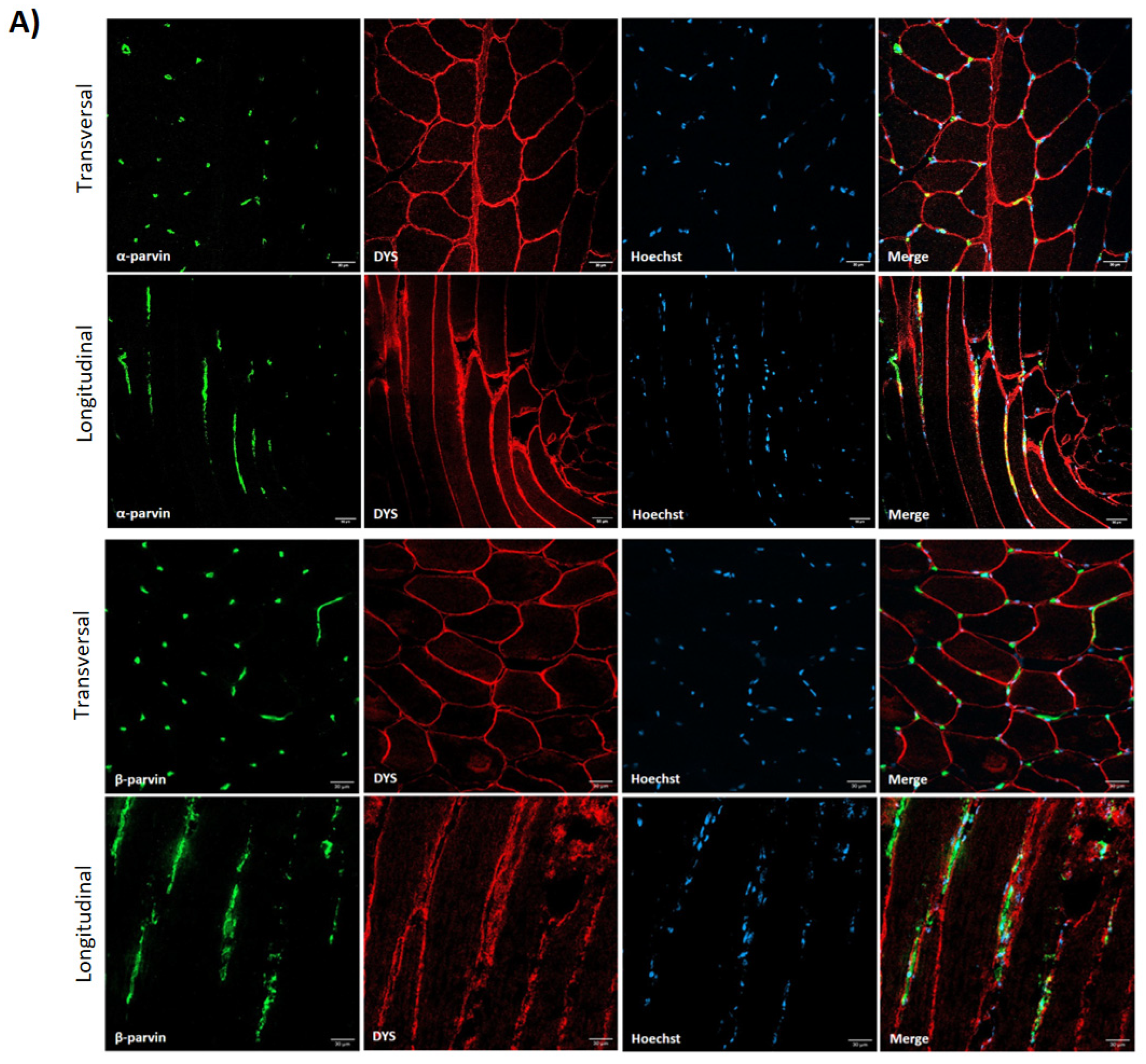
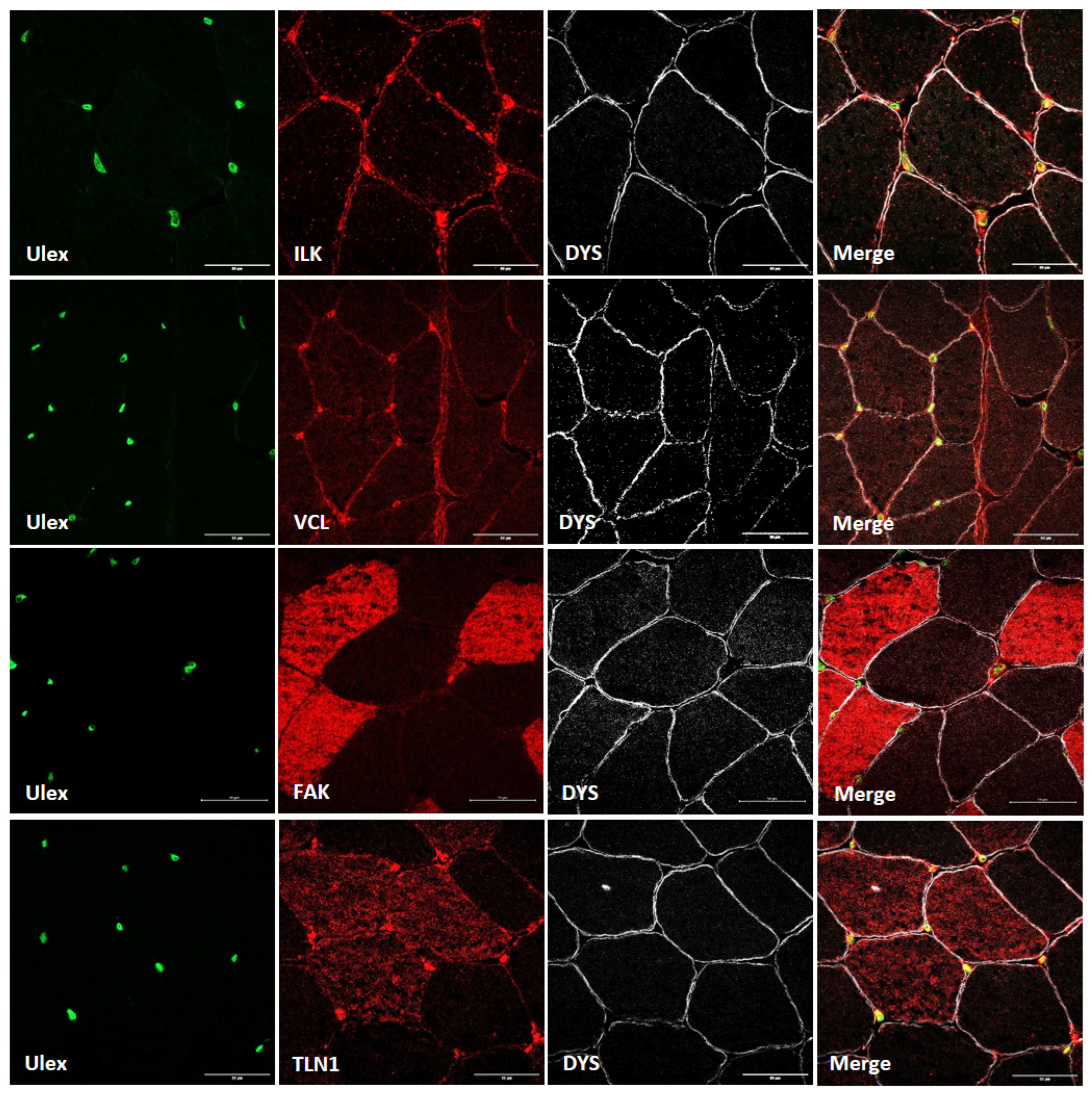
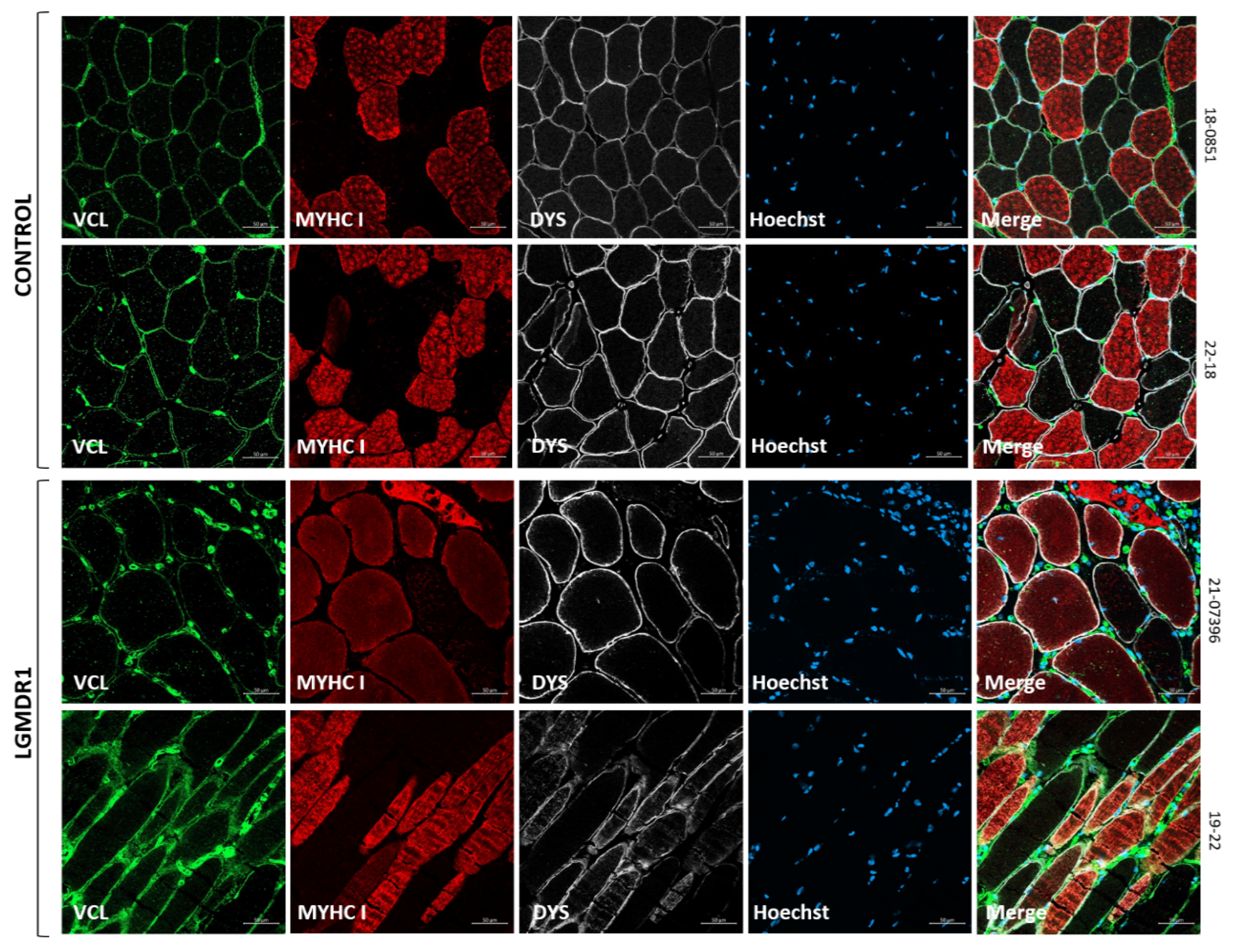
3.1. Costamere Proteins Are Fiber-Type-Dependent and Also Present in the Endothelial Tissue
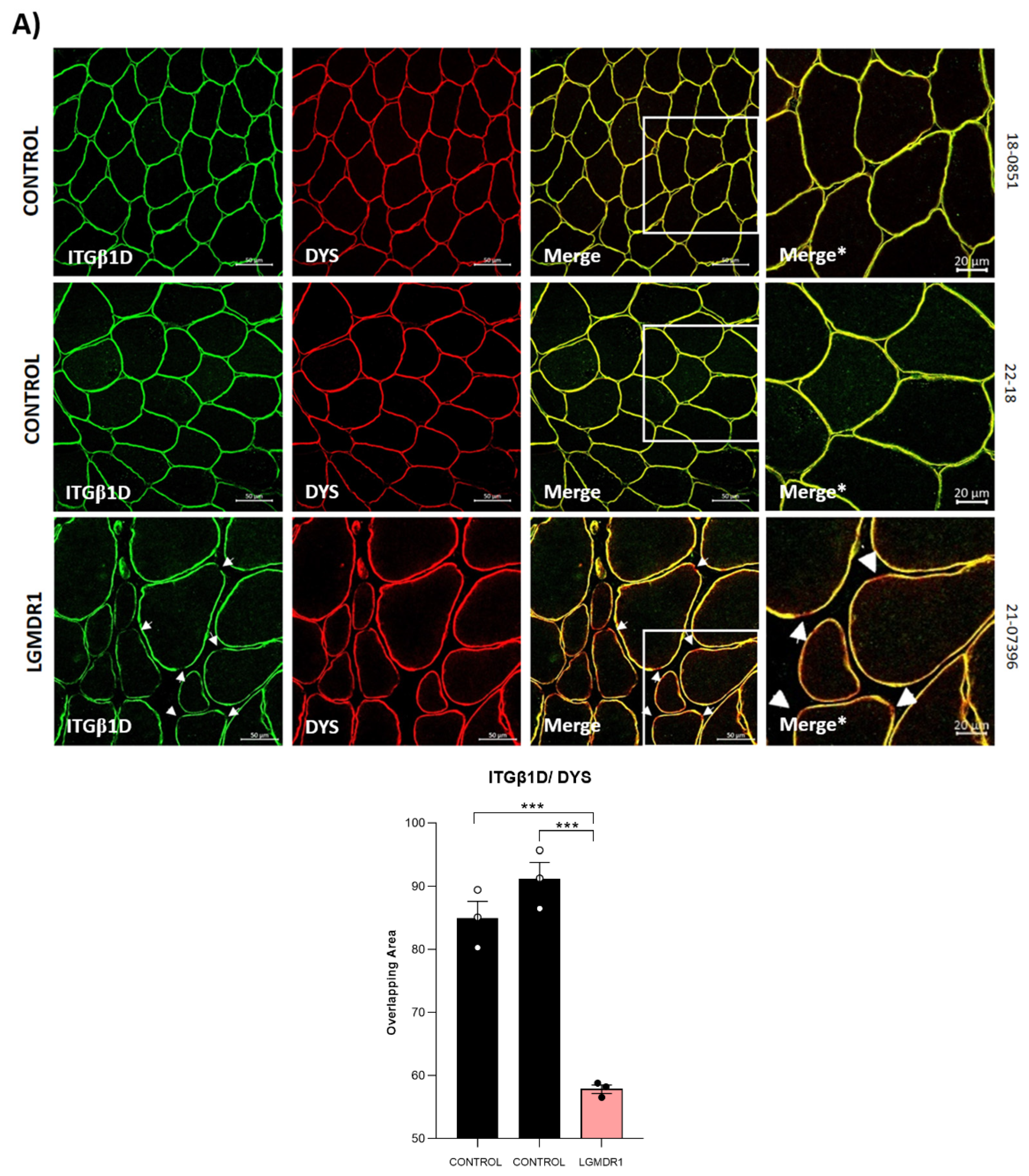
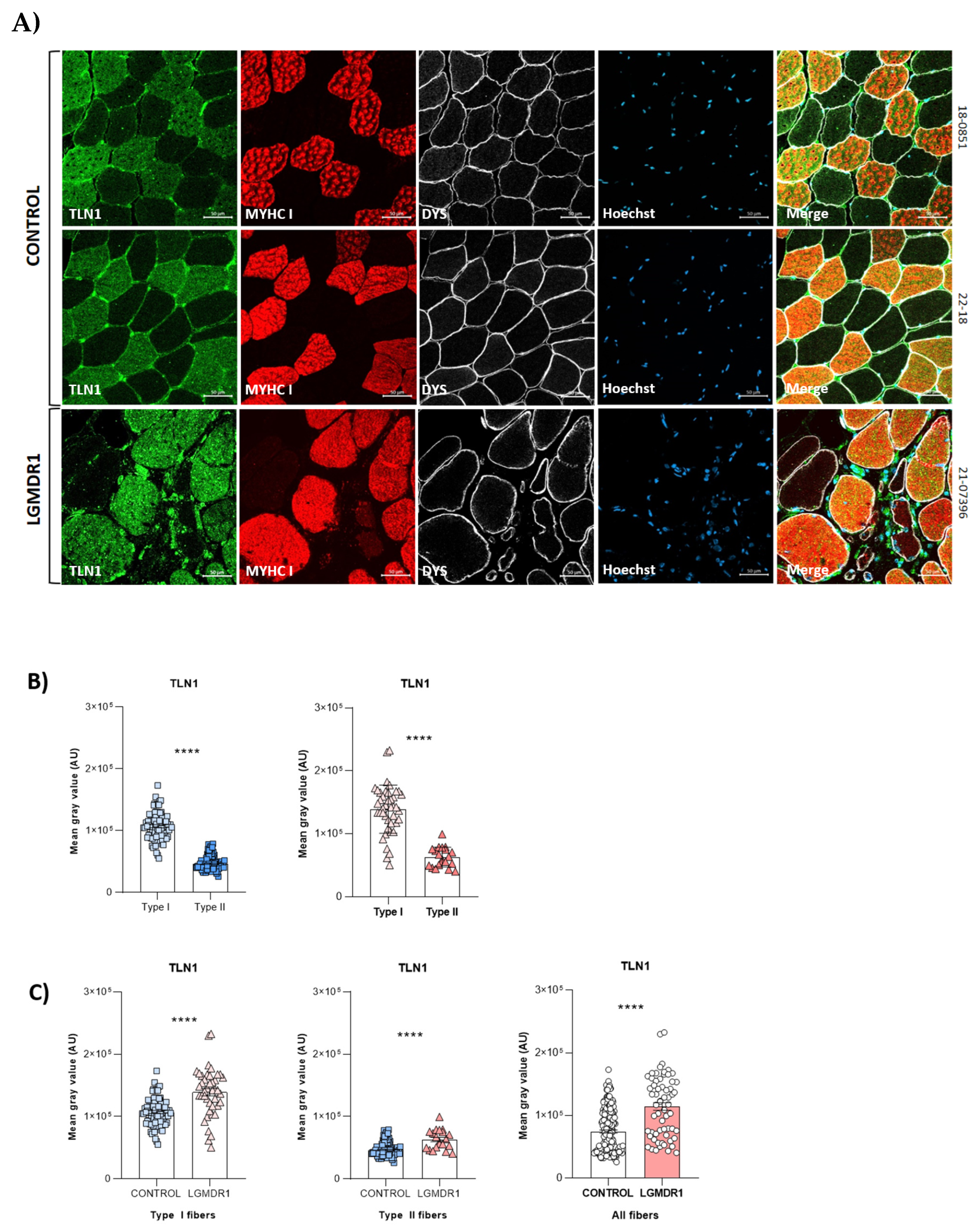
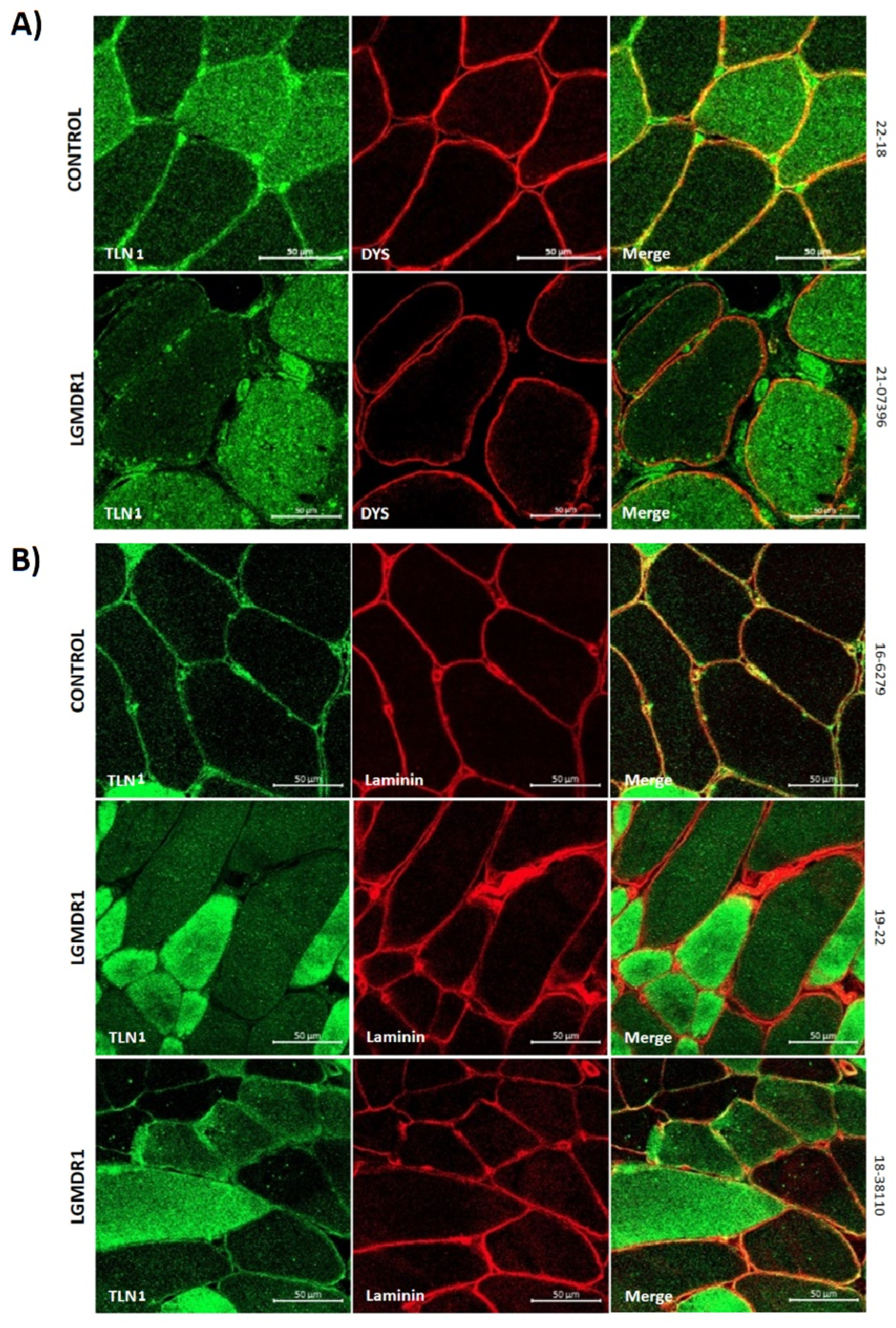
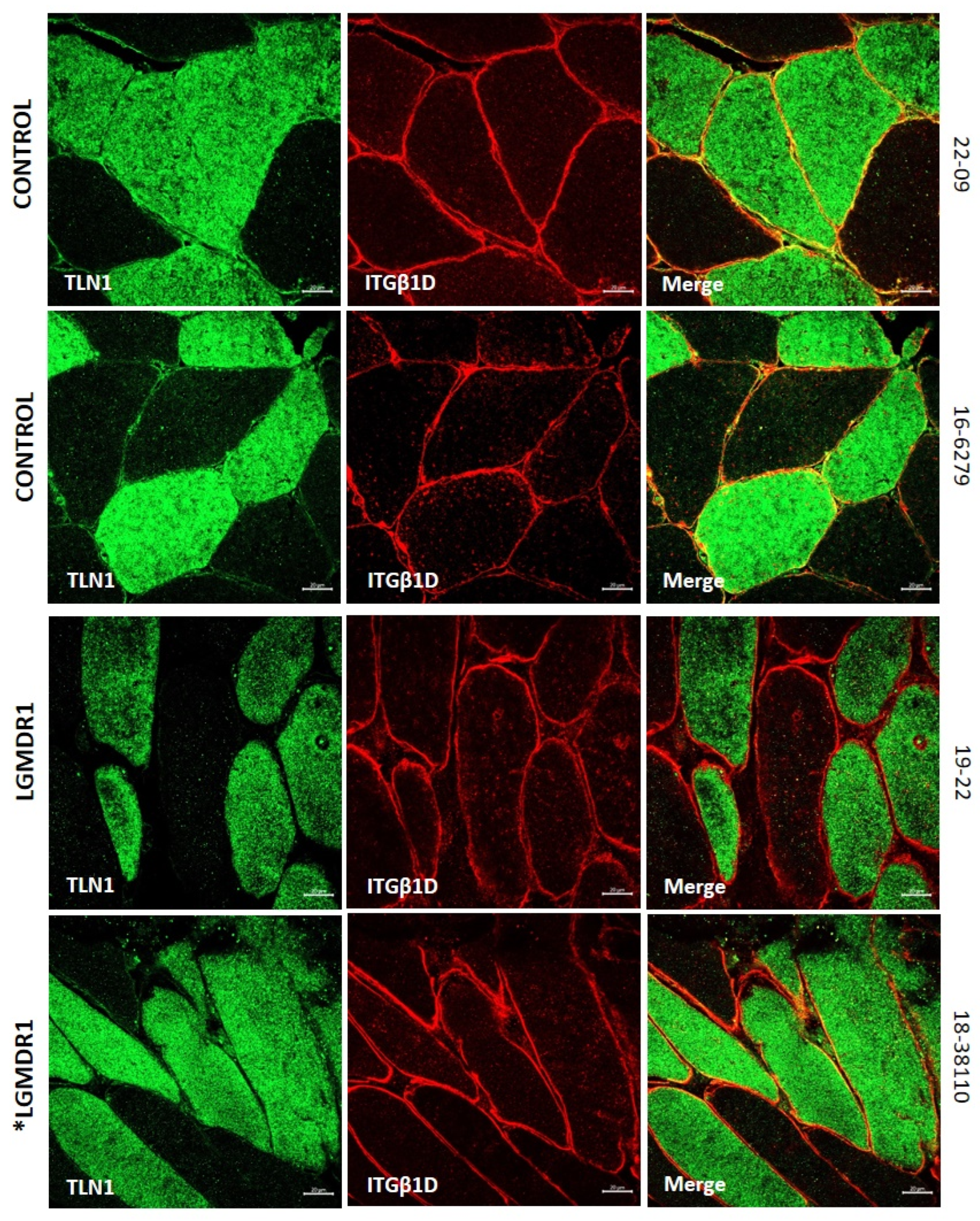
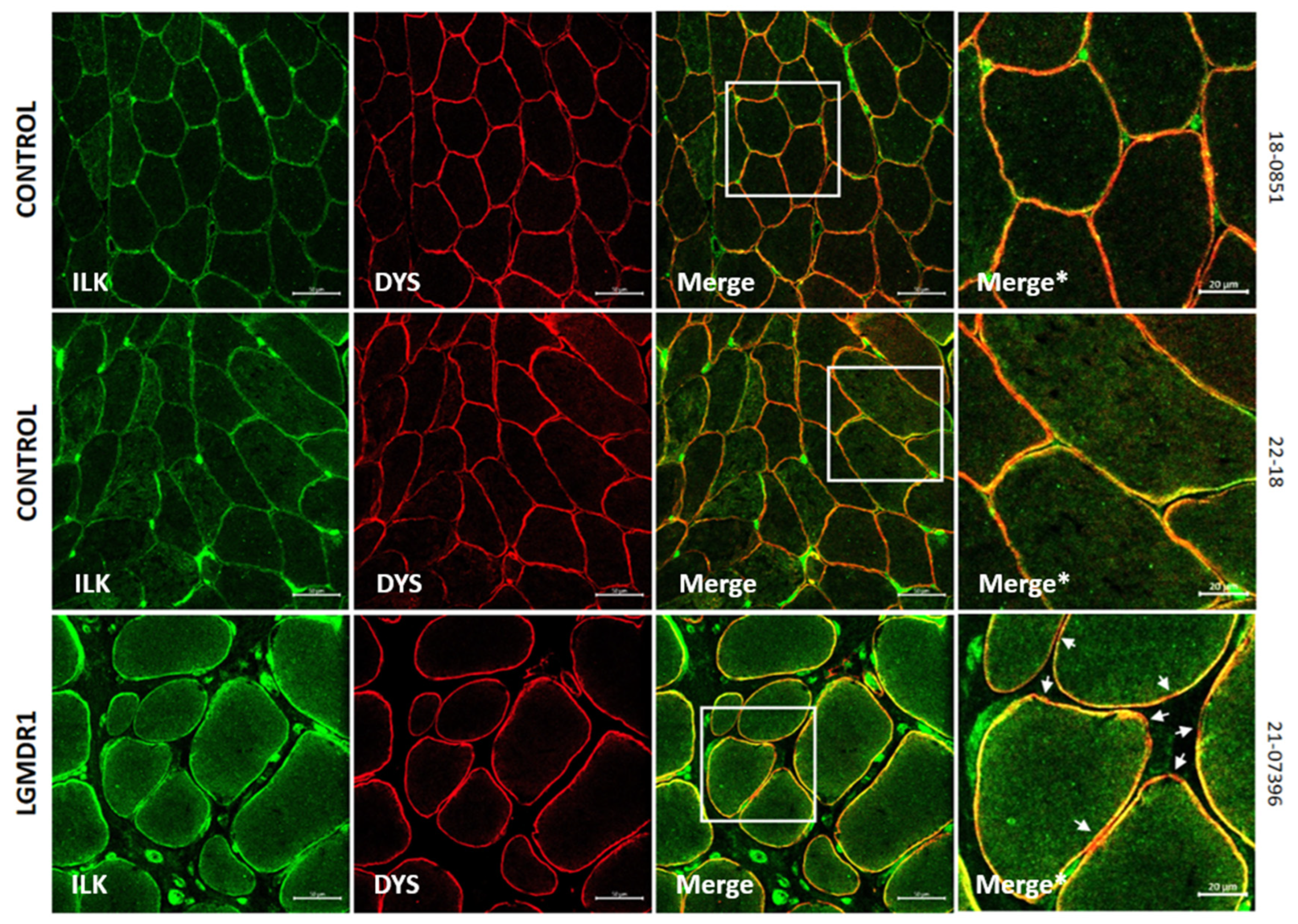
3.2. Abnormal Distribution of ITGβ1D and TLN1 in LGMDR1 Patients’ Muscle
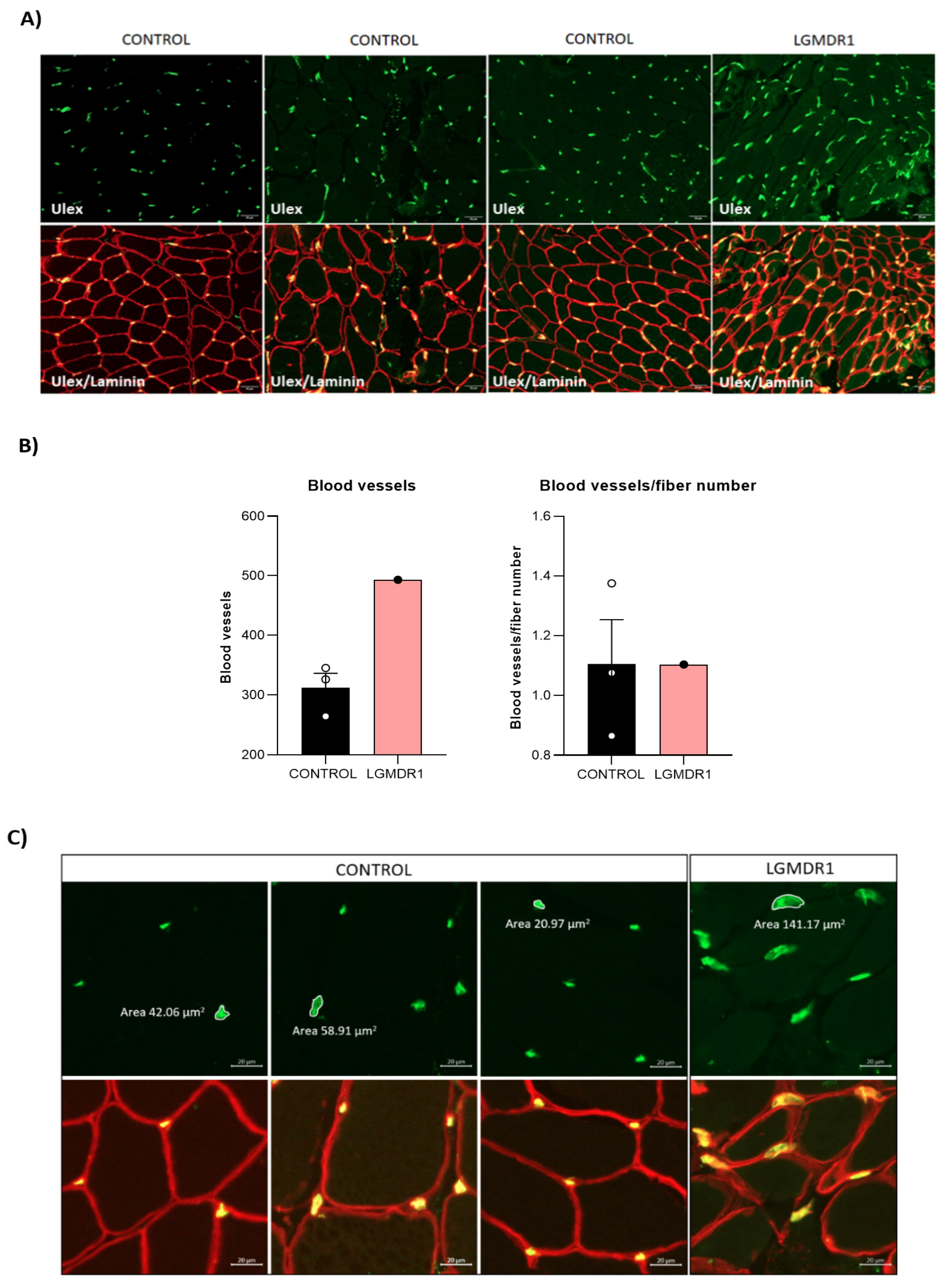
3.3. Abnormal Blood Vessel Morphology in LGMDR1 Patients’ Muscles
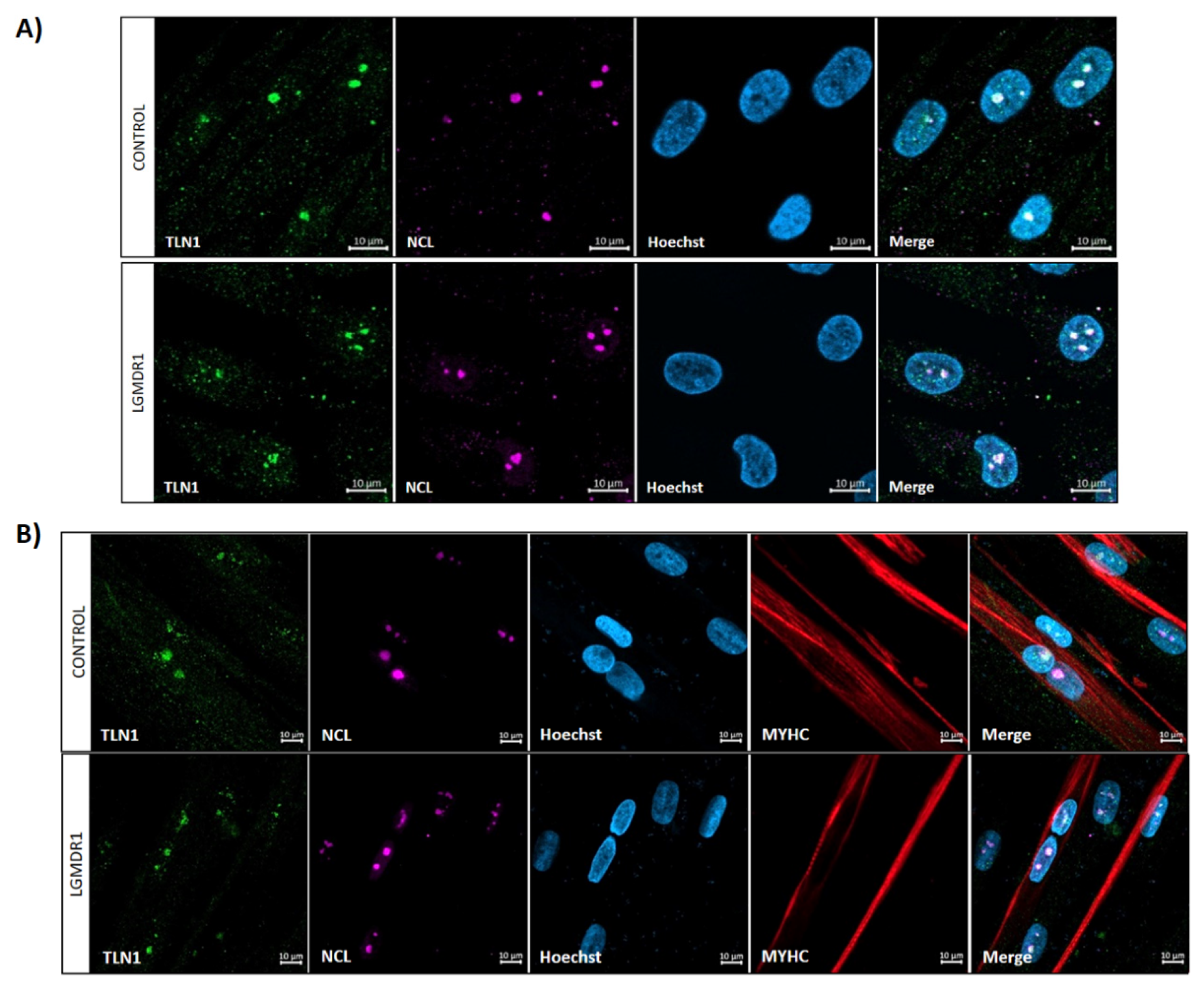
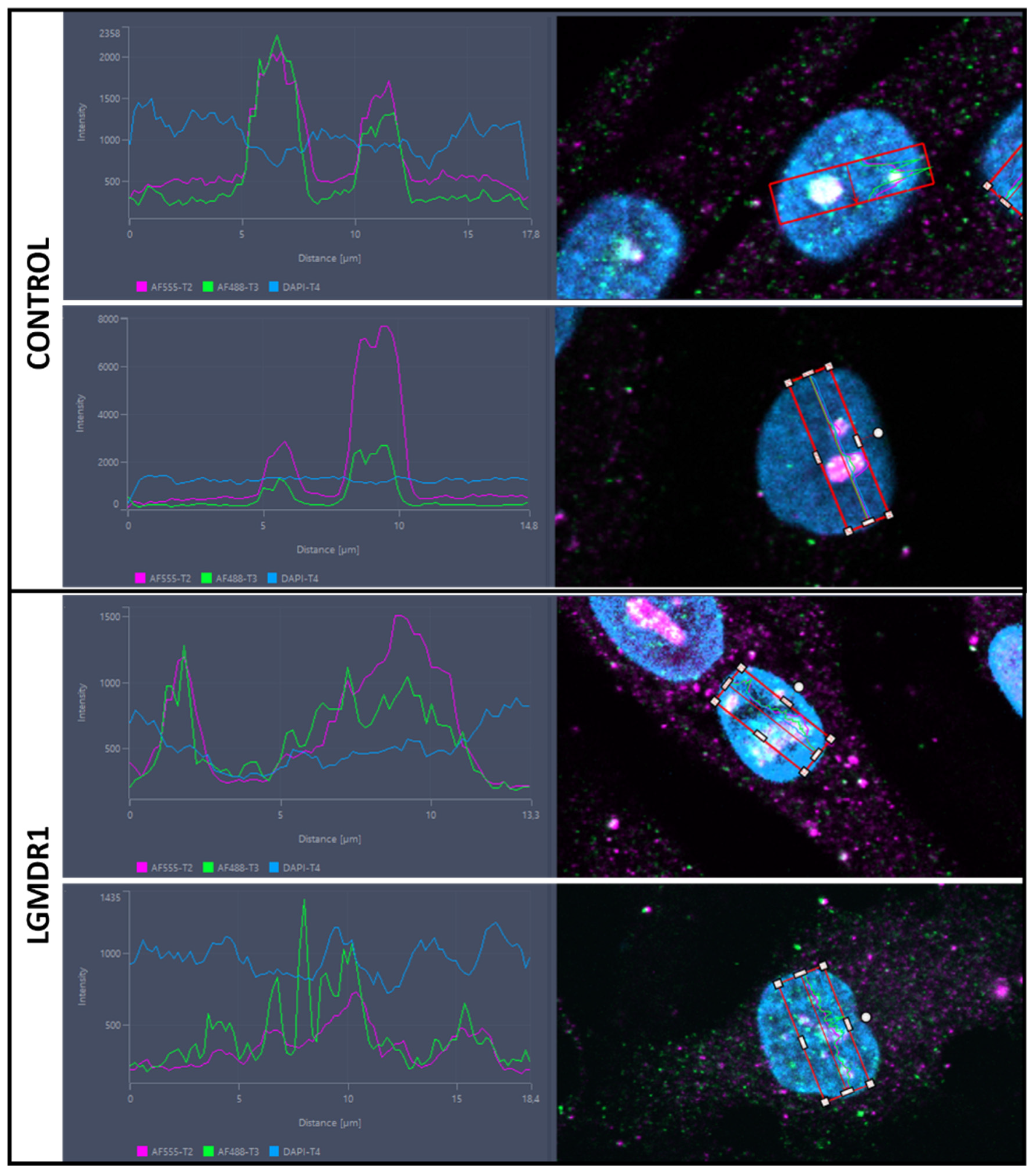
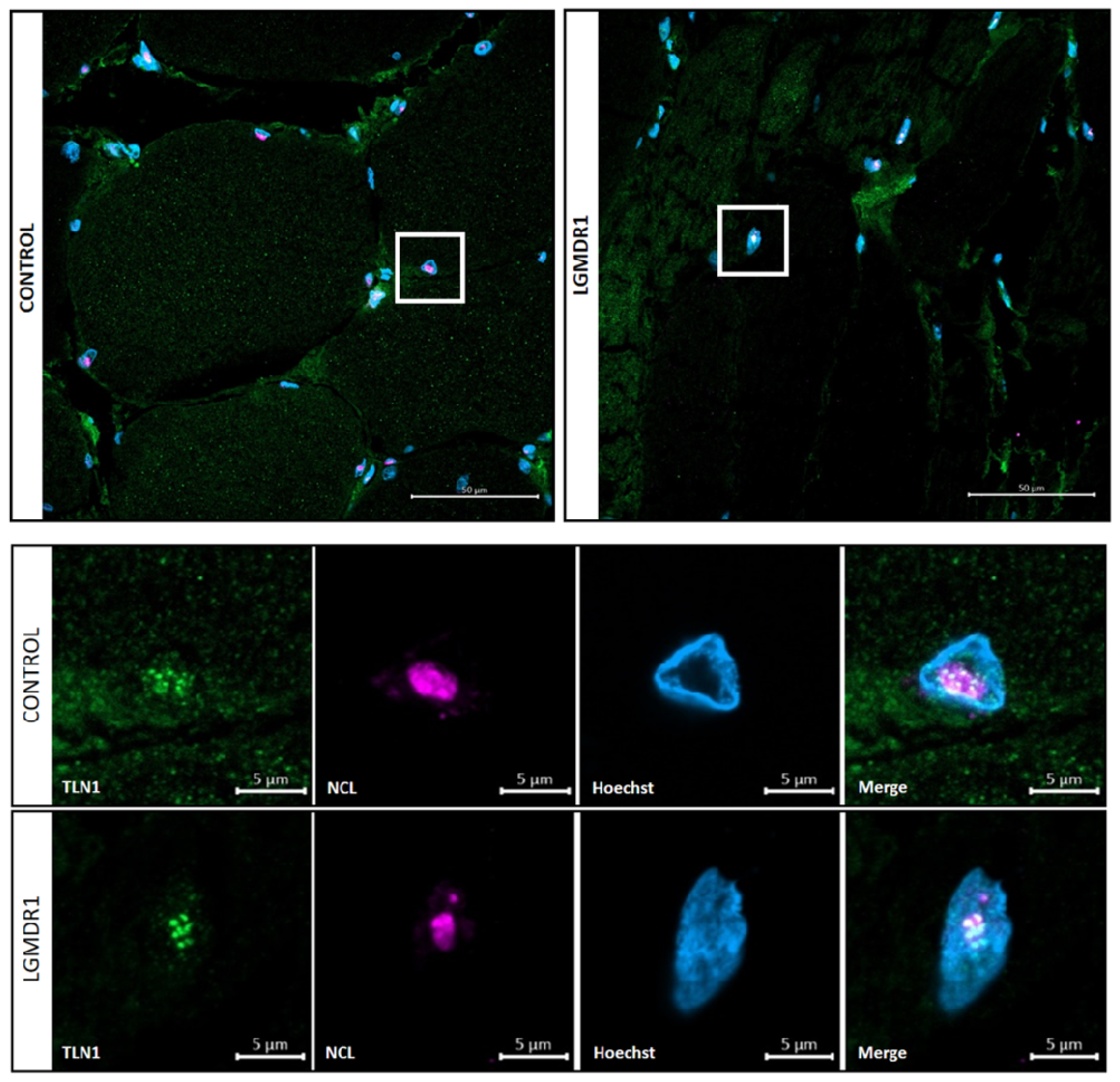
3.4. TLN1 Localizes in the Nucleolus in CD56− Cells, Myoblasts, Myotubes and Muscle
3.5. Abnormal Nuclear Morphology in LGMDR1 Patient Samples
3.6. Centrosome Organization Is Impaired in LGMDR1 Cells
3.7. Aberrant Mitosis in LGMDR1 CD56− Cells
4. Discussion
4.1. Fiber-Type Distribution in Healthy Muscle
4.2. Vascular Endothelium Alteration in LGMDR1
4.3. Implications of Altered ITGβ1D Distribution in LGMDR1
- (a)
- Impaired structure and function of muscle fibers
- (b)
- Nuclear alterations in muscle and cells
- (c)
- Centrosome alterations in muscle cells
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fardeau, M.; Hillaire, D.; Mignard, C.; Feingold, N.; Feingold, J.; Mignard, D.; De Ubeda, B.; Collin, H.; Tomé, F.M.S.; Richard, I.; et al. Juvenile Limb-Girdle Muscular Dystrophy Clinical, Histopathological and Genetic Data from a Small Community Living in the Reunion Island. Brain 1996, 119, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, M.; Sáenz, A.; Roudaut, C.; Poza, J.J.; Urtizberea, J.A.; Cobo, A.M.; Richard, I.; García Bragado, F.; Leturcq, F.; Kaplan, J.C.; et al. Limb-Girdle Muscular Dystrophy in Guipuzcoa (Basque Country, Spain). Brain 1998, 121, 1735–1747. [Google Scholar] [CrossRef]
- Sorimachi, H.; Imajoh-Ohmi, S.; Emori, Y.; Kawasaki, H.; Ohno, S.; Minami, Y.; Suzuki, K. Molecular Cloning of a Novel Mammalian Calcium-Dependent Protease Distinct from Both m- and μ-Types. Specif. Expr. MRNA Skelet. Muscle. J. Biol. Chem. 1989, 264, 20106–20111. [Google Scholar] [CrossRef]
- Sorimachi, H.; Toyama-Sorimachi, N.; Saido, T.C.; Kawasaki, H.; Sugita, H.; Miyasaka, M.; Arahata, K.; Ishiura, S.; Suzuki, K. Muscle-Specific Calpain, P94, Is Degraded by Autolysis Immediately after Translation, Resulting in Disappearance from Muscle. J. Biol. Chem. 1993, 268, 10593–10605. [Google Scholar] [CrossRef]
- Sorimachi, H.; Kinbara, K.; Kimura, S.; Takahashi, M.; Ishiura, S.; Sasagawa, N.; Sorimachi, N.; Shimada, H.; Tagawa, K.; Maruyama, K.; et al. Muscle-Specific Calpain, P94, Responsible for Limb Girdle Muscular Dystrophy Type 2A, Associates with Connectin through IS2, a P94-Specific Sequence. J. Biol. Chem. 1995, 270, 31158–31162. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Tidball, J.G.; Spencer, M.J. Null Mutation of Calpain 3 (P94) in Mice Causes Abnormal Sarcomere Formation In Vivo and In Vitro. Hum. Mol. Genet. 2004, 13, 1373–1388. [Google Scholar] [CrossRef]
