
A Simple and Scalable Assay for Multiplexed Flow Cytometric Profiling of Surface Markers on Small Extracellular Vesicles
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Background on EVs
1.2. Challenges in Current Methods for EVs Protein Profiling
1.3. The Rationale and Aim of Our Study
2. Materials and Methods
2.1. Cell Cultures
2.2. Metabolic Labelling of sEVs
2.3. CFSE Labelling of sEVs
2.4. sEVs Isolation by Differential Ultracentrifugation
2.5. NanoFACS Analysis of Fluorescent sEVs
2.6. Bodipy-Exo and CFSE-sEVs Immunostaining
2.7. Western Blot
2.8. Statistical Analysis
3. Results
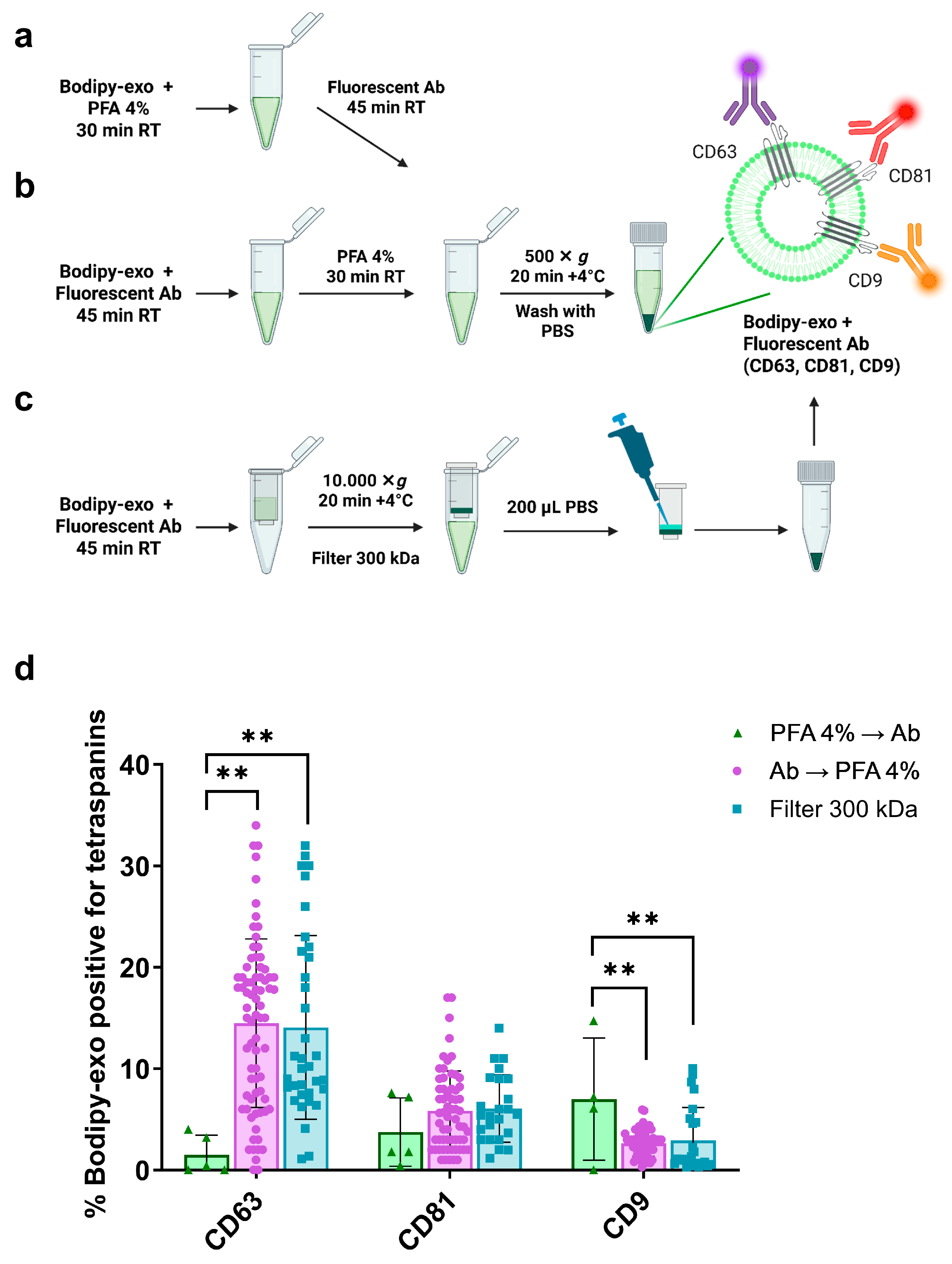
3.1. Comparison of Different Strategies for Bodipy-Exo Immunostaining
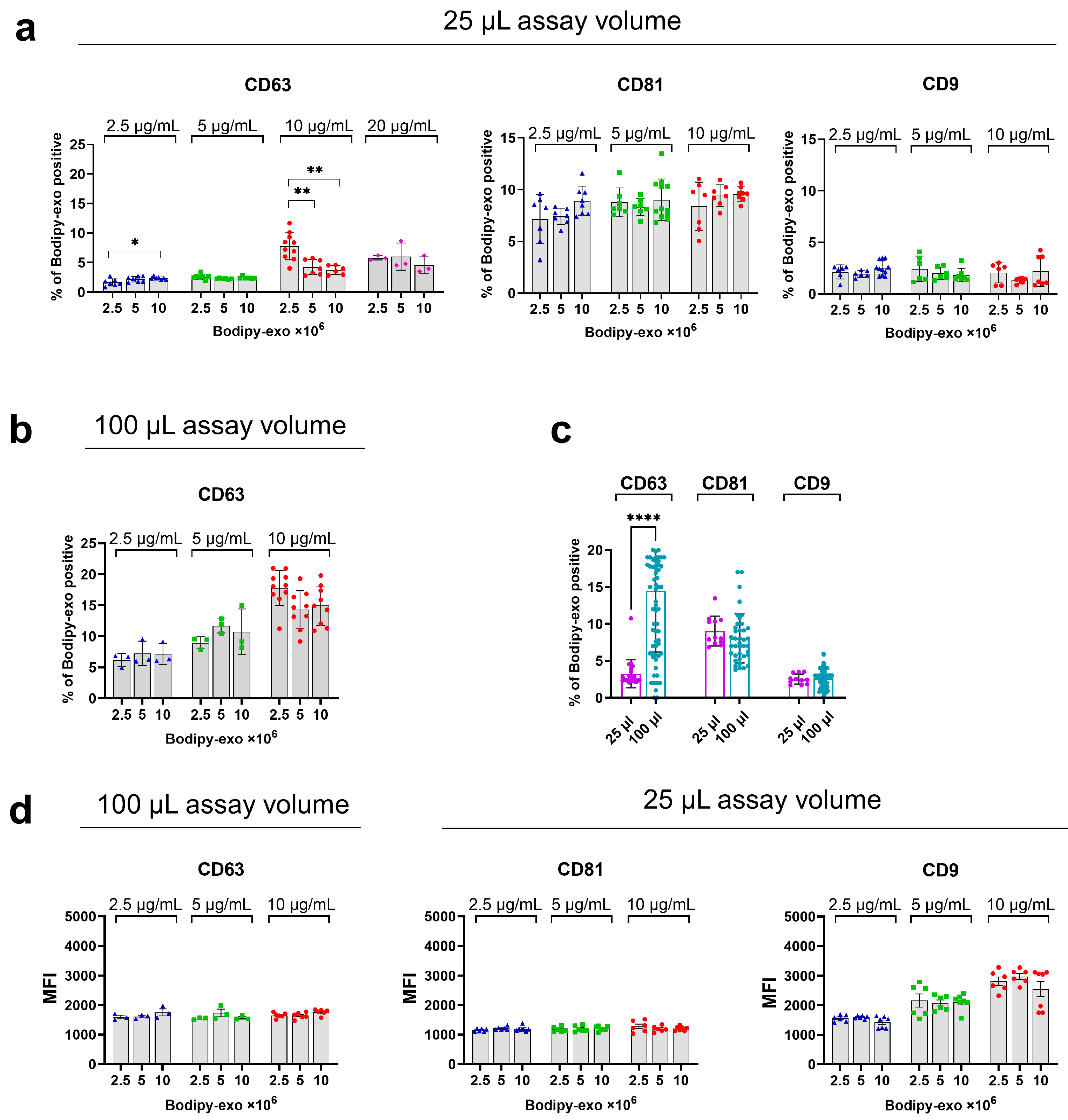
3.2. Optimization of Bodipy-Exo and Tetraspanin Antibody Concentration
3.3. Validation of Signal Specificity and Background Reduction
3.4. CFSE Staining Does Not Interfere with Tetraspanins Antibody Labelling
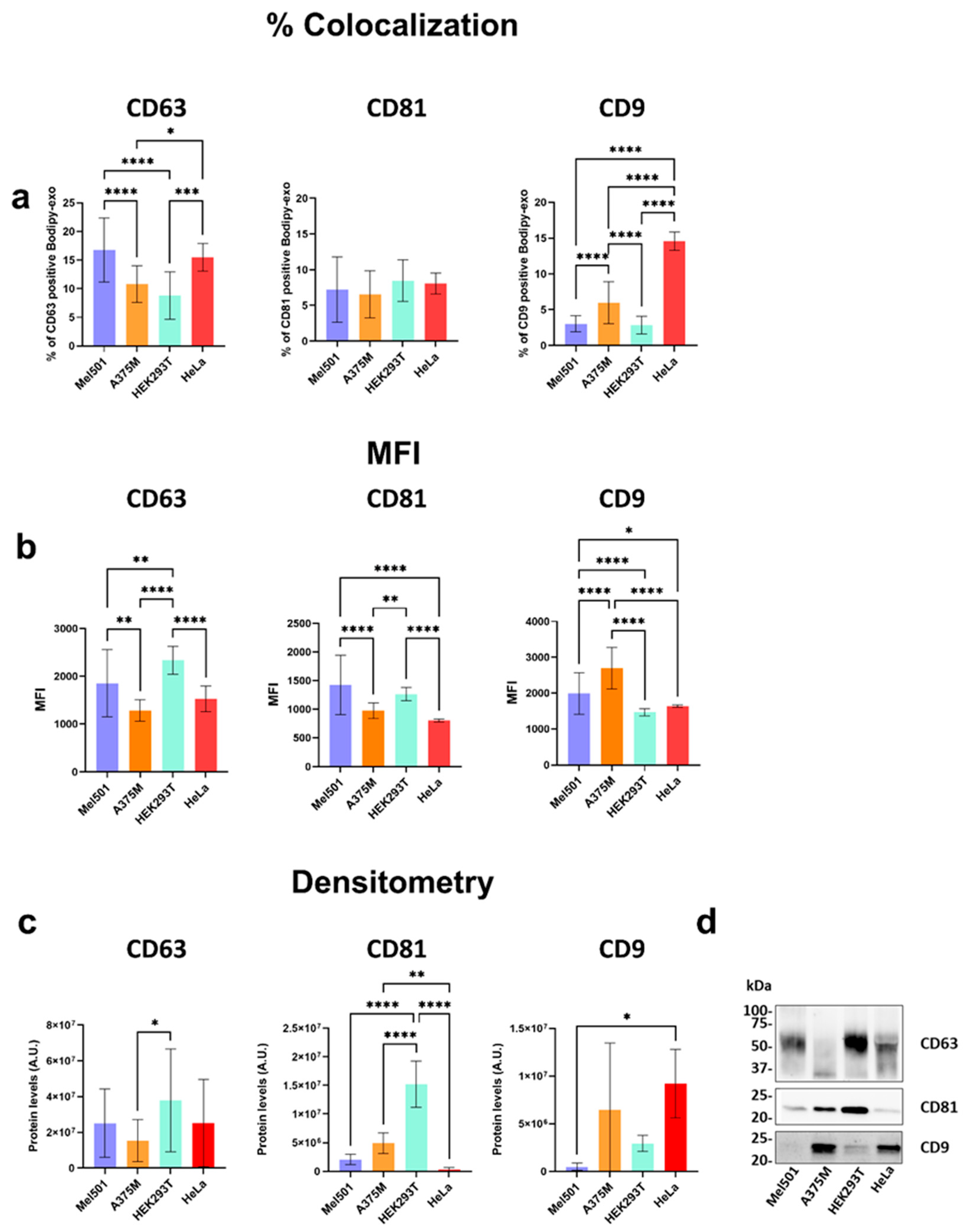
3.5. Characterization of Tetraspanins Expressions in sEVs from Different Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrugger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Shiba, T.; Yoshida, T.; Bolidong, D.; Kato, K.; Sato, Y.; Mochizuki, M.; Seto, T.; Kawashiri, S.; Hanayama, R. Precise analysis of single small extracellular vesicles using flow cytometry. Sci. Rep. 2024, 14, 7465. [Google Scholar] [CrossRef]
- Martin-Jaular, L.; Nevo, N.; Schessner, J.P.; Tkach, M.; Jouve, M.; Dingli, F.; Loew, D.; Witwer, K.W.; Ostrowski, M.; Borner, G.H.H.; et al. Unbiased proteomic profiling of host cell extracellular vesicle composition and dynamics upon HIV-1 infection. EMBO J. 2021, 40, e105492. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Arkesteijn, G.J.A.; Bremer, M.; Cimorelli, M.; Dignat-George, F.; Giebel, B.; Gorgens, A.; Hendrix, A.; Kuiper, M.; Lacroix, R.; et al. A compendium of single extracellular vesicle flow cytometry. J. Extracell. Vesicles 2023, 12, e12299. [Google Scholar] [CrossRef]
