

Chromosomal Instability Is Associated with cGAS–STING Activation in EGFR-TKI Refractory Non-Small-Cell Lung Cancer
 , , , ,
, , , ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Study Design and Acquisition of Tumor Samples and Clinical Data
2.2. DNA and RNA Extraction
2.3. Molecular Pathway Analysis
2.4. Computational Methods for Analysis of RNA-Seq Data and CIN Score
2.5. Vector Engineering and Transfection
2.6. Imaging of Spindle Morphology and CIN
2.7. Immunoblotting
2.8. Colony-Formation Assay
2.9. In Vivo Tumor Growth Inhibition Assay
2.10. Immunohistochemical Staining
2.11. Statistical Analysis
3. Results
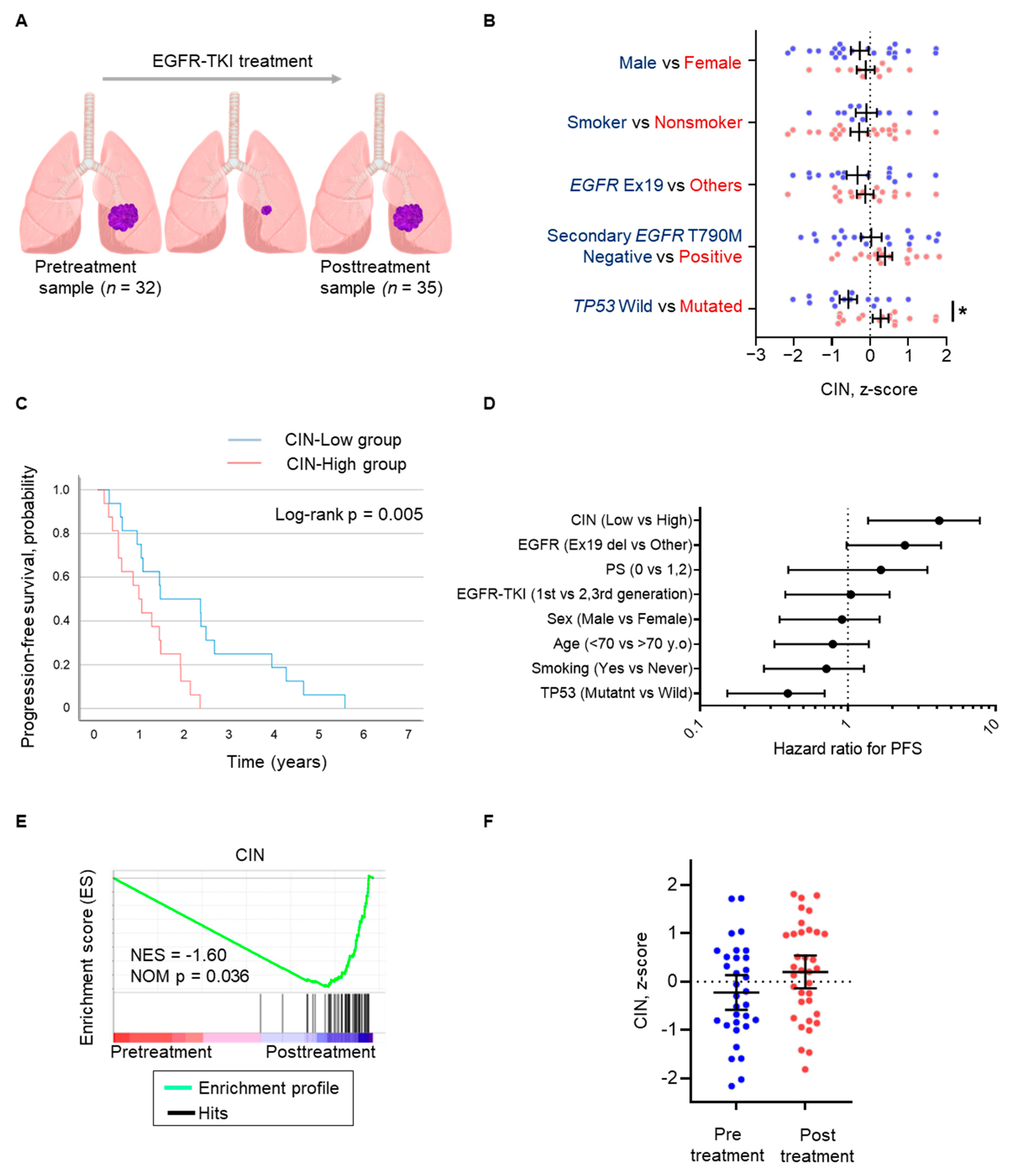
3.1. Influence of CIN on Outcomes of EGFR-Mutated NSCLC Treated with EGFR-TKIs
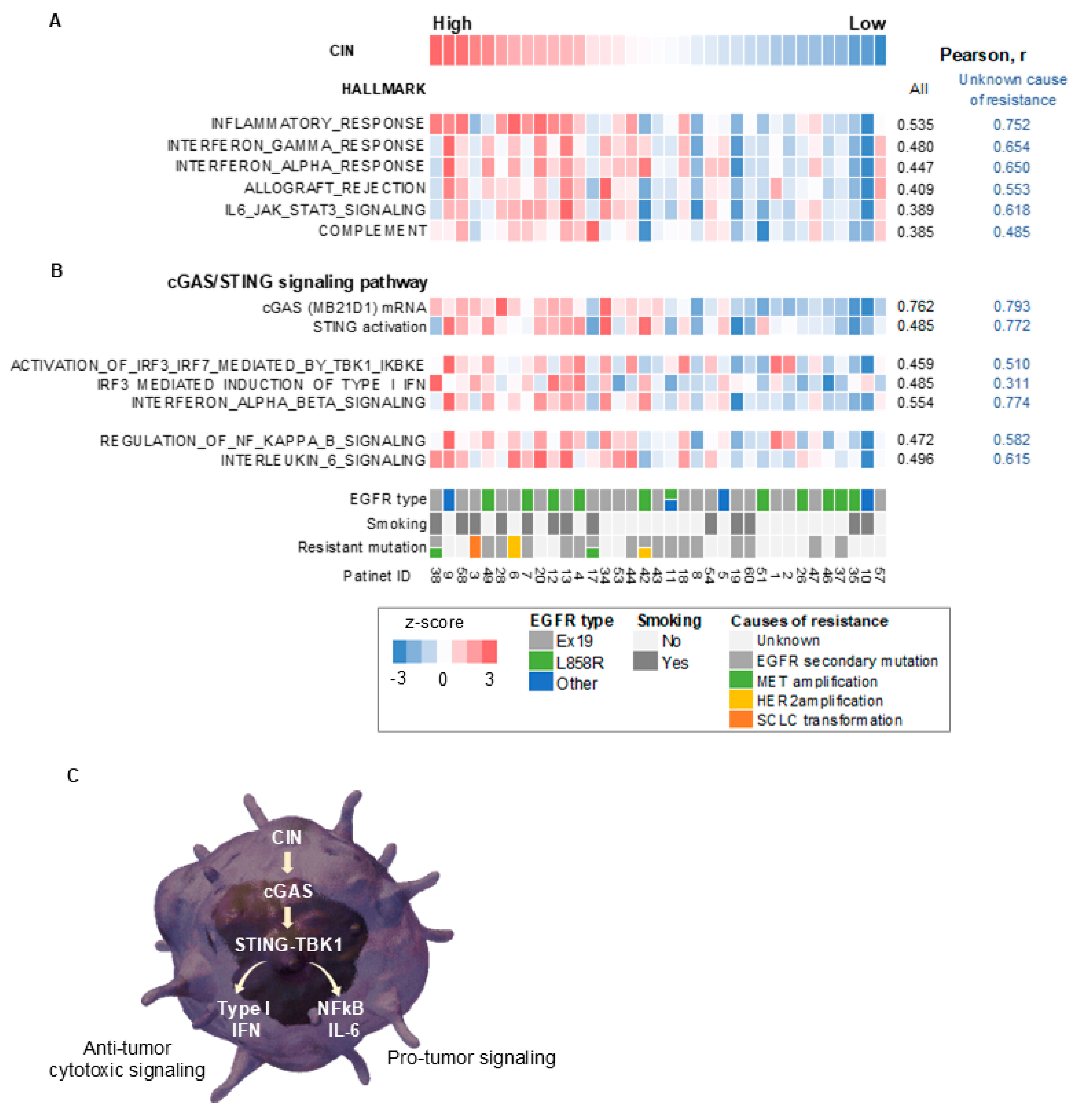
3.2. CIN Activates an Innate Immune Response in EGFR-Mutated NSCLC
3.3. Chromosomal Instability Induces EGFR-TKI Refractoriness
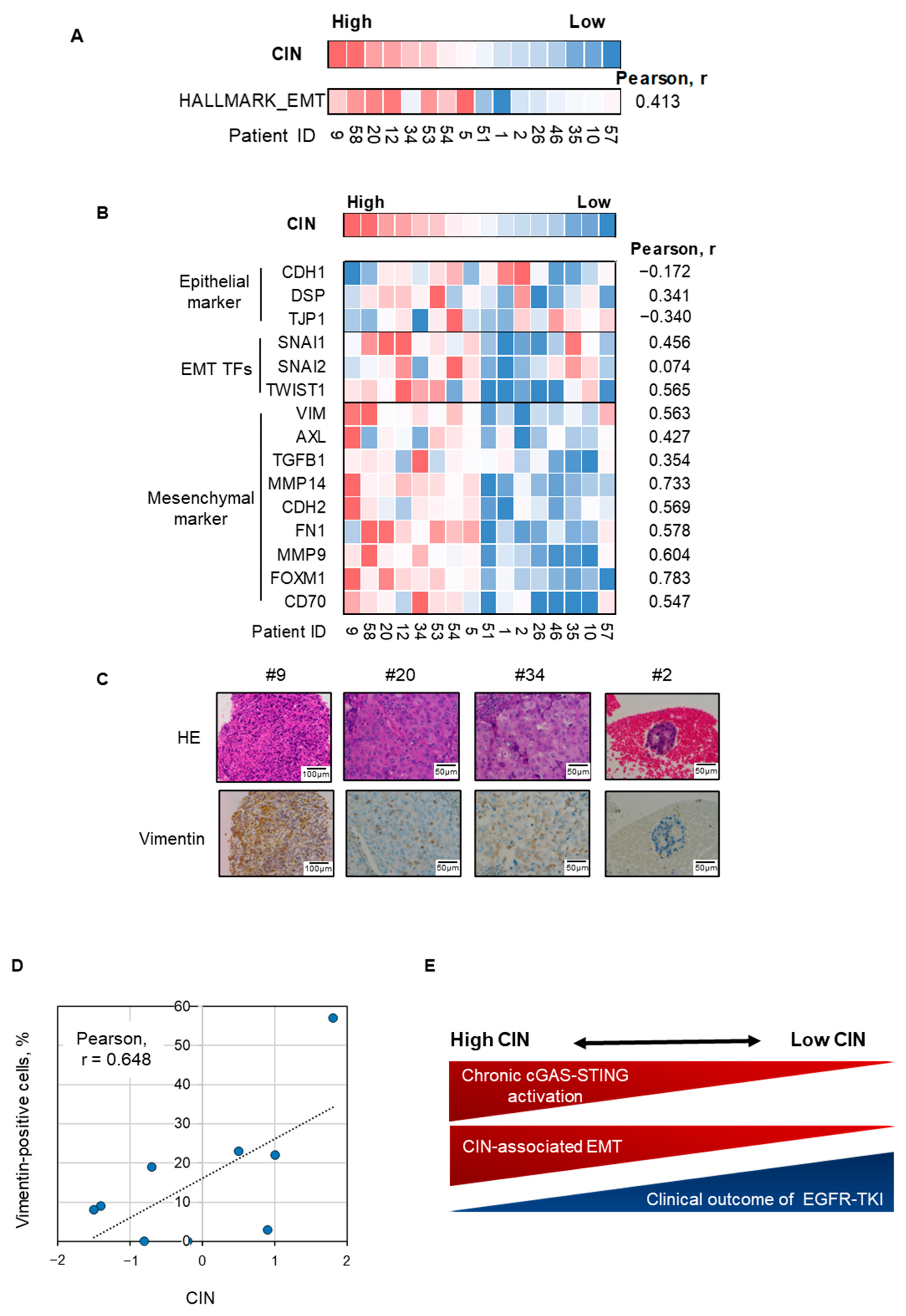
3.4. EMT Is Associated with Activated cGAS–STING Signaling in Chromosomally Unstable NSCLC with EGFR-Activating Mutation
3.5. Epithelial–Mesenchymal Plasticity Observed in Chromosomally Unstable NSCLC Tumor Harboring EGFR-Activating Mutation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Ponce Aix, S.; Paz-Ares, L.; Chiu, C.H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Vokes, N.I.; Chambers, E.; Nguyen, T.; Coolidge, A.; Lydon, C.A.; Le, X.; Sholl, L.; Heymach, J.V.; Nishino, M.; Van Allen, E.M.; et al. Concurrent TP53 Mutations Facilitate Resistance Evolution in EGFR-Mutant Lung Adenocarcinoma. J. Thorac. Oncol. 2022, 17, 779–792. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493-503. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Sun, H.; Diao, L.; Tong, P.; Liu, D.; Li, L.; Fan, Y.; Poteete, A.; Lim, S.O.; Howells, K.; et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with β-blockers. Sci. Transl. Med. 2017, 9, eaao4307. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef]