- Sáenz, A.; Azpitarte, M.; Armañanzas, R.; Leturcq, F.; Alzualde, A.; Inza, I.; García-Bragado, F.; De la Herran, G.; Corcuera, J.; Cabello, A.; et al. Gene Expression Profiling in Limb-Girdle Muscular Dystrophy 2A. PLoS ONE 2008, 3, e3750. [Google Scholar] [CrossRef]
- Pardo, J.V.; Siliciano, J.D.; Craig, S.W. A Vinculin-Containing Cortical Lattice in Skeletal Muscle: Transverse Lattice Elements (“costameres”) Mark Sites of Attachment between Myofibrils and Sarcolemma. Proc. Natl. Acad. Sci. USA 1983, 80, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Danowski, B.A.; Imanaka-Yoshida, K.; Sanger, J.M.; Sanger, J.W. Costameres Are Sites of Force Transmission to the Substratum in Adult Rat Cardiomyocytes. J. Cell Biol. 1992, 118, 1411–1420. [Google Scholar] [CrossRef]
- Trimarchi, F.; Favaloro, A.; Fulle, S.; Magaudda, L.; Puglielli, C.; Di Mauro, D. Culture of Human Skeletal Muscle Myoblasts: Timing Appearance and Localization of Dystrophin-Glycoprotein Complex and Vinculin-Talin-Integrin Complex. Cells Tissues Organs 2006, 183, 87–98. [Google Scholar] [CrossRef]
- Jaka, O.; Casas-Fraile, L.; Azpitarte, M.; Aiastui, A.; López de Munain, A.; Sáenz, A. FRZB and Melusin, Overexpressed in LGMD2A, Regulate Integrin Β1D Isoform Replacement Altering Myoblast Fusion and the Integrin-Signalling Pathway. Expert. Rev. Mol. Med. 2017, 19, e2. [Google Scholar] [CrossRef]
- Pickett-Heaps, J.D. Preprophase Microtubule Bands in Some Abnormal Mitotic Cells of Wheat. J. Cell Sci. 1969, 4, 397–420. [Google Scholar] [CrossRef] [PubMed]
- Bornens, M. Centrosome Composition and Microtubule Anchoring Mechanisms. Curr. Opin. Cell Biol. 2002, 14, 25–34. [Google Scholar] [CrossRef]
- O’Toole, E.; Greenan, G.; Lange, K.I.; Srayko, M.; Müller-Reichert, T. The Role of γ-Tubulin in Centrosomal Microtubule Organization. PLoS ONE 2012, 7, e29795. [Google Scholar] [CrossRef] [PubMed]
- Tassin, A.M.; Maro, B.; Bornens, M. Fate of Microtubule-Organizing Centers during Myogenesis In Vitro. J. Cell Biol. 1985, 100, 35–46. [Google Scholar] [CrossRef]
- Bugnard, E.; Zaal, K.J.M.; Ralston, E. Reorganization of Microtubule Nucleation during Muscle Differentiation. Cell Motil. Cytoskelet. 2005, 60, 1–13. [Google Scholar] [CrossRef]
- de Andrade Rosa, I.; Corrêa, S.; Costa, M.L.; Mermelstein, C. The Scaffolding Protein Calpain-3 Has Multiple Distributions in Embryonic Chick Muscle Cells and It Is Essential for the Formation of Muscle Fibers. Tissue Cell 2020, 67, 101436. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Kustermann, M.; Ernhofer, B.; Hoger, H.; Bittner, R.E.; Schmidt, W.M. Proteins Implicated in Muscular Dystrophy and Cancer Are Functional Constituents of the Centrosome. Life Sci. Alliance 2022, 5, e202201367. [Google Scholar] [CrossRef]
- Peng, H.; Ong, Y.M.; Shah, W.A.; Holland, P.C.; Carbonetto, S. Integrins Regulate Centrosome Integrity and Astrocyte Polarization Following a Wound. Dev. Neurobiol. 2013, 73, 333–353. [Google Scholar] [CrossRef]
- Barker, A.R.; McIntosh, K.V.; Dawe, H.R. Centrosome Positioning in Non-Dividing Cells. Protoplasma 2016, 253, 1007–1021. [Google Scholar] [CrossRef]
- Satir, P. Landmarks in Cilia Research from Leeuwenhoek to US. Cell Motil. 1995, 32, 90–94. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of Structure and Function of Mammalian Cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef]