- Woud, W.W.; van der Pol, E.; Mul, E.; Hoogduijn, M.J.; Baan, C.C.; Boer, K.; Merino, A. An imaging flow cytometry-based methodology for the analysis of single extracellular vesicles in unprocessed human plasma. Commun. Biol. 2022, 5, 633. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Bostancioglu, R.B.; Welsh, J.A.; Zickler, A.M.; Murke, F.; Corso, G.; Felldin, U.; Hagey, D.W.; Evertsson, B.; Liang, X.M.; et al. Systematic Methodological Evaluation of a Multiplex Bead-Based Flow Cytometry Assay for Detection of Extracellular Vesicle Surface Signatures. Front. Immunol. 2018, 9, 1326. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Mathieu, M.; Nevo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D.; et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021, 12, 4389. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gorgens, A.; Mussack, V.; Pepeldjiyska, E.; Hartz, A.S.; Rank, A.; Schmohl, J.; Kramer, D.; Andaloussi, S.E.; Pfaffl, M.W.; et al. Description and optimization of a multiplex bead-based flow cytometry method (MBFCM) to characterize extracellular vesicles in serum samples from patients with hematological malignancies. Cancer Gene Ther. 2022, 29, 1600–1615. [Google Scholar] [CrossRef] [PubMed]
- Giovanazzi, A.; van Herwijnen, M.J.C.; Kleinjan, M.; van der Meulen, G.N.; Wauben, M.H.M. Surface protein profiling of milk and serum extracellular vesicles unveils body fluid-specific signatures. Sci. Rep. 2023, 13, 8758. [Google Scholar] [CrossRef]
- Barreca, V.; Boussadia, Z.; Polignano, D.; Galli, L.; Tirelli, V.; Sanchez, M.; Falchi, M.; Bertuccini, L.; Iosi, F.; Tatti, M.; et al. Metabolic labelling of a subpopulation of small extracellular vesicles using a fluorescent palmitic acid analogue. J. Extracell. Vesicles 2023, 12, e12392. [Google Scholar] [CrossRef] [PubMed]
- Coscia, C.; Parolini, I.; Sanchez, M.; Biffoni, M.; Boussadia, Z.; Zanetti, C.; Fiani, M.L.; Sargiacomo, M. Generation, Quantification, and Tracing of Metabolically Labeled Fluorescent Exosomes. In Lentiviral Vectors and Exosomes as Gene and Protein Delivery Tools; Federico, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 217–235. [Google Scholar]
