

Cancer Vulnerabilities Through Targeting the ATR/Chk1 and ATM/Chk2 Axes in the Context of DNA Damage
 , , and
, , and
Abstract

1. Introduction
2. DNA Damage and Repair Mechanisms
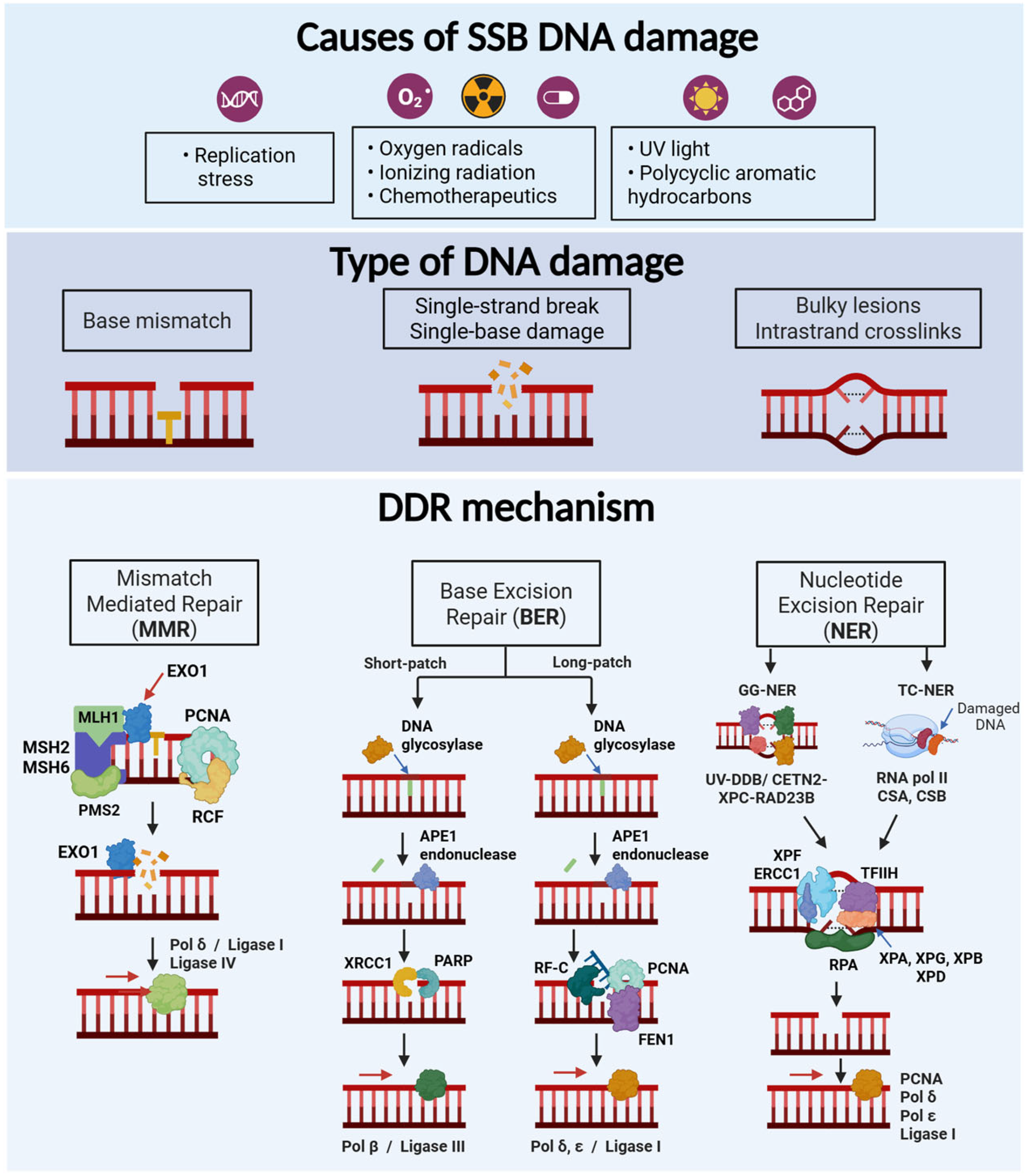
2.1. Main Types of DNA Damage
2.2. Mechanisms of DNA Damage Response
2.2.1. Single Strand Breaks
Mismatch Repair (MMR)
Base Excision Repair (BER)
Nucleotide Excision Repair (NER)
3. Double Strand Breaks (DSB)
3.1. Non- Homologous End Joining (NHEJ)
3.2. Homologous Recombination Repair (HRR)
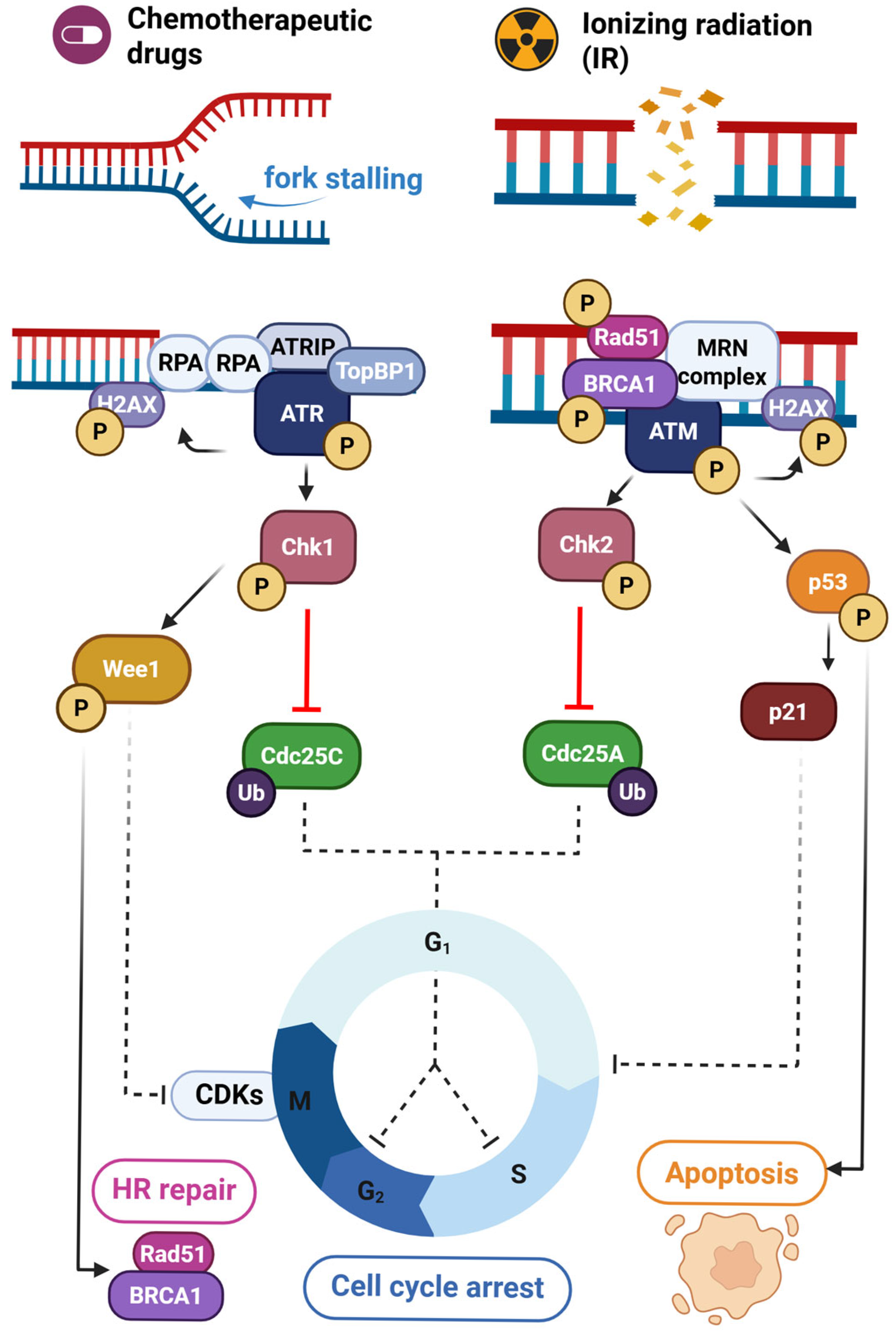
4. The ATR/Chk1 and ATM/Chk2 Axes in DNA Damage Repair
4.1. ATR/Chk1 Signaling
4.2. ATM/Chk2 Signaling
4.3. Cross-Talk Between ATR/Chk1 and ATM/Chk2
4.4. ATR Inhibitors
4.5. ATM Inhibitors
4.6. Chk1/2 Inhibitors
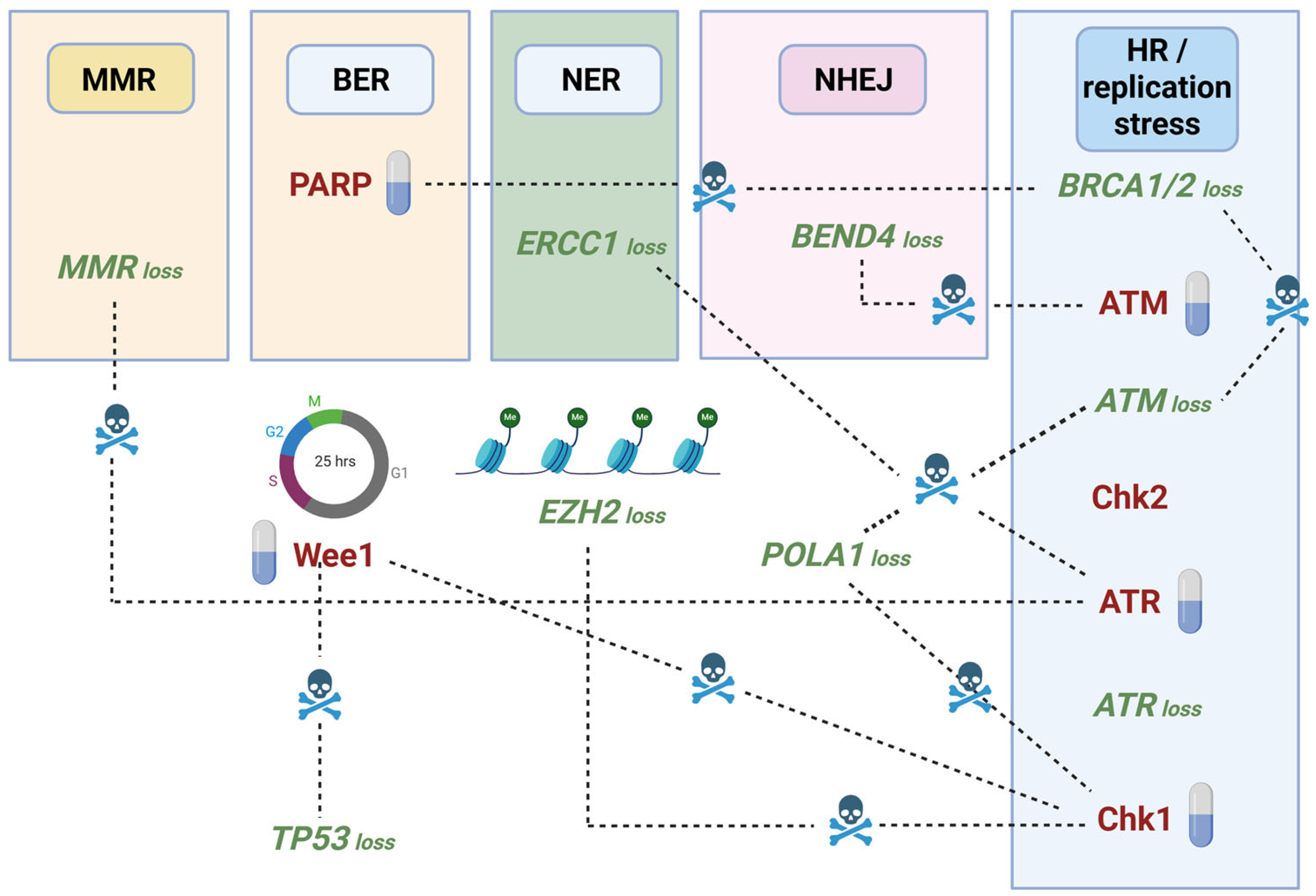
5. Synthetic Lethality Approaches to Target the ATR/Chk1 and ATM/Chk2 Axes
6. Synergistic Anticancer Effects Elicited by Combining ATM/Chk2 or ATR/Chk1 Inhibition with Immunotherapy
7. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological Response of Cancer Cells to Radiation Treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef]
- Boshuizen, J.; Peeper, D.S. Rational Cancer Treatment Combinations: An Urgent Clinical Need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Zenke, F.T.; Curtin, N.J. DNA Damage Response Inhibitors in Cancer Therapy: Lessons from the Past, Current Status and Future Implications. Nat. Rev. Drug Discov. 2025, 24, 19–39. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Zhang, H.; Kreis, J.; Schelhorn, S.E.; Dahmen, H.; Grombacher, T.; Zühlsdorf, M.; Zenke, F.T.; Guan, Y. Mapping Combinatorial Drug Effects to DNA Damage Response Kinase Inhibitors. Nat. Commun. 2023, 14, 8310. [Google Scholar] [CrossRef]
- Choi, W.; Lee, E.S. Therapeutic Targeting of DNA Damage Response in Cancer. Int. J. Mol. Sci. 2022, 23, 1701. [Google Scholar] [CrossRef]
- Qian, J.; Liao, G.; Chen, M.; Peng, R.W.; Yan, X.; Du, J.; Huang, R.; Pan, M.; Lin, Y.; Gong, X.; et al. Advancing Cancer Therapy: New Frontiers in Targeting DNA Damage Response. Front. Pharmacol. 2024, 15, 1474337. [Google Scholar] [CrossRef]
- Zhou, B.B.S.; Elledge, S.J. The DNA Damage Response: Putting Checkpoints in Perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Maremonti, E.; Brede, D.A.; Olsen, A.K.; Eide, D.M.; Berg, E.S. Ionizing Radiation, Genotoxic Stress, and Mitochondrial DNA Copy-Number Variation in Caenorhabditis Elegans: Droplet Digital PCR Analysis. Mutat. Res. Toxicol. Environ. Mutagen. 2020, 858–860, 503277. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing Radiation-Induced Metabolic Oxidative Stress and Prolonged Cell Injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Kciuk, M.; Marciniak, B.; Mojzych, M.; Kontek, R. Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies. Int. J. Mol. Sci. 2020, 21, 7264. [Google Scholar] [CrossRef]
- Cadet, J.; Richard Wagner, J. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Sriraman, A.; Debnath, T.K.; Xhemalce, B.; Miller, K.M. Making It or Breaking It: DNA Methylation and Genome Integrity. Essays Biochem. 2020, 64, 687. [Google Scholar] [CrossRef]
- Bordin, D.L.; Lirussi, L.; Nilsen, H. Cellular Response to Endogenous DNA Damage: DNA Base Modifications in Gene Expression Regulation. DNA Repair 2021, 99, 103051. [Google Scholar] [CrossRef]
- Thompson, P.S.; Cortez, D. New Insights into Abasic Site Repair and Tolerance. DNA Repair 2020, 90, 102866. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Brown, M.W.; Kim, Y.; Williams, G.M.; Huck, J.D.; Surtees, J.A.; Finkelstein, I.J. Dynamic DNA Binding Licenses a Repair Factor to Bypass Roadblocks in Search of DNA Lesions. Nat. Commun. 2016, 7, 10607. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. Postreplicative Mismatch Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA Mismatch Repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Cannavo, E.; Gerrits, B.; Marra, G.; Schlapbach, R.; Jiricny, J. Characterization of the Interactome of the Human MutL Homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2007, 282, 2976–2986. [Google Scholar] [CrossRef]