- Yuan, S.; Li, J.; Diener, D.R.; Choma, M.A.; Rosenbaum, J.L.; Sun, Z. Target-of-Rapamycin Complex 1 (Torc1) Signaling Modulates Cilia Size and Function through Protein Synthesis Regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 2021–2026. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, T.; Larsen, L.J.; Pedersen, L.B.; Christensen, S.T.; Møller, L.B. TSC1 and TSC2 Regulate Cilia Length and Canonical Hedgehog Signaling via Different Mechanisms. Cell. Mol. Life Sci. 2018, 75, 2663–2680. [Google Scholar] [CrossRef] [PubMed]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular Signalling by Primary Cilia in Development, Organ Function and Disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Jin, X.; Mohieldin, A.M.; Muntean, B.S.; Green, J.A.; Shah, J.V.; Mykytyn, K.; Nauli, S.M. Cilioplasm Is a Cellular Compartment for Calcium Signaling in Response to Mechanical and Chemical Stimuli. Cell. Mol. Life Sci. 2014, 71, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Rico, A.; Guembelzu, G.; Palomo, V.; Martínez, A.; Aiastui, A.; Casas-fraile, L.; Valls, A.; de Munain, A.L.; Sáenz, A. Allosteric Modulation of GSK-3β as a New Therapeutic Approach in Limb Girdle Muscular Dystrophy R1 Calpain 3-related. Int. J. Mol. Sci. 2021, 22, 7367. [Google Scholar] [CrossRef]
- Ding, F.; Huang, D.; Wang, M.; Peng, J. An 86 Amino Acids Motif in CAPN3 Is Essential for Formation of the Nucleolus-Localized Def-CAPN3 Complex. Biochem. Biophys. Res. Commun. 2022, 623, 66–73. [Google Scholar] [CrossRef]
- Schmidt, W.M.; Uddin, M.H.; Dysek, S.; Moser-Thier, K.; Pirker, C.; Höger, H.; Ambros, I.M.; Ambros, P.F.; Berger, W.; Bittner, R.E. DNA Damage, Somatic Aneuploidy, and Malignant Sarcoma Susceptibility in Muscular Dystrophies. PLoS Genet. 2011, 7, e1002042. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K. A new program for investigating adult human skeletal muscle grown aneurally in tissue culture. Neurology 1975, 25, 58–67. [Google Scholar] [CrossRef]
- Teixidó-Travesa, N.; Roig, J.; Lüders, J. The Where, When and How of Microtubule Nucleation—One Ring to Rule Them All. J. Cell Sci. 2012, 125, 4445–4456. [Google Scholar] [CrossRef] [PubMed]
- Tovey, C.A.; Conduit, P.T. Microtubule Nucleation by γ-Tubulin Complexes and Beyond. Essays Biochem. 2018, 62, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef]
- Mansour, H.; de Tombe, P.P.; Samarel, A.M.; Russell, B. Restoration of Resting Sarcomere Length After Uniaxial Static Strain Is Regulated by Protein Kinase Cε and Focal Adhesion Kinase. Circ. Res. 2004, 94, 642–649. [Google Scholar] [CrossRef]
- Ervasti, J.M. Costameres: The Achilles’ Heel of Herculean Muscle. J. Biol. Chem. 2003, 278, 13591–13594. [Google Scholar] [CrossRef]
- Hodges, B.L.; Kaufman, S.J. Developmental Regulation and Functional Significance of Alternative Splicing of NCAM and 7 1 Integrin in Skeletal Muscle. BAM-PADOVA 1996, 6, 437–448. [Google Scholar]
- Pardo, J.V.; Siliciano, J.D.; Craig, S.W. Vinculin Is a Component of an Extensive Network of Myofibril-Sarcolemma Attachment Regions in Cardiac Muscle Fibers. J. Cell Biol. 1983, 97, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, G.; Cutroneo, G.; Rizzo, G.; Arco, A.; Santoro, G.; Bramanti, P.; Vitetta, A.G.; Pisani, A.; Trimarchi, F.; Favaloro, A. Sarcoglycan and Integrin Localization in Normal Human Skeletal Muscle: A Confocal Laser Scanning Microscope Study. Eur. J. Histochem. 2004, 48, 245–252. [Google Scholar]