- Tirelli, V.; Grasso, F.; Barreca, V.; Polignano, D.; Gallinaro, A.; Cara, A.; Sargiacomo, M.; Fiani, M.L.; Sanchez, M. Flow cytometric procedures for deep characterization of nanoparticles. Biol. Methods Protoc. 2025, 10, bpaf019. [Google Scholar] [CrossRef]
- Kormelink, T.G.; Arkesteijn, G.J.; Nauwelaers, F.A.; van den Engh, G.; Nolte-‘t Hoen, E.N.; Wauben, M.H. Prerequisites for the analysis and sorting of extracellular vesicle subpopulations by high-resolution flow cytometry. Cytom. Part A J. Int. Soc. Anal. Cytol. 2015, 89, 135–147. [Google Scholar] [CrossRef]
- Puzar Dominkus, P.; Stenovec, M.; Sitar, S.; Lasic, E.; Zorec, R.; Plemenitas, A.; Zagar, E.; Kreft, M.; Lenassi, M. PKH26 labeling of extracellular vesicles: Characterization and cellular internalization of contaminating PKH26 nanoparticles. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1350–1361. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Maire, C.L.; Reimer, R.; Duhrsen, L.; Kolbe, K.; Holz, M.; Schneider, E.; Rissiek, A.; Babayan, A.; Hille, C.; et al. Imaging flow cytometry facilitates multiparametric characterization of extracellular vesicles in malignant brain tumours. J. Extracell. Vesicles 2019, 8, 1588555. [Google Scholar] [CrossRef]
- Burn, O.K.; Mair, F.; Ferrer-Font, L. Combinatorial antibody titrations for high-parameter flow cytometry. Cytom. Part A J. Int. Soc. Anal. Cytol. 2024, 105, 388–393. [Google Scholar] [CrossRef]
- Kalina, T.; Lundsten, K.; Engel, P. Relevance of Antibody Validation for Flow Cytometry. Cytom. Part A J. Int. Soc. Anal. Cytol. 2020, 97, 126–136. [Google Scholar] [CrossRef]
- Trummer, A.; De Rop, C.; Tiede, A.; Ganser, A.; Eisert, R. Isotype controls in phenotyping and quantification of microparticles: A major source of error and how to evade it. Thromb. Res. 2008, 122, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Inglis, H.C.; Danesh, A.; Shah, A.; Lacroix, J.; Spinella, P.C.; Norris, P.J. Techniques to improve detection and analysis of extracellular vesicles using flow cytometry. Cytom. Part A J. Int. Soc. Anal. Cytol. 2015, 87, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Gorgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8, 1587567. [Google Scholar] [CrossRef]