- Fishel, R. Mismatch Repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLalpha in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef]
- Prindle, M.J.; Loeb, L.A. DNA Polymerase Delta in DNA Replication and Genome Maintenance. Environ. Mol. Mutagen. 2012, 53, 666–682. [Google Scholar] [CrossRef]
- Schmidt, M.H.M.; Pearson, C.E. Disease-Associated Repeat Instability and Mismatch Repair. DNA Repair 2016, 38, 117–126. [Google Scholar] [CrossRef]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized Base Damage and Single-Strand Break Repair in Mammalian Genomes: Role of Disordered Regions and Posttranslational Modifications in Early Enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-Excision Repair of Oxidative DNA Damage by DNA Glycosylases. Mutat. Res. 2005, 591, 45–59. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base Excision Repair and Its Implications to Cancer Therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Çaǧlayan, M.; Horton, J.K.; Dai, D.P.; Stefanick, D.F.; Wilson, S.H. Oxidized Nucleotide Insertion by Pol β Confounds Ligation during Base Excision Repair. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Frosina, G.; Fortini, P.; Rossi, O.; Carrozzino, F.; Raspaglio, G.; Cox, L.S.; Lane, D.P.; Abbondandolo, A.; Dogliotti, E. Two Pathways for Base Excision Repair in Mammalian Cells. J. Biol. Chem. 1996, 271, 9573–9578. [Google Scholar] [CrossRef]
- Campalans, A.; Kortulewski, T.; Amouroux, R.; Menoni, H.; Vermeulen, W.; Radicella, J.P. Distinct Spatiotemporal Patterns and PARP Dependence of XRCC1 Recruitment to Single-Strand Break and Base Excision Repair. Nucleic Acids Res. 2013, 41, 3115–3129. [Google Scholar] [CrossRef]
- Fousteri, M.; Mullenders, L.H.F. Transcription-Coupled Nucleotide Excision Repair in Mammalian Cells: Molecular Mechanisms and Biological Effects. Cell Res. 2008, 18, 73–84. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-Excision Repair of Oxidative DNA Damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef]
- Gillet, L.C.J.; Schärer, O.D. Molecular Mechanisms of Mammalian Global Genome Nucleotide Excision Repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef]
- Aboussekhra, A.; Biggerstaff, M.; Shivji, M.K.K.; Vilpo, J.A.; Moncollin, V.; Podust, V.N.; Protić, M.; Hübscher, U.; Egly, J.M.; Wood, R.D. Mammalian DNA Nucleotide Excision Repair Reconstituted with Purified Protein Components. Cell 1995, 80, 859–868. [Google Scholar] [CrossRef]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, S.; Hanaoka, F. A Multistep Damage Recognition Mechanism for Global Genomic Nucleotide Excision Repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, F.; Hennecke, U.; Carell, T.; Cramer, P. CPD Damage Recognition by Transcribing RNA Polymerase II. Science 2007, 315, 859–862. [Google Scholar] [CrossRef]
- Okuda, M.; Kinoshita, M.; Kakumu, E.; Sugasawa, K.; Nishimura, Y. Structural Insight into the Mechanism of TFIIH Recognition by the Acidic String of the Nucleotide Excision Repair Factor XPC. Structure 2015, 23, 1827–1837. [Google Scholar] [CrossRef]
- Volker, M.; Moné, M.J.; Karmakar, P.; Van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.J.; Van Driel, R.; Van Zeeland, A.A.; Mullenders, L.H.F. Sequential Assembly of the Nucleotide Excision Repair Factors in Vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Kokic, G.; Chernev, A.; Tegunov, D.; Dienemann, C.; Urlaub, H.; Cramer, P. Structural Basis of TFIIH Activation for Nucleotide Excision Repair. Nat. Commun. 2019, 10, 2885. [Google Scholar] [CrossRef]
- Sugasawa, K.; Akagi, J.-i.; Nishi, R.; Iwai, S.; Hanaoka, F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Mol. Cell 2009, 36, 642–653. [Google Scholar] [CrossRef]