- Korthuis, R.J. Skeletal Muscle Circulation. In Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Publishers: San Rafael, CA, USA, 2011; Volume 3, pp. 1–144. [Google Scholar] [CrossRef]
- Di Tomaso, M.V.; Liddle, P.; Lafon-Hughes, L.; Laura, A.; Folle, G. Chromatin Damage Patterns Shift According to Eu/ Heterochromatin Replication. In The Mechanisms of DNA Replication; IntechOpen: London, UK, 2013. [Google Scholar]
- Reverte, C.G.; Benware, A.; Jones, C.W.; LaFlamme, S.E. Perturbing Integrin Function Inhibits Microtubule Growth from Centrosomes, Spindle Assembly, and Cytokinesis. J. Cell Biol. 2006, 174, 491–497. [Google Scholar] [CrossRef]
- Ng, D.C.H.; Ho, U.Y.; Grounds, M.D. Cilia, Centrosomes and Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 9605. [Google Scholar] [CrossRef]
- Park, R.S.; Légier, G.M.F.; Cartwright, J., Jr.; Goldstein, M.A. Perinuclear Microtubules in Postnatal Rat Heart. J. Morphol. 1984, 179, 13–19. [Google Scholar] [CrossRef]
- Espigat-Georger, A.; Dyachuk, V.; Chemin, C.; Emorine, L.; Merdes, A. Nuclear Alignment in Myotubes Requires Centrosome Proteins Recruited by Nesprin-1. J. Cell Sci. 2016, 129, 4227–4237. [Google Scholar] [CrossRef] [PubMed]
- Roman, W.; Gomes, E.R. Nuclear Positioning in Skeletal Muscle. Semin. Cell Dev. Biol. 2018, 82, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Khodjakov, A.; Cole, R.W.; Oakley, B.R.; Rieder, C.L. Centrosome-Independent Mitotic Spindle Formation in Vertebrates. Curr. Biol. 2000, 10, 59–67. [Google Scholar] [CrossRef]
- Mathes, S.; Vanmunster, M.; Bloch, W.; Suhr, F. Evidence for Skeletal Muscle Fiber Type-Specific Expressions of Mechanosensors. Cell. Mol. Life Sci. 2019, 76, 2987–3004. [Google Scholar] [CrossRef]
- Andresen, B.; de Marees, M.; Schiffer, T.; Bloch, W.; Suhr, F. Skeletal Muscle Fiber Type-Specific Expressions of Mechanosensors Integrin-Linked Kinase, Talin, and Vinculin and Their Modulation by Loading and Environmental Conditions in Humans. FASEB J. 2022, 36, e22458. [Google Scholar] [CrossRef] [PubMed]
- Graham, Z.A.; Gallagher, P.M.; Cardozo, C.P. Focal Adhesion Kinase and Its Role in Skeletal Muscle. J. Muscle Res. Cell Motil. 2015, 36, 305–315. [Google Scholar] [CrossRef]
- Turner, C.E.; Kramarcy, N.; Sealock, R.; Burridge, K. Localization of Paxillin, a Focal Adhesion Protein, to Smooth Muscle Dense Plaques, and the Myotendinous and Neuromuscular Junctions of Skeletal Muscle. Exp. Cell Res. 1991, 192, 651–655. [Google Scholar] [CrossRef]
- Draeger, A.; Amos, W.B.; Ikebe, M.; Small, J.V. The Cytoskeletal and Contractile Apparatus of Smooth Muscle: Contraction Bands and Segmentation of the Contractile Elements. J. Cell Biol. 1990, 111, 2463–2473. [Google Scholar] [CrossRef]
- Javerzat, S.; Franco, M.; Herbert, J.; Platonova, N.; Peille, A.-L.; Pantesco, V.; De Vos, J.; Assou, S.; Bicknell, R.; Bikfalvi, A.; et al. Correlating Global Gene Regulation to Angiogenesis in the Developing Chick Extra-Embryonic Vascular System. PLoS ONE 2009, 4, e7856. [Google Scholar] [CrossRef]