- Welsh, J.A.; Van Der Pol, E.; Arkesteijn, G.J.A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles 2020, 9, 1713526. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Telford, B.; Musich, T.A.; McKinnon, K.; Clayborne, C.; Braig, Z.; Rosner, A.; Demberg, T.; Watson, D.C.; Karpova, T.S.; et al. Labeling Extracellular Vesicles for Nanoscale Flow Cytometry. Sci. Rep. 2017, 7, 1878. [Google Scholar] [CrossRef] [PubMed]
- Pospichalova, V.; Svoboda, J.; Dave, Z.; Kotrbova, A.; Kaiser, K.; Klemova, D.; Ilkovics, L.; Hampl, A.; Crha, I.; Jandakova, E.; et al. Simplified protocol for flow cytometry analysis of fluorescently labeled exosomes and microvesicles using dedicated flow cytometer. J. Extracell. Vesicles 2015, 4, 25530. [Google Scholar] [CrossRef]
- Loconte, L.; Arguedas, D.; El, R.; Zhou, A.; Chipont, A.; Guyonnet, L.; Guerin, C.; Piovesana, E.; Vazquez-Ibar, J.L.; Joliot, A.; et al. Detection of the interactions of tumour derived extracellular vesicles with immune cells is dependent on EV-labelling methods. J. Extracell. Vesicles 2023, 12, e12384. [Google Scholar] [CrossRef]
- Tertel, T.; Schoppet, M.; Stambouli, O.; Al-Jipouri, A.; James, P.F.; Giebel, B. Imaging flow cytometry challenges the usefulness of classically used extracellular vesicle labeling dyes and qualifies the novel dye Exoria for the labeling of mesenchymal stromal cell-extracellular vesicle preparations. Cytotherapy 2022, 24, 619–628. [Google Scholar] [CrossRef]
- Fan, Y.; Pionneau, C.; Cocozza, F.; Boëlle, P.Y.; Chardonnet, S.; Charrin, S.; Théry, C.; Zimmermann, P.; Rubinstein, E. Differential proteomics argues against a general role for CD9, CD81 or CD63 in the sorting of proteins into extracellular vesicles. J. Extracell. Vesicles 2023, 12, e12352. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef]