- De Laat, W.L.; Appeldoorn, E.; Sugasawa, K.; Weterings, E.; Jaspers, N.G.J.; Hoeijmakers, J.H.J. DNA-Binding Polarity of Human Replication Protein A Positions Nucleases in Nucleotide Excision Repair. Genes Dev. 1998, 12, 2598–2609. [Google Scholar] [CrossRef]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of Dual Incision and Repair Synthesis in Human Nucleotide Excision Repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA Polymerases, Recruited by Different Mechanisms, Carry out NER Repair Synthesis in Human Cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef]
- Paul-Konietzko, K.; Thomale, J.; Arakawa, H.; Iliakis, G. DNA Ligases I and III Support Nucleotide Excision Repair in DT40 Cells with Similar Efficiency. Photochem. Photobiol. 2015, 91, 1173–1180. [Google Scholar] [CrossRef]
- Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells 2020, 9, 1853. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.G.W.; Walter, J.C.; Loparo, J.J. Two-Stage Synapsis of DNA Ends during Non-Homologous End Joining. Mol. Cell 2016, 61, 850. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA End-Joining for Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2017, 293, 10512. [Google Scholar] [CrossRef]
- Deriano, L.; Roth, D.B. Modernizing the Nonhomologous End-Joining Repertoire: Alternative and Classical NHEJ Share the Stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524. [Google Scholar] [CrossRef]
- Simoneau, A.; Zou, L. An Extending ATR-CHK1 Circuitry: The Replication Stress Response and Beyond. Curr. Opin. Genet. Dev. 2021, 71, 92–98. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA Damage Checkpoint Kinases in Cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Buscemi, G.; Perego, P.; Carenini, N.; Nakanishi, M.; Chessa, L.; Chen, J.; Khanna, K.K.; Delia, D. Activation of ATM and Chk2 Kinases in Relation to the Amount of DNA Strand Breaks. Oncogene 2004, 23, 7691–7700. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Besnard, E.; Fisher, D.; Piette, J.; Dulić, V. Chk1 Is Dispensable for G2 Arrest in Response to Sustained DNA Damage When the ATM/P53/P21 Pathway Is Functional. Oncogene 2011, 30, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A Mitosis-Specific and R Loop-Driven ATR Pathway Promotes Faithful Chromosome Segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef]
- Petermann, E.; Caldecott, K.W. Evidence That the ATR/Chk1 Pathway Maintains Normal Replication Fork Progression during Unperturbed S Phase. Cell Cycle 2006, 5, 2203–2209. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 Kinase Signaling Mechanism That Limits Origin Firing during Unperturbed DNA Replication. Proc. Natl. Acad. Sci. USA 2019, 116, 13374–13383. [Google Scholar] [CrossRef]
- Smith, J.; Mun Tho, L.; Xu, N.; Gillespie, D.A. The ATM–Chk2 and ATR–Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs Kinases-the Lessons from the Mouse Models: Inhibition ≠ Deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-Dependent Phosphorylation of MDMX Contribute to P53 Activation after DNA Damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, D. Repair Pathway Choice for Double-Strand Breaks. Essays Biochem. 2020, 64. [Google Scholar] [CrossRef]
- Mirman, Z.; de Lange, T. 53BP1: A DSB Escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Belal, H.; Ying Ng, E.F.; Meitinger, F. 53BP1-Mediated Activation of the Tumor Suppressor P53. Curr. Opin. Cell Biol. 2024, 91. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kuk, M.U.; Kim, J.W.; Lee, Y.H.; Lee, Y.S.; Choy, H.E.; Park, S.C.; Park, J.T. ATM Mediated-P53 Signaling Pathway Forms a Novel Axis for Senescence Control. Mitochondrion 2020, 55, 54–63. [Google Scholar] [CrossRef]