- Sit, B.; Gutmann, D.; Iskratsch, T. Costameres, Dense Plaques and Podosomes: The Cell Matrix Adhesions in Cardiovascular Mechanosensing. J. Muscle Res. Cell Motil. 2019, 40, 197–209. [Google Scholar] [CrossRef]
- Yamaji, S.; Suzuki, A.; Sugiyama, Y.; Koide, Y.I.; Yoshida, M.; Kanamori, H.; Mohri, H.; Ohno, S.; Ishigatsubo, Y. A Novel Integrin-Linked Kinase-Binding Protein, Affixin, Is Involved in the Early Stage of Cell-Substrate Interaction. J. Cell Biol. 2001, 153, 1251–1264. [Google Scholar] [CrossRef]
- Matsuda, C.; Kameyama, K.; Tagawa, K.; Ogawa, M.; Suzuki, A.; Yamaji, S.; Okamoto, H.; Nishino, I.; Hayashi, Y.K. Dysferlin Interacts with Affixin (β-Parvin) at the Sarcolemma. J. Neuropathol. Exp. Neurol. 2005, 64, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, J.L.; Gkretsi, V.; Wu, C. Assembly and Signaling of Adhesion Complexes. Curr. Top. Dev. Biol. 2005, 68, 183–225. [Google Scholar]
- Budatha, M.; Zhang, J.; Schwartz, M.A. Fibronectin-Mediated Inflammatory Signaling Through Integrin A5 in Vascular Remodeling. J. Am. Heart Assoc. 2021, 10, e021160. [Google Scholar] [CrossRef] [PubMed]
- Krahn, M.; Goicoechea, M.; Hanisch, F.; Groen, E.; Bartoli, M.; Pécheux, C.; Garcia-Bragado, F.; Leturcq, F.; Jeannet, P.Y.; Lobrinus, J.A.; et al. Eosinophilic Infiltration Related to CAPN3 Mutations: A Pathophysiological Component of Primary Calpainopathy? Clin. Genet. 2011, 80, 398–402. [Google Scholar] [CrossRef]
- Pitter, B.; Werner, A.-C.; Montanez, E. Parvins Are Required for Endothelial Cell–Cell Junctions and Cell Polarity During Embryonic Blood Vessel Formation. Arter. Thromb. Vasc. Biol. 2018, 38, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Immunopathogenesis of Inflammatory Myopathies. Ann. Neurol. 1995, 37, 74–86. [Google Scholar] [CrossRef]
- Thompson, C.; Piguet, V.; Choy, E. The Pathogenesis of Dermatomyositis. Br. J. Dermatol. 2018, 179, 1256–1262. [Google Scholar] [CrossRef]
- Palecek, S.P.; Schmidt, C.E.; Lauffenburger, D.A.; Horwitz, A.F. Integrin Dynamics on the Tail Region of Migrating Fibroblasts. J. Cell Sci. 1996, 109, 941–952. [Google Scholar] [CrossRef]
- Schwander, M.; Leu, M.; Stumm, M.; Dorchies, O.M.; Ruegg, U.T.; Schittny, J.; Müller, U. Β1 Integrins Regulate Myoblast Fusion and Sarcomere Assembly. Dev. Cell 2003, 4, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Chen, C.S.; Ingber, D.E. Demonstration of Mechanical Connections between Integrins, Cytoskeletal Filaments, and Nucleoplasm That Stabilize Nuclear Structure. Proc. Natl. Acad. Sci. USA 1997, 94, 849–854. [Google Scholar] [CrossRef]
- Thomas, C.H.; Collier, J.H.; Sfeir, C.S.; Healy, K.E. Engineering Gene Expression and Protein Synthesis by Modulation of Nuclear Shape. Proc. Natl. Acad. Sci. USA 2002, 99, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cook, P.C.; Zindy, E.; Williams, C.J.; Jowitt, T.A.; Streuli, C.H.; MacDonald, A.S.; Redondo-Muñoz, J. Integrin A4β1 Controls G9a Activity That Regulates Epigenetic Changes and Nuclear Properties Required for Lymphocyte Migration. Nucleic Acids Res. 2015, 44, 3031–3044. [Google Scholar] [CrossRef] [PubMed]
- Zwerger, M.; Ho, C.Y.; Lammerding, J. Nuclear Mechanics in Disease. Annu. Rev. Biomed. Eng. 2011, 13, 397–428. [Google Scholar] [CrossRef]