- Han, C.; Kang, H.; Yi, J.; Kang, M.; Lee, H.; Kwon, Y.; Jung, J.; Lee, J.; Park, J. Single-vesicle imaging and co-localization analysis for tetraspanin profiling of individual extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12047. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Andronico, L.A.; Jung, S.R.; Chen, H.; Fujimoto, B.; Vojtech, L.; Chiu, D.T. High-Throughput Counting and Superresolution Mapping of Tetraspanins on Exosomes Using a Single-Molecule Sensitive Flow Technique and Transistor-like Semiconducting Polymer Dots. Angew. Chem. 2021, 60, 13470–13475. [Google Scholar] [CrossRef]
- Willms, E.; Cabanas, C.; Mager, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, L.; Cheng, H.; Zhang, Q.; Wang, S.; Zhong, W.; He, C.; Wei, Q. Unlocking the Potential of Extracellular Vesicles in Cardiovascular Disease. J. Cell. Mol. Med. 2025, 29, e70407. [Google Scholar] [CrossRef] [PubMed]
- Grange, C.; Bussolati, B. Extracellular vesicles in kidney disease. Nat. Rev. Nephrol. 2022, 18, 499–513. [Google Scholar] [CrossRef]
- Viktorsson, K.; Haag, P.; Shah, C.H.; Franzen, B.; Arapi, V.; Holmsten, K.; Sandstrom, P.; Lewensohn, R.; Ullen, A. Profiling of extracellular vesicles of metastatic urothelial cancer patients to discover protein signatures related to treatment outcome. Mol. Oncol. 2022, 16, 3620–3641. [Google Scholar] [CrossRef]
- Zanganeh, S.; Abbasgholinejad, E.; Doroudian, M.; Esmaelizad, N.; Farjadian, F.; Benhabbour, S.R. The Current Landscape of Glioblastoma Biomarkers in Body Fluids. Cancers 2023, 15, 3804. [Google Scholar] [CrossRef]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.B.; Kumar, R.; et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar] [CrossRef]
- Cruz, C.G.; Sodawalla, H.M.; Mohanakumar, T.; Bansal, S. Extracellular Vesicles as Biomarkers in Infectious Diseases. Biology 2025, 14, 182. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, S. Plasma extracellular vesicle: A novel biomarker for neurodegenerative disease diagnosis. Extracell. Vesicles Circ. Nucl. Acids 2024, 5, 569–573. [Google Scholar] [CrossRef]
- Gonul, C.P.; Karacicek, B.; Genc, S. Neuron-Derived Extracellular Vesicles: Emerging Biomarkers and Functional Mediators in Alzheimer’s Disease, with Comparative Insights Into Neurodevelopment and Aging. Dev. Neurobiol. 2025, 85, e22984. [Google Scholar] [CrossRef] [PubMed]