- Adams, C.J.; Graham, A.L.; Jansson, M.; Coutts, A.S.; Edelmann, M.; Smith, L.; Kessler, B.; La Thangue, N.B. ATM and Chk2 Kinase Target the P53 Cofactor Strap. EMBO Rep. 2008, 9, 1222. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A Checkpoint Pathway Guards against Radioresistant DNA Synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Petsalaki, E.; Zachos, G. An ATM-CHK2-INCENP Pathway Prevents Chromatin Breakage by Regulating the Abscission Checkpoint. Mol. Cell. Oncol. 2021, 8, 1877999. [Google Scholar] [CrossRef]
- Niwa, Y.; Kamimura, K.; Ogawa, K.; Oda, C.; Tanaka, Y.; Horigome, R.; Ohtsuka, M.; Miura, H.; Fujisawa, K.; Yamamoto, N.; et al. Cyclin D1 Binding Protein 1 Responds to DNA Damage through the ATM-CHK2 Pathway. J. Clin. Med. 2022, 11. [Google Scholar] [CrossRef]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-Dependent Phosphorylation and Activation of ATM in Response to UV Treatment or Replication Fork Stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and Cell Cycle-Dependent Regulation of ATR in Response to DNA Double-Strand Breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA Damage Response: Next-Generation Anti-Cancer Therapeutic Strategies. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of Tumor Responses to DNA Damaging Therapy by the Selective ATR Inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.R.; Lord, S.; El Badri, S.; Harman, R.; Holmes, J.; Kamzi, F.; Maughan, T.; McIntosh, D.; Mukherjee, S.; Ooms, A.; et al. CHARIOT: A Phase I Study of Berzosertib with Chemoradiotherapy in Oesophageal and Other Solid Cancers Using Time to Event Continual Reassessment Method. Br. J. Cancer 2024, 130, 467–475. [Google Scholar] [CrossRef]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus Gemcitabine versus Gemcitabine Alone in Platinum-Resistant High-Grade Serous Ovarian Cancer: A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet. Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembé, A.B.; Cho, H.; HKim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase i Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Wethington, S.L.; Shah, P.D.; Martin, L.; Tanyi, J.L.; Latif, N.; Morgan, M.; Torigian, D.A.; Rodriguez, D.; Smith, S.A.; Dean, E.; et al. Combination ATR (Ceralasertib) and PARP (Olaparib) Inhibitor (CAPRI) Trial in Acquired PARP Inhibitor-Resistant Homologous Recombination-Deficient Ovarian Cancer. Clin. Cancer Res. 2023, 29, 2800–2807. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lucking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bomer, U.; Moosmayer, D.; Eberspacher, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Noor, N.R.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Llorca-Cardenosa, M.J.; Aronson, L.I.; Krastev, D.B.; Nieminuszczy, J.; Alexander, J.; Song, F.; Dylewska, M.; Broderick, R.; Brough, R.; Zimmermann, A.; et al. SMG8/SMG9 Heterodimer Loss Modulates SMG1 Kinase to Drive ATR Inhibitor Resistance. Cancer Res. 2022, 82, 3962–3973. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-Wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef]
- Hargreaves, C.E.; Salatino, S.; Sasson, S.C.; Charlesworth, J.E.G.; Bateman, E.; Patel, A.M.; Anzilotti, C.; Broxholme, J.; Knight, J.C.; Patel, S.Y. Decreased ATM Function Causes Delayed DNA Repair and Apoptosis in Common Variable Immunodeficiency Disorders. J. Clin. Immunol. 2021, 41, 1315–1330. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.B.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C.M. Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef]
- Jin, M.H.; Oh, D.Y. ATM in DNA Repair in Cancer. Pharmacol. Ther. 2019, 203. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Wei, Y.C.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM Kinase Inhibitor KU-60019 Radiosensitizes Glioma Cells, Compromises Insulin, AKT and ERK Prosurvival Signaling, and Inhibits Migration and Invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef]