- Belkin, A.M.; Zhidkova, N.I.; Balzac, F.; Altruda, F.; Tomatis, D.; Maier, A.; Tarone, G.; Koteliansky, V.E.; Burridge, K. Β1D Integrin Displaces the Β1A Isoform in Striated Muscles: Localization at Junctional Structures and Signaling Potential in Nonmuscle Cells. J. Cell Biol. 1996, 132, 211–226. [Google Scholar] [CrossRef]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Spencer, M.J. Regulation of the M-Cadherin-β-Catenin Complex by Calpain 3 during Terminal Stages of Myogenic Differentiation. Mol. Cell Biol. 2006, 26, 8437–8447. [Google Scholar] [CrossRef]
- Papageorgiou, A.P.; Heymans, S. Interactions between the Extracellular Matrix and Inflammation during Viral Myocarditis. Immunobiology 2012, 217, 503–510. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Popovich, P.G. Extracellular Matrix Regulation of Inflammation in the Healthy and Injured Spinal Cord. Exp. Neurol. 2014, 258, 24–34. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Bogdani, M.; Bollyky, J.B.; Hull, R.L.; Wight, T.N. The Role of Hyaluronan and the Extracellular Matrix in Islet Inflammation and Immune Regulation. Curr. Diab. Rep. 2012, 12, 471–480. [Google Scholar] [CrossRef]
- Yun, S.; Budatha, M.; Dahlman, J.E.; Coon, B.G.; Ryan, T.; Langer, R.; Anderson, D.G.; Baillie, G.; Martin, A. Interaction between integrin α5 and PDE4D regulates endothelial inflammatory signalling. Nat. Cell Biol. 2016, 18, 1043–1053. [Google Scholar] [CrossRef]
- Yurdagul, A.; Green, J.; Albert, P.; McInnis, M.C.; Mazar, A.P.; Orr, A.W. A5Β1 Integrin Signaling Mediates Oxidized Low-Density Lipoprotein-Induced Inflammation and Early Atherosclerosis. Arter. Thromb. Vasc. Biol. 2014, 34, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Ginsberg, M.H.; Shattil, S.J.; Deckmyn, H.; Schwartz, M.A. Matrix-Specific Suppression of Integrin Activation in Shear Stress Signaling. Mol. Biol. Cell 2006, 17, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Tejeda-Muñoz, N.; Morselli, M.; Moriyama, Y.; Sheladiya, P.; Pellegrini, M.; De Robertis, E.M. Canonical Wnt Signaling Induces Focal Adhesion and Integrin Beta-1 Endocytosis. iScience 2022, 25, 104123. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Zent, R.; Grant, R.; Rees, D.J.G.; Hynes, R.O.; Ginsberg, M.H. The Talin Head Domain Binds to Integrin β Subunit Cytoplasmic Tails and Regulates Integrin Activation. J. Biol. Chem. 1999, 274, 28071–28074. [Google Scholar] [CrossRef]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; De Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin Binding to Integrin β Tails: A Final Common Step in Integrin Activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and Paxillin Dynamics at Focal Adhesions in the Protrusions of Migrating Cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef]
- Bellis, S.L.; Miller, J.T.; Turner, C.E. Characterization of Tyrosine Phosphorylation of Paxillin in Vitro by Focal Adhesion Kinase. J. Biol. Chem. 1995, 270, 17437–17441. [Google Scholar] [CrossRef]
- Boppart, M.D.; Mahmassani, Z.S. Integrin Signaling: Linking Mechanical Stimulation to Skeletal Muscle Hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef]
