

Effects of Sodium–Glucose Cotransporter 2 Inhibitors on Calcium Homeostasis: Where We Stand Now
 , , , , ,
, , , , , 
 ,
,  ,
, 
Abstract
1. Introduction
1.1. Diabetes and Bone Fragility
1.2. Effect of Antidiabetic Drug on Bone Health
2. Methods
Search Strategy
3. The Controversial Role of SGLT2i on Fracture Risk
3.1. Preclinical Studies
3.2. Clinical Studies
4. Discussion and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2hPG | Postprandial blood glucose |
| AGEs | Advanced glycation end products |
| BAD | Bcl-2 agonist of cell death |
| BMAT | Bone Marrow Adipose Tissue |
| BMD | Bone mineral density |
| BMI | Body Mass Index |
| CANVAS | CANagliflozin cardioVascular Assessment Study Program |
| CANVAS-R | CANagliflozin cardioVascular Renal Assessment Study Program |
| CTX | Carboxy-terminal telopeptide of collagen type 1 |
| FBG | Fasting blood glucose |
| FGF-23 | Fibroblast Growth Factor 23 |
| HbA1c | Hemoglobin A1C |
| IGFR-1 | Insulin-like growth factor 1 receptor |
| PTH | Parathyrod Hormone |
| SGLT2i | Sodium-glucose cotransporter-2 inhibitors |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
References
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://www.diabetesatlas.org (accessed on 12 May 2025).
- Russo, G.T.; Manicardi, V.; Rossi, M.C.; Orsi, E.; Solini, A. Sex- and gender-differences in chronic long-term complications of type 1 and type 2 diabetes mellitus in Italy. Nutr. Metab. Cardiovasc. Dis. NMCD 2022, 32, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xie, Q.; Pan, X.; Zhang, R.; Zhang, X.; Peng, G.; Zhang, Y.; Shen, S.; Tong, N. Type 2 diabetes mellitus in adults: Pathogenesis, prevention and therapy. Signal Transduct. Target. Ther. 2024, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, I.; Catalano, A.; Gennari, L.; Gaudio, A. Osteoporosis and Fragility Fractures in Type 2 Diabetes. J. Diabetes Res. 2020, 2020, 9342696. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, C.; Colangelo, L.; Santori, R.; Renella, M.; Mastrantonio, M.; Minisola, S.; Pepe, J. The Interplay Between Bone and Glucose Metabolism. Front. Endocrinol. 2020, 11, 122. [Google Scholar] [CrossRef]
- Joshi, A.; Varthakavi, P.; Chadha, M.; Bhagwat, N. A study of bone mineral density and its determinants in type 1 diabetes mellitus. J. Osteoporos. 2013, 2013, 397814. [Google Scholar] [CrossRef]
- Thrailkill, K.; Lumpkin, C.; Bunn, R.; Kemp, S.; Fowlkes, J. Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E735–E745. [Google Scholar] [CrossRef]
- Mieczkowska, A.; Mansur, S.; Irwin, N.; Flatt, P.; Chappard, D.; Mabilleau, G. Alteration of the bone tissue material properties in type 1 diabetes mellitus: A Fourier transform infrared microspectroscopy study. Bone 2015, 76, 31–39. [Google Scholar] [CrossRef]
- Uchida, T.; Nakamura, T.; Hashimoto, N.; Matsuda, T.; Kotani, K.; Sakaue, H.; Kido, Y.; Hayashi, Y.; Nakayama, K.I.; White, M.F.; et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat. Med. 2005, 11, 175–182. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.; Eder, A.; Mao, M.; Bast, R.C.; Boyd, D.; Mills, G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999, 18, 6635–6640. [Google Scholar] [CrossRef]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol. 2017, 13, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, G.; D’Amato, G.; De Santis, S.; Grano, M.; Faienza, M.F. Mechanisms of altered bone remodeling in children with type 1 diabetes. World J. Diabetes 2021, 12, 997–1009. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, G.F.; Zhao, P.P.; Xiao, W.J.; Karasik, D.; Xu, Y.J.; Zheng, H.F. The paradox of bone mineral density and fracture risk in type 2 diabetes. Endocrine 2024, 85, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Romero-Díaz, C.; Duarte-Montero, D.; Gutiérrez-Romero, S.A.; Mendivil, C.O. Diabetes and Bone Fragility. Diabetes Ther. 2021, 12, 71–86. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tencerova, M.; Figeac, F.; Ditzel, N.; Taipaleenmaki, H.; Nielsen, T.K.; Kassem, M. High-fat diet-induced obesity promotes expansion of bone marrow adipose tissue and impairs skeletal stem cell functions in mice. J. Bone Miner. Res. 2018, 33, 1154–1165. [Google Scholar] [CrossRef]