- Gong, K.; Guo, G.; Gerber, D.E.; Gao, B.; Peyton, M.; Huang, C.; Minna, J.D.; Hatanpaa, K.J.; Kernstine, K.; Cai, L.; et al. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J. Clin. Investig. 2018, 128, 2500–2518. [Google Scholar] [CrossRef]
- Ibusuki, R.; Iwama, E.; Shimauchi, A.; Tsutsumi, H.; Yoneshima, Y.; Tanaka, K.; Okamoto, I. TP53 gain-of-function mutations promote osimertinib resistance via TNF-α-NF-κB signaling in EGFR-mutated lung cancer. NPJ Precis. Oncol. 2024, 8, 60. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Lin, P.; Chen, Y.; Xu, J.; Huang, X.; Wen, W.; Zhang, L.; Kong, W.; Zhao, Z.; Ye, Y.; Bao, Z.; et al. A multicenter-retrospective cohort study of chromosome instability in lung cancer: Clinical characteristics and prognosis of patients harboring chromosomal instability detected by metagenomic next-generation sequencing. J. Thorac. Dis. 2023, 15, 112–122. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Lanng, K.R.B.; Lauridsen, E.L.; Jakobsen, M.R. The balance of STING signaling orchestrates immunity in cancer. Nat. Immunol. 2024, 25, 1144–1157. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sweis, R.F.; Kasper, S.; Hamid, O.; Bhatia, S.; Dummer, R.; Stradella, A.; Long, G.V.; Spreafico, A.; Shimizu, T.; et al. Combination of the STING Agonist MIW815 (ADU-S100) and PD-1 Inhibitor Spartalizumab in Advanced/Metastatic Solid Tumors or Lymphomas: An Open-Label, Multicenter, Phase Ib Study. Clin. Cancer Res. 2023, 29, 110–121. [Google Scholar] [CrossRef]