- Zimmermann, A.; Zenke, F.T.; Chiu, L.Y.; Dahmen, H.; Pehl, U.; Fuchss, T.; Grombacher, T.; Blume, B.; Vassilev, L.T.; Blaukat, A. A New Class of Selective ATM Inhibitors as Combination Partners of DNA Double-Strand Break Inducing Cancer Therapies. Mol. Cancer Ther. 2022, 21, 859–870. [Google Scholar] [CrossRef]
- Waqar, S.N.; Robinson, C.; Olszanski, A.J.; Spira, A.; Hackmaster, M.; Lucas, L.; Sponton, L.; Jin, H.; Hering, U.; Cronier, D.; et al. Phase I Trial of ATM Inhibitor M3541 in Combination with Palliative Radiotherapy in Patients with Solid Tumors. Invest. New Drugs 2022, 40, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The Brain-Penetrant Clinical ATM Inhibitor AZD1390 Radiosensitizes and Improves Survival of Preclinical Brain Tumor Models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [PubMed]
- Walls, G.M.; Oughton, J.B.; Chalmers, A.J.; Brown, S.; Collinson, F.; Forster, M.D.; Franks, K.N.; Gilbert, A.; Hanna, G.G.; Hannaway, N.; et al. CONCORDE: A Phase I Platform Study of Novel Agents in Combination with Conventional Radiotherapy in Non-Small-Cell Lung Cancer. Clin. Transl. Radiat. Oncol. 2020, 25, 61–66. [Google Scholar] [CrossRef]
- Jucaite, A.; Stenkrona, P.; Cselényi, Z.; De Vita, S.; Buil-Bruna, N.; Varnäs, K.; Savage, A.; Varrone, A.; Johnström, P.; Schou, M.; et al. Brain Exposure of the ATM Inhibitor AZD1390 in Humans-a Positron Emission Tomography Study. Neuro. Oncol. 2021, 23, 687–696. [Google Scholar] [CrossRef]
- Merry, C.; Fu, K.; Wang, J.; Yeh, I.J.; Zhang, Y. Targeting the Checkpoint Kinase Chk1 in Cancer Therapy. Cell Cycle 2010, 9, 279–283. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; De Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-Grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a Cell Cycle Checkpoint Kinase 1 and 2 Inhibitor, in BRCA Wild-Type Recurrent High-Grade Serous Ovarian Cancer: A First-in-Class Proof-of-Concept Phase 2 Study. Lancet. Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lee, J.-m.; Gao, B.; Miller, R.; Lee, J.Y.; Colombo, N.; Vergote, I.; Credille, K.M.; Young, S.R.; McNeely, S.; et al. A Phase 2 Study of Prexasertib (LY2606368) in Platinum Resistant or Refractory Recurrent Ovarian Cancer. Gynecol. Oncol. 2022, 167, 213–225. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Özgüroğlu, M.; Han, J.Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients with Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.-J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013-e1824. [Google Scholar] [CrossRef] [PubMed]
- Laroche-Clary, A.; Lucchesi, C.; Rey, C.; Verbeke, S.; Bourdon, A.; Chaire, V.; Algéo, M.P.; Cousin, S.; Toulmonde, M.; Vélasco, V.; et al. CHK1 Inhibition in Soft-Tissue Sarcomas: Biological and Clinical Implications. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I Study of the Checkpoint Kinase 1 Inhibitor GDC-0575 in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef]
- Han, J.H.-J.; Kim, K.-T.; Im, J.; Park, S.; Choi, M.K.; Kim, I.; Nam, K.-Y.; Yoon, J. Abstract 1461: PHI-101, a Potent and Novel Inhibitor of CHK2 in Ovarian and Breast Cancer Cells. Cancer Res. 2021, 81, 1461. [Google Scholar] [CrossRef]
- Park, S.J.; Chang, S.J.; Suh, D.H.; Kong, T.W.; Song, H.; Kim, T.H.; Kim, J.W.; Kim, H.S.; Lee, S.J. A Phase IA Dose-Escalation Study of PHI-101, a New Checkpoint Kinase 2 Inhibitor, for Platinum-Resistant Recurrent Ovarian Cancer. BMC Cancer 2022, 22, 28. [Google Scholar] [CrossRef]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 Is a Potent and Selective Inhibitor of CHK2 That Potentiates the Cytotoxicity of PARP Inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. BRCAness, Homologous Recombination Deficiencies, and Synthetic Lethality. Cancer Res. 2023, 83, 1173–1174. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Mavroeidi, D.; Georganta, A.; Panagiotou, E.; Syrigos, K.; Souliotis, V.L. Targeting ATR Pathway in Solid Tumors: Evidence of Improving Therapeutic Outcomes. Int. J. Mol. Sci. 2024, 25, 2767. [Google Scholar] [CrossRef]