- Figeacm, F.; Tencerova, M.; Ali, D.; Andersen, T.L.; Appadoo, D.R.C.; Kerckhofs, G.; Ditzel, N.; Kowal, J.M.; Rauch, A.; Kassem, M. Impaired Bone Fracture Healing in Type 2 Diabetes Is Caused by Defective Functions of Skeletal Progenitor Cells. Stem Cells 2022, 40, 149–164. [Google Scholar] [CrossRef]
- Tencerova, M.; Frost, M.; Figeac, F.; Nielsen, T.K.; Ali, D.; Lauterlein, J.L.; Andersen, T.L.; Haakonsson, A.K.; Rauch, A.; Madsen, J.S.; et al. Obesity-associated hypermetabolism and accelerated senescence of bone marrow stromal stem cells suggest a potential mechanism for bone fragility. Cell Rep. 2019, 27, 2050–2062.e6. [Google Scholar] [CrossRef]
- Teissier, T.; Temkin, V.; Pollak, R.D.; Cox, L.S. Crosstalk Between Senescent Bone Cells and the Bone Tissue Microenvironment Influences Bone Fragility During Chronological Age and in Diabetes. Front. Physiol. 2022, 13, 812157. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Janghorbani, M.; Van Dam, R.M.; Willett, W.C.; Hu, F.B. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol. 2007, 166, 495–505. [Google Scholar] [CrossRef]
- Poundarik, A.; Wu, P.; Evis, Z.; Sroga, G.E.; Ural, A.; Rubin, M.; Vashishth, D. A direct role of collagen glycation in bone fracture. J. Mech. Behav. Biomed. Mater. 2015, 52, 120–130. [Google Scholar] [CrossRef]
- McCarthy, A.; Etcheverry, S.; Bruzzone, L.; Lettieri, G.; Barrio, D.; Cortizo, A. Non-enzymatic glycosylation of a type I collagen matrix: Effects on osteoblastic development and oxidative stress. BMC Cell Biol. 2001, 2, 16. [Google Scholar] [CrossRef]
- Glantschnig, H.; Fisher, J.; Wesolowski, G.; Rodan, G.; Reszka, A. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003, 10, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; He, X.; Farmer, P.; Boden, S.; Kozlowski, M.; Rubin, J.; Nanes, M.S. Inhibition of osteoblast differentiation by tumor necrosis factor-α. Endocrinology 2000, 141, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar] [CrossRef]
- Williams, G.A.; Wang, Y.; Callon, K.E.; Watson, M.; Lin, J.-M.; Lam, J.B.B.; Costa, J.L.; Orpe, A.; Broom, N.; Naot, D.; et al. In vitro and in vivo effects of adiponectin on bone. Endocrinology 2009, 150, 3603–3610. [Google Scholar] [CrossRef]
- Tamura, T.; Yoneda, M.; Yamane, K.; Nakanishi, S.; Nakashima, R.; Okubo, M.; Kohno, N. Serum leptin and adiponectin are positively associated with bone mineral density at the distal radius in patients with type 2 diabetes mellitus. Metabolism 2007, 56, 623–628. [Google Scholar] [CrossRef]
- Pritchard, J.M.; Giangregorio, L.M.; Atkinson, S.A.; Beattie, K.A.; Inglis, D.; Ioannidis, G.; Punthakee, Z.; Adachi, J.D.; Papaioannou, A. Association of larger holes in the trabecular bone at the distal radius in postmenopausal women with T2D compared to controls. Arthritis Care Res. 2012, 64, 83–91. [Google Scholar] [CrossRef]
- Burghardt, A.J.; Issever, A.S.; Schwartz, A.V.; Davis, K.A.; Masharani, U.; Majumdar, S.; Link, T.M. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2010, 95, 5045–5055. [Google Scholar] [CrossRef]
- Eller-Vainicher, C.; Cairoli, E.; Grassi, G.; Grassi, F.; Catalano, A.; Merlotti, D.; Falchetti, A.; Gaudio, A.; Chiodini, I.; Gennari, L. Pathophysiology and Management of Type 2 Diabetes Mellitus Bone Fragility. J. Diabetes Res. 2020, 2020, 7608964. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Melton, L.J.; Leibson, C.L.; Achenbach, S.J.; Therneau, T.M.; Khosla, S. Fracture risk in type 2 diabetes: Update of a population-based study. JBMM 2008, 23, 1334–1342. [Google Scholar] [CrossRef]