- Moser, J.C.; Alistar, A.; Cohen, E.; Garmey, E.; Kazmi, S.; Mooneyham, T.; Sun, L.; Yap, T.; Mahalingam, D. 618 Phase 1 clinical trial evaluating the safety, biologic and anti-tumor activity of the novel STING agonist IMSA101 administered both as monotherapy and in combination with PD-(L) 1 checkpoint inhibitors. J. Immunother. Cancer 2023, 11. [Google Scholar] [CrossRef]
- Li, J.; Hubisz, M.J.; Earlie, E.M.; Duran, M.A.; Hong, C.; Varela, A.A.; Lettera, E.; Deyell, M.; Tavora, B.; Havel, J.J.; et al. Non-cell-autonomous cancer progression from chromosomal instability. Nature 2023, 620, 1080–1088. [Google Scholar] [CrossRef]
- Yonesaka, K.; Tanizaki, J.; Maenishi, O.; Haratani, K.; Kawakami, H.; Tanaka, K.; Hayashi, H.; Sakai, K.; Chiba, Y.; Tsuya, A.; et al. HER3 Augmentation via Blockade of EGFR/AKT Signaling Enhances Anticancer Activity of HER3-Targeting Patritumab Deruxtecan in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 390–403. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef]
- Lu, X.; Wang, S.; Hua, X.; Chen, X.; Zhan, M.; Hu, Q.; Cao, L.; Wu, Z.; Zhang, W.; Zuo, X.; et al. Targeting the cGAS-STING Pathway Inhibits Peripheral T-cell Lymphoma Progression and Enhances the Chemotherapeutic Efficacy. Adv. Sci. 2024, 11, e2306092. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L.; et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci. Transl. Med. 2020, 12, eaaz4589. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Jin, Y.; Bao, H.; Le, X.; Fan, X.; Tang, M.; Shi, X.; Zhao, J.; Yan, J.; Xu, Y.; Quek, K.; et al. Distinct co-acquired alterations and genomic evolution during TKI treatment in non-small-cell lung cancer patients with or without acquired T790M mutation. Oncogene 2020, 39, 1846–1859. [Google Scholar] [CrossRef]