- Spitzberg, J.D.; Ferguson, S.; Yang, K.S.; Peterson, H.M.; Carlson, J.C.T.; Weissleder, R. Multiplexed analysis of EV reveals specific biomarker composition with diagnostic impact. Nat. Commun. 2023, 14, 1239. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cai, N.; Niu, Q.; Tian, Y.; Hu, Y.; Yan, X. Quantitative assessment of lipophilic membrane dye-based labelling of extracellular vesicles by nano-flow cytometry. J. Extracell. Vesicles 2023, 12, e12351. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, Z.; Melvin, B.C.; Basu Ray, N.; Ikezu, S.; Ikezu, T. Comparison of nanoimaging and nanoflow based detection of extracellular vesicles at a single particle resolution. J. Extracell. Biol. 2024, 3, e70016. [Google Scholar] [CrossRef]
- Wolf, B.; Piksa, M.; Beley, I.; Patoux, A.; Besson, T.; Cordier, V.; Voedisch, B.; Schindler, P.; Stollner, D.; Perrot, L.; et al. Therapeutic antibody glycosylation impacts antigen recognition and immunogenicity. Immunology 2022, 166, 380–407. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polignano, D.; Barreca, V.; Sanchez, M.; Sargiacomo, M.; Fiani, M.L. A Simple and Scalable Assay for Multiplexed Flow Cytometric Profiling of Surface Markers on Small Extracellular Vesicles. Cells 2025, 14, 989. https://doi.org/10.3390/cells14130989
Polignano D, Barreca V, Sanchez M, Sargiacomo M, Fiani ML. A Simple and Scalable Assay for Multiplexed Flow Cytometric Profiling of Surface Markers on Small Extracellular Vesicles. Cells. 2025; 14(13):989. https://doi.org/10.3390/cells14130989
Chicago/Turabian StylePolignano, Deborah, Valeria Barreca, Massimo Sanchez, Massimo Sargiacomo, and Maria Luisa Fiani. 2025. "A Simple and Scalable Assay for Multiplexed Flow Cytometric Profiling of Surface Markers on Small Extracellular Vesicles" Cells 14, no. 13: 989. https://doi.org/10.3390/cells14130989
APA StylePolignano, D., Barreca, V., Sanchez, M., Sargiacomo, M., & Fiani, M. L. (2025). A Simple and Scalable Assay for Multiplexed Flow Cytometric Profiling of Surface Markers on Small Extracellular Vesicles. Cells, 14(13), 989. https://doi.org/10.3390/cells14130989