- Hopkins, J.L.; Lan, L.; Zou, L. DNA Repair Defects in Cancer and Therapeutic Opportunities. Genes Dev. 2022, 36, 278. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Willmore, E.; Prendergast, L.; Curtin, N.J. ATR, CHK1 and WEE1 Inhibitors Cause Homologous Recombination Repair Deficiency to Induce Synthetic Lethality with PARP Inhibitors. Br. J. Cancer 2024, 131, 905–917. [Google Scholar] [CrossRef]
- Biegała, Ł.; Gajek, A.; Szymczak-Pajor, I.; Marczak, A.; Śliwińska, A.; Rogalska, A. Targeted Inhibition of the ATR/CHK1 Pathway Overcomes Resistance to Olaparib and Dysregulates DNA Damage Response Protein Expression in BRCA2MUT Ovarian Cancer Cells. Sci. Rep. 2023, 13, 22659. [Google Scholar] [CrossRef]
- Haciefendi, A.; Eskiler, G.G. The Suppression of ATR/Chk1 Pathway by Elimusertib ATR Inhibitor in Triple Negative Breast Cancer Cells. Am. J. Transl. Res. 2023, 15, 4902. [Google Scholar]
- Sofianidi, A.; Dumbrava, E.E.; Syrigos, K.N.; Nasrazadani, A. Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers 2024, 16, 1139. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia Telangiectasia and Rad3-Related Inhibitors and Cancer Therapy: Where We Stand. J. Hematol. Oncol. 2019, 12, 1–8. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR Inhibition Induces Synthetic Lethality and Overcomes Chemoresistance in TP53- or ATM-Defective Chronic Lymphocytic Leukemia Cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Wang, M.; Ran, X.; Leung, W.; Kawale, A.; Saxena, S.; Ouyang, J.; Patel, P.S.; Dong, Y.; Yin, T.; Shu, J.; et al. ATR Inhibition Induces Synthetic Lethality in Mismatch Repair-Deficient Cells and Augments Immunotherapy. Genes Dev. 2023, 37, 929–943. [Google Scholar] [CrossRef]
- Schneider, H.E.; Schmitt, L.M.; Job, A.; Lankat-Buttgereit, B.; Gress, T.; Buchholz, M.; Gallmeier, E. Synthetic Lethality between ATR and POLA1 Reveals a Potential New Target for Individualized Cancer Therapy. Neoplasia 2024, 57, 101038. [Google Scholar] [CrossRef]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J. Clin. Invest. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Chen, Y.; Zheng, W.; Demanelis, K.; Liu, Y.; Connelly, J.A.; Wang, H.; Li, S.; Wang, Q.J. Synthetic Lethal Combination of CHK1 and WEE1 Inhibition for Treatment of Castration-Resistant Prostate Cancer. Oncogene 2024, 43, 789–803. [Google Scholar] [CrossRef]
- León, T.E.; Rapoz-D’Silva, T.; Bertoli, C.; Rahman, S.; Magnussen, M.; Philip, B.; Farah, N.; Richardson, S.E.; Ahrabi, S.; Guerra-Assunção, J.A.; et al. EZH2-Deficient T-Cell Acute Lymphoblastic Leukemia Is Sensitized to CHK1 Inhibition through Enhanced Replication Stress. Cancer Discov. 2020, 10, 998–1017. [Google Scholar] [CrossRef]
- Chang, T.Y.; Yan, Y.; Yu, Z.Y.; Rathore, M.; Lee, N.Z.; Tseng, H.J.; Cheng, L.H.; Huang, W.J.; Zhang, W.; Chan, E.R.; et al. Combined HDAC8 and Checkpoint Kinase Inhibition Induces Tumor-Selective Synthetic Lethality in Preclinical Models. J. Clin. Invest. 2024, 134. [Google Scholar] [CrossRef]
- Chen, C.C.; Kass, E.M.; Yen, W.F.; Ludwig, T.; Moynahan, M.E.; Chaudhuri, J.; Jasin, M. ATM Loss Leads to Synthetic Lethality in BRCA1 BRCT Mutant Mice Associated with Exacerbated Defects in Homology-Directed Repair. Proc. Natl. Acad. Sci. USA 2017, 114, 7665–7670. [Google Scholar] [CrossRef]
- Yao, Y.; Lv, H.; Zhang, M.; Li, Y.; Herman, J.G.; Brock, M.V.; Gao, A.; Wang, Q.; Fuks, F.; Zhang, L.; et al. Epigenetic Silencing of BEND4, a Novel DNA Damage Repair Gene, Is a Synthetic Lethal Marker for ATM Inhibitor in Pancreatic Cancer. Front. Med. 2024, 18, 721–734. [Google Scholar] [CrossRef]
- Oh, K.S.; Nam, A.R.; Bang, J.H.; Seo, H.R.; Kim, J.M.; Yoon, J.; Kim, T.Y.; Oh, D.Y. A Synthetic Lethal Strategy Using PARP and ATM Inhibition for Overcoming Trastuzumab Resistance in HER2-Positive Cancers. Oncogene 2022, 41, 3939–3952. [Google Scholar] [CrossRef]