- Chmielecki, J.; Gray, J.E.; Cheng, Y.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat. Commun. 2023, 14, 1070. [Google Scholar] [CrossRef]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 2021, 56, 2427–2439.e2424. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, Q.; Jiang, T.; Wang, W.; Zhao, J.J. Targeting tumor-associated macrophages with STING agonism improves the antitumor efficacy of osimertinib in a mouse model of EGFR-mutant lung cancer. Front. Immunol. 2023, 14, 1077203. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Evans, R.; Green, R.; An, Z.; Deng, J.; Treacy, C.; Mustapha, R.; Monypenny, J.; Costoya, C.; Lawler, K.; et al. Osimertinib and anti-HER3 combination therapy engages immune dependent tumor toxicity via STING activation in trans. Cell Death Dis. 2022, 13, 274. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.Y.; Shen, Y.J.; Zhang, C.C.; Qian, L.Q.; Wang, H.; Wang, K.; Jin, H.Z.; Zhu, X.R.; Ding, Z.P.; Zhang, Q.; et al. Combination of radiotherapy and PD-L1 blockade induces abscopal responses in EGFR-mutated lung cancer through activating CD8. Transl. Oncol. 2024, 48, 102074. [Google Scholar] [CrossRef] [PubMed]
- Barriga, F.M.; Tsanov, K.M.; Ho, Y.J.; Sohail, N.; Zhang, A.; Baslan, T.; Wuest, A.N.; Del Priore, I.; Meškauskaitė, B.; Livshits, G.; et al. MACHETE identifies interferon-encompassing chromosome 9p21.3 deletions as mediators of immune evasion and metastasis. Nat. Cancer 2022, 3, 1367–1385. [Google Scholar] [CrossRef]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; Fang, J.M.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Lorenzoni, P.R.; Mombach, J.C.M.; Hashimoto, R.F. LncRNA PTENP1/miR-21/PTEN Axis Modulates EMT and Drug Resistance in Cancer: Dynamic Boolean Modeling for Cell Fates in DNA Damage Response. Int. J. Mol. Sci. 2024, 25, 8264. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef]