- Hour, T.C.; Lan Nhi, N.T.; Lai, I.J.; Chuu, C.P.; Lin, P.C.; Chang, H.W.; Su, Y.F.; Chen, C.H.; Chen, Y.K. Kaempferol-Enhanced Migration and Differentiation of C2C12 Myoblasts via ITG1B/FAK/Paxillin and IGF1R/AKT/MTOR Signaling Pathways. Mol. Nutr. Food Res. 2024, 68, e2300685. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wu, C.; Liu, Z.; Zhao, S.; Cheng, X.; Huang, J. SULF1 Regulates Malignant Progression of Colorectal Cancer by Modulating ARSH via FAK/PI3K/AKT/MTOR Signaling. Cancer Cell Int. 2024, 24, 201. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Tedrow, J.R.; Burgkart, R. Viscoelastic Properties of the Cell Nucleus. Biochem. Biophys. Res. Commun. 2000, 269, 781–786. [Google Scholar] [CrossRef]
- Rowat, A.C.; Lammerding, J.; Herrmann, H.; Aebi, U. Towards an Integrated Understanding of the Structure and Mechanics of the Cell Nucleus. BioEssays 2008, 30, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.J.; Hästbacka, H.S.E.; Puustinen, M.C.; Pessa, J.C.; Luoto, J.C.; Sundström, E.; Goult, B.T.; Jacquemet, G.; Henriksson, E.; Sistonen, L. Nuclear talin-1 provides a bridge between cell adhesion and gene expression. iScience 2025, 28, 111745. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. Disruption of the Nuclear Organisation of P53 in Response to DNA Stress. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef]
- Yang, K.; Yang, J.; Yi, J. Nucleolar Stress: Hallmarks, Sensing Mechanism and Diseases. Cell Stress 2018, 2, 125–140. [Google Scholar] [CrossRef]
- Goudarzi, K.M.; Nistér, M.; Lindström, M.S. MTOR Inhibitors Blunt the P53 Response to Nucleolar Stress by Regulating RPL11 and MDM2 Levels. Cancer Biol. Ther. 2014, 15, 1499–1514. [Google Scholar] [CrossRef]
- Rico, A.; Valls, A.; Guembelzu, G.; Azpitarte, M.; Aiastui, A.; Zufiria, M.; Jaka, O.; López de Munain, A.; Sáenz, A. Altered Expression of Proteins Involved in Metabolism in LGMDR1 Muscle Is Lost in Cell Culture Conditions. Orphanet J. Rare Dis. 2023, 18, 315. [Google Scholar] [CrossRef]
- LaFlamme, S.E.; Mathew-Steiner, S.; Singh, N.; Colello-Borges, D.; Nieves, B. Integrin and Microtubule Crosstalk in the Regulation of Cellular Processes. Cell. Mol. Life Sci. 2018, 75, 4177–4185. [Google Scholar] [CrossRef]
- Taranum, S.; Vaylann, E.; Meinke, P.; Abraham, S.; Yang, L.; Neumann, S.; Karakesisoglou, I.; Wehnert, M.; Noegel, A.A. LINC Complex Alterations in DMD and EDMD/CMT Fibroblasts. Eur. J. Cell Biol. 2012, 91, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Oakley, B.R. An Abundance of Tubulins. Trends Cell Biol. 2000, 10, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Robinson, M.L.; Lambert, L.; Srivastava, O.P. Lens-Specific ΒA3/A1-Conditional Knockout Mice: Phenotypic Characteristics and Calpain Activation Causing Protein Degradation and Insolubilization. PLoS ONE 2023, 18, e0281386. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, K.M.; Anstine, L.J.; Keri, R.A. Centrosome Aberrations as Drivers of Chromosomal Instability in Breast Cancer. Endocrinology 2021, 162, bqab208. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biopsy Number | Gender | Status | Mutation 1 | Mutation 2 | Muscle | Age # | Age at Onset | Functional Status | Clinical Information |
|---|---|---|---|---|---|---|---|---|---|
| 22-09 | Male | Control | - | - | Biceps | 47 | - | - | - |
| 22-18 | Male | Control | - | - | Semitendinosus | 24 | - | - | - |