- Ratz, L.; Brambillasca, C.; Bartke, L.; Huetzen, M.A.; Goergens, J.; Leidecker, O.; Jachimowicz, R.D.; van de Ven, M.; Proost, N.; Siteur, B.; et al. Combined Inhibition of EZH2 and ATM Is Synthetic Lethal in BRCA1-Deficient Breast Cancer. Breast Cancer Res. 2022, 24, 41. [Google Scholar] [CrossRef]
- Shi, C.; Qin, K.; Lin, A.; Jiang, A.; Cheng, Q.; Liu, Z.; Zhang, J.; Luo, P. The Role of DNA Damage Repair (DDR) System in Response to Immune Checkpoint Inhibitor (ICI) Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 268. [Google Scholar] [CrossRef]
- Murray, C.E.; Kornepati, A.V.R.; Ontiveros, C.; Liao, Y.; de la Peña Avalos, B.; Rogers, C.M.; Liu, Z.; Deng, Y.; Bai, H.; Kari, S.; et al. Tumour-Intrinsic PDL1 Signals Regulate the Chk2 DNA Damage Response in Cancer Cells and Mediate Resistance to Chk1 Inhibitors. Mol. Cancer 2024, 23, 242. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.Y.L.; Peng, G.; Yap, T.A. A Tale of Two Checkpoints: ATR Inhibition and PD-(L)1 Blockade. Annu. Rev. Med. 2022, 73, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Hardaker, E.L.; Sanseviero, E.; Karmokar, A.; Taylor, D.; Milo, M.; Michaloglou, C.; Hughes, A.; Mai, M.; King, M.; Solanki, A.; et al. The ATR Inhibitor Ceralasertib Potentiates Cancer Checkpoint Immunotherapy by Regulating the Tumor Microenvironment. Nat. Commun. 2024, 15. [Google Scholar] [CrossRef]
- Taniguchi, H.; Chakraborty, S.; Takahashi, N.; Banerjee, A.; Caeser, R.; Zhan, Y.A.; Tischfield, S.E.; Chow, A.; Nguyen, E.M.; Villalonga, Á.Q.; et al. ATR Inhibition Activates Cancer Cell CGAS/STING-Interferon Signaling and Promotes Antitumor Immunity in Small-Cell Lung Cancer. Sci. Adv. 2024, 10, eado4618. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR Inhibitor AZD6738 Enhances the Antitumor Activity of Radiotherapy and Immune Checkpoint Inhibitors by Potentiating the Tumor Immune Microenvironment in Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8, e000340. [Google Scholar] [CrossRef]
- Li, C.; Wang, B.; Tu, J.; Liu, C.; Wang, Y.; Chen, J.; Huang, Y.; Liu, B.; Yuan, X. ATM Inhibition Enhance Immunotherapy by Activating STING Signaling and Augmenting MHC Class I. Cell Death Dis. 2024, 15, 519. [Google Scholar] [CrossRef]
- Jin, W.J.; Zangl, L.M.; Hyun, M.; Massoud, E.; Schroeder, K.; Alexandridis, R.A.; Morris, Z.S. ATM Inhibition Augments Type I Interferon Response and Antitumor T-Cell Immunity When Combined with Radiation Therapy in Murine Tumor Models. J. Immunother. cancer 2023, 11, e007474. [Google Scholar] [CrossRef]
- Chen, J.L.Y.; Pan, C.K.; Lin, L.C.; Huang, Y.S.; Huang, T.H.; Yang, S.J.; Kuo, S.H.; Lin, Y.L. Combination of Ataxia Telangiectasia and Rad3-Related Inhibition with Ablative Radiotherapy Remodels the Tumor Microenvironment and Enhances Immunotherapy Response in Lung Cancer. Cancer Immunol. Immunother. 2024, 74, 8. [Google Scholar] [CrossRef]
- Do, K.T.; Manuszak, C.; Thrash, E.; Giobbie-Hurder, A.; Hu, J.; Kelland, S.; Powers, A.; de Jonge, A.; Shapiro, G.I.; Severgnini, M. Immune Modulating Activity of the CHK1 Inhibitor Prexasertib and Anti-PD-L1 Antibody LY3300054 in Patients with High-Grade Serous Ovarian Cancer and Other Solid Tumors. Cancer Immunol. Immunother. 2021, 70, 2991–3000. [Google Scholar] [CrossRef]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 Axis Promotes CGAS/STING Signaling in ARID1A-Deficient Tumors. J. Clin. Invest. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Concannon, K.; Morris, B.B.; Gay, C.M.; Byers, L.A. Combining Targeted DNA Repair Inhibition and Immune-Oncology Approaches for Enhanced Tumor Control. Mol. Cell 2023, 83, 660–680. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Treatment | Type of Cancer | Phase | Identifier | |
|---|---|---|---|---|---|