- Ma, F.; Li, B.; Liu, S.Y.; Iyer, S.S.; Yu, Y.; Wu, A.; Cheng, G. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 2015, 194, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Al-Rawi, D.H.; Lettera, E.; Li, J.; DiBona, M.; Bakhoum, S.F. Targeting chromosomal instability in patients with cancer. Nat. Rev. Clin. Oncol. 2024, 21, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Di Bona, M.; Bakhoum, S.F. Micronuclei and Cancer. Cancer Discov. 2024, 14, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yonesaka, K.; Kurosaki, T.; Tanizaki, J.; Kawakami, H.; Tanaka, K.; Maenishi, O.; Takamura, S.; Sakai, K.; Chiba, Y.; Teramura, T.; et al. Chromosomal Instability Is Associated with cGAS–STING Activation in EGFR-TKI Refractory Non-Small-Cell Lung Cancer. Cells 2025, 14, 447. https://doi.org/10.3390/cells14060447
Yonesaka K, Kurosaki T, Tanizaki J, Kawakami H, Tanaka K, Maenishi O, Takamura S, Sakai K, Chiba Y, Teramura T, et al. Chromosomal Instability Is Associated with cGAS–STING Activation in EGFR-TKI Refractory Non-Small-Cell Lung Cancer. Cells. 2025; 14(6):447. https://doi.org/10.3390/cells14060447
Chicago/Turabian StyleYonesaka, Kimio, Takashi Kurosaki, Junko Tanizaki, Hisato Kawakami, Kaoru Tanaka, Osamu Maenishi, Shiki Takamura, Kazuko Sakai, Yasutaka Chiba, Takeshi Teramura, and et al. 2025. "Chromosomal Instability Is Associated with cGAS–STING Activation in EGFR-TKI Refractory Non-Small-Cell Lung Cancer" Cells 14, no. 6: 447. https://doi.org/10.3390/cells14060447
APA StyleYonesaka, K., Kurosaki, T., Tanizaki, J., Kawakami, H., Tanaka, K., Maenishi, O., Takamura, S., Sakai, K., Chiba, Y., Teramura, T., Goto, H., Otsuka, E., Okida, H., Funabashi, M., Hashimoto, Y., Hirotani, K., Kamai, Y., Kagari, T., Nishio, K., ... Hayashi, H. (2025). Chromosomal Instability Is Associated with cGAS–STING Activation in EGFR-TKI Refractory Non-Small-Cell Lung Cancer. Cells, 14(6), 447. https://doi.org/10.3390/cells14060447