| 18-08051 | Female | Control | - | - | Quadriceps | 46 | - | - | - |
| 23-10 | Male | Control | - | - | Deltoid | 75 | - | - | - |
| 16-6279 | Male | Control | - | - | Quadriceps | 35 | - | - | - |
| 19-22 | Female | LGMDR1 | p.(Arg788Serfs*14) | Complete deletion of CANP3 gene | Biceps | 23 | 23 | Ambulant | Benign |
| 21-07396 | Male | LGMDR1 | p.(Arg490Trp) | p.(Arg490Trp) | Tibialis anterior | 46 | 33 | Ambulant | Mild facial weakness. Proximal weakness. Bilateral scapular winging. Bilateral atrophy of biceps and pectoral muscles. |
| B09-83 | Female | LGMDR1 | DelEx2-6 | DelEx2-6 | Quadriceps | 12 | 12 | Ambulant | Proximal weakness. Not able to climb stairs. |
| 18-38110 | Female | LGMDR1 | p.(Arg489Gln) | c.1116-2A>C | Biceps | 47 | Fifth decade | Ambulant | Initial symptoms were myalgia and fatigue, with persistently elevated CK, X9 (1800 UI/l). No muscle weakness at 52 years old. Unspecific slight changes in the biopsy. |
| 97-168 | Male | LGMDR1 | p.(Ser479Gly) | c.1992+1G>T | N.A. | 41 | 20 | Ambulant | Muscle weakness of the pelvic and scapular girdles. |
| Biopsy N. | Status | Muscle | Age | Mutation 1 | Mutation 2 |
|---|---|---|---|---|---|
| 15-12 | CONTROL | Deltoid | 36 | - | - |
| 13-07 | CONTROL | Deltoid | 36 | - | - |
| 13-09 | CONTROL | Vastus lateralis | 37 | - | - |
| 22-18 | CONTROL | Semitendinosus | 24 | - | - |
| 23-10 | CONTROL | Deltoid | 75 | - | - |
| 10-39 | LGMDR1 | Deltoid | 29 | p.(Lys254del) | p.(X822Leuext62X) |
| 09-21 | LGMDR1 | Biceps | 19 | p.(His690Argfs*9) | p.(His690Argfs*9) |
| 09-25 | LGMDR1 | Deltoid | 28 | p.(Lys254Glu) | p.(Pro637HisfsX25) |
| Exp05 | LGMDR1 | Deltoid | 13 | p.(Arg788SersX14) | p.(Arg788SersX14) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valls, A.; Ruiz-Roldán, C.; Immanuel, J.; Alonso-Martín, S.; Gallardo, E.; Fernández-Torrón, R.; Bonilla, M.; Lersundi, A.; Hernández-Laín, A.; Domínguez-González, C.; et al. The Role of Integrin β1D Mislocalization in the Pathophysiology of Calpain 3-Related Limb–Girdle Muscular Dystrophy. Cells 2025, 14, 446. https://doi.org/10.3390/cells14060446
Valls A, Ruiz-Roldán C, Immanuel J, Alonso-Martín S, Gallardo E, Fernández-Torrón R, Bonilla M, Lersundi A, Hernández-Laín A, Domínguez-González C, et al. The Role of Integrin β1D Mislocalization in the Pathophysiology of Calpain 3-Related Limb–Girdle Muscular Dystrophy. Cells. 2025; 14(6):446. https://doi.org/10.3390/cells14060446
Chicago/Turabian StyleValls, Andrea, Cristina Ruiz-Roldán, Jenita Immanuel, Sonia Alonso-Martín, Eduard Gallardo, Roberto Fernández-Torrón, Mario Bonilla, Ana Lersundi, Aurelio Hernández-Laín, Cristina Domínguez-González, and et al. 2025. "The Role of Integrin β1D Mislocalization in the Pathophysiology of Calpain 3-Related Limb–Girdle Muscular Dystrophy" Cells 14, no. 6: 446. https://doi.org/10.3390/cells14060446
APA StyleValls, A., Ruiz-Roldán, C., Immanuel, J., Alonso-Martín, S., Gallardo, E., Fernández-Torrón, R., Bonilla, M., Lersundi, A., Hernández-Laín, A., Domínguez-González, C., Vílchez, J. J., Iruzubieta, P., López de Munain, A., & Sáenz, A. (2025). The Role of Integrin β1D Mislocalization in the Pathophysiology of Calpain 3-Related Limb–Girdle Muscular Dystrophy. Cells, 14(6), 446. https://doi.org/10.3390/cells14060446