| ATRi | Berzosertib (M6620, VX-970) | Cisplatin and capecitabine | Esophageal adenocarcinoma Squamous cell adenocarcinoma | I | NCT03641547 |
| Topotecan | Small cell lung cancer | I/II | NCT02487095 | ||
| Gemcitabine | Ovarian serous tumor Recurrent fallopian tube carcinoma Recurrent ovarian carcinoma | II | NCT02595892 | ||
| Carboplatin, gemcitabine, cisplatin, etoposide, and irinotecan | Advanced solid tumors | I | NCT02157792 | ||
| Cisplatin and gemcitabine | Urothelial carcinoma | I/II | NCT02567409 | ||
| Monotherapy | Advanced solid tumors | II | NCT03718091 | ||
| Ceralasertib (AZD6738) | Paclitaxel | Refractory cancer | I | NCT02630199 | |
| Olaparib | High-grade serous ovarian carcinoma | I | NCT03462342 | ||
| Small cell lung cancer | II | NCT03428607 | |||
| X-ray radiotherapy | Solid tumor refractory to conventional treatment | I | NCT02223923 | ||
| Durmalumab and olaparib | Bile duct cancer | II | NCT04298021 | ||
| Monotherapy | Leukemia Myelodysplastic syndrome | I | NCT03770429 | ||
| Elimusertib (BAY 1895344) | Monotherapy | Advanced solid tumors Non-Hodgkins’ lymphoma | I | NCT03188965 | |
| Relapsed or refractory solid tumors | I/II | NCT05071209 | |||
| Pembrolizumab | Advanced solid tumors | I | NCT04095273 | ||
| Radiation therapy and pembrolizumab | Head and neck squamous cell carcinoma | I | NCT04576091 | ||
| Leucovorin, fluorouracil, and irinotecan | Advanced or metastatic cancers of the stomach and intestines | I | NCT04535401 | ||
| Cisplatin and gemcitabine | Advanced solid tumors | I | NCT04491942 | ||
| Tuvusertib (M1774) | Niraparib | Metastatic or aocally Advanced unresectable solid tumors | I | NCT04170153 | |
| Avelumab | ARID1 A-mutated endometrial cancer | II | NCT06518564 | ||
| Cemiplimab | Non-small cell lung cancer | I/II | NCT05882734 | ||
| ATMi | AZD0156 | Olaparib and irinotecan | Advanced solid tumors | I | NCT02588105 |
| AZD1390 | Radiotherapy and durmalumav | Non-small cell lung cancer | I | NCT04550104 | |
| Soft tissue sarcoma | I | NCT05116254 | |||
| Radiotherapy | Brain cancer | I | NCT03423628 | ||
| Stereotactic body radiotherapy | Metastatic solid tumors | I | NCT05678010 | ||
| Monotherapy | Grade 4 glioblastoma | I | NCT05182905 | ||
| Chk1/2i | Prexasertib (LY-2606368) | Olaparib | Solid tumors | I | NCT03057145 |
| Monotherapy | Ovarian cancer Breast cancer Prostate cancer | II | NCT02203513 | ||
| Ovarian cancer | II | NCT03414047 | |||
| Neoplasms | I | NCT02514603 | |||
| Cisplatin, cetuximab, fluorouracil, LY3023414, leucovorin | Solid tumors | I | NCT02124148 | ||
| Mitoxantrone, etoposide, and cytarabine | Acute myeloid leukemia Myelodysplastic syndromes | I | NCT03735446 | ||
| Chk1i | GDC-0575 | Gemcitabine | Lymphoma Solid tumors | I | NCT01564251 |
| Chk2i | PHI-101 | Monotherapy | Peritoneal, fallopian or ovarian cancer | I | NCT04678102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez, A.; Artola, M.; Leon, S.; Otegui, N.; Jimeno, A.; Serrano, D.; Calvo, A. Cancer Vulnerabilities Through Targeting the ATR/Chk1 and ATM/Chk2 Axes in the Context of DNA Damage. Cells 2025, 14, 748. https://doi.org/10.3390/cells14100748
Fernandez A, Artola M, Leon S, Otegui N, Jimeno A, Serrano D, Calvo A. Cancer Vulnerabilities Through Targeting the ATR/Chk1 and ATM/Chk2 Axes in the Context of DNA Damage. Cells. 2025; 14(10):748. https://doi.org/10.3390/cells14100748
Chicago/Turabian StyleFernandez, Anell, Maider Artola, Sergio Leon, Nerea Otegui, Aroa Jimeno, Diego Serrano, and Alfonso Calvo. 2025. "Cancer Vulnerabilities Through Targeting the ATR/Chk1 and ATM/Chk2 Axes in the Context of DNA Damage" Cells 14, no. 10: 748. https://doi.org/10.3390/cells14100748
APA StyleFernandez, A., Artola, M., Leon, S., Otegui, N., Jimeno, A., Serrano, D., & Calvo, A. (2025). Cancer Vulnerabilities Through Targeting the ATR/Chk1 and ATM/Chk2 Axes in the Context of DNA Damage. Cells, 14(10), 748. https://doi.org/10.3390/cells14100748