- Palermo, A.; D’Onofrio, L.; Eastell, R.; Schwartz, A.V.; Pozzilli, P.; Napoli, N. Oral anti-diabetic drugs and fracture risk, cut to the bone: Safe or dangerous? A narrative review. Osteoporos. Int. 2015, 26, 2073–2089. [Google Scholar] [CrossRef]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia 2005, 48, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.V.; Sellmeyer, D.E. Thiazolidinedione therapy gets complicated: Is bone loss the price of improved insulin resistance? Diabetes Care 2007, 30, 1670–1671. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, S.K.; Jo, K.J.; Song, D.-Y.; Lim, D.-M.; Park, K.-Y.; Bonewald, L.F.; Kim, B.-J. Exendin-4 increases bone mineral density in type 2 diabetic OLETF rats potentially through the down-regulation of SOST/sclerostin in osteocytes. Life Sci. 2013, 92, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Sheng, H.; Zhang, M.; Bu, L.; Yang, P.; Li, L.; Li, F.; Sheng, C.; Han, Y.; Qu, S.; et al. Risk of bone fractures associated with glucagon-like peptide-1 receptor agonists’ treatment: A meta-analysis of randomized controlled trials. Endocrine 2015, 48, 107–115. [Google Scholar] [CrossRef]
- Mamza, J.; Marlin, C.; Wang, C.; Chokkalingam, K.; Idris, I. DPP-4 inhibitor therapy and bone fractures in people with Type 2 diabetes—A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2016, 116, 288–298. [Google Scholar] [CrossRef]
- Lv, F.; Cai, X.; Lin, C.; Yang, W.; Ji, L. Effects of Semaglutide and Tirzepatide on Bone Metabolism in Type 2 Diabetic Mice. Pharmaceuticals 2024, 17, 1655. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- American Diabetes Association Professional Practice Committee. Standard of Care in Diabetes 2025. Diabetes Care 2024, 48, S1–S352. [Google Scholar]
- Vallon, V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu. Rev. Med. 2015, 66, 255–270. [Google Scholar] [CrossRef]
- Saisho, Y. SGLT2 inhibitors: The star in the treatment of type 2 diabetes? Diseases 2020, 8, 14. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. CANVAS Program Collaborative Group: Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- US Food and Drug Administration: Highlights of Prescribing Information: Invokana, 2018. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-revises-label-diabetes-drug-canagliflozin-invokana-invokamet (accessed on 10 August 2021).
- Health Canada: Invokana. Product Monograph. Available online: https://pdf.hres.ca/dpd_pm/00038913.PDF (accessed on 10 August 2021).
- Thrailkill, K.M.; Clay Bunn, R.; Nyman, J.S.; Rettiganti, M.R.; Cockrell, G.E.; Wahl, E.C.; Uppuganti, S.; Lumpkin, C.K., Jr.; Fowlkes, J.L. SGLT2 inhibitor therapy improves blood glucose but does not prevent diabetic bone disease in diabetic DBA/2J male mice. Bone 2016, 82, 101–107, Erratum in Bone 2017, 105, 316. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thrailkill, K.M.; Nyman, J.S.; Bunn, R.C.; Uppuganti, S.; Thompson, K.L.; Lumpkin, C.J.; Kalaitzoglou, E.; Fowlkes, J.L. The impact of SGLT2 inhibitors, compared with insulin, on diabetic bone disease in a mouse model of type 1 diabetes. Bone 2016, 94, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Gerber, C.; Wang, X.; David, V.; Quaggin, S.E.; Isakova, T.; Martin, A. Long-Term Effects of Sglt2 Deletion on Bone and Mineral Metabolism in Mice. JBMR Plus 2021, 5, e10526. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kalanski, S.; Pradhan, S.; Hon, A.; Xia, Y.; Safvati, N.; Rivera, J.C.; Lu, M.; Demer, L.L.; Tintut, Y. Effects of Empagliflozin on Vascular and Skeletal Mineralization in Hyperlipidemic Mice. Vasc. Pharmacol. 2024, 155, 107376. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H. Canagliflozin: Clinical efficacy and safety. In Proceedings of the Endocrinology and Metabolic Drugs Advisory Committee Meeting, Silver Spring, MD, USA, 11 December 2013. [Google Scholar]
- Jabbour, S.; Seufert, J.; Scheen, A.; Bailey, C.J.; Karup, C.; Langkilde, A.M. Dapagliflozin in patients with type 2 diabetes mellitus: A pooled analysis of safety data from phase IIb/III clinical trials. Diabetes Obes Metab. 2018, 20, 620–628. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Skeith, M.D.; Healey, L.A.; Cutler, R.E. Effect of phloridzin on uric acid excretion in man. Am. J. Physiol. Content 1970, 219, 1080–1082. [Google Scholar] [CrossRef]
- Taylor, S.I.; Blau, J.E.; Rother, K.I. Possible adverse effects of SGLT2 inhibitors on bone. Lancet Diabetes Endocrinol. 2015, 3, 8–10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bilezikian, J.P.; Watts, N.B.; Usiskin, K.; Polidori, D.; Fung, A.; Sullivan, D.; Rosenthal, N. Evaluation of bone mineral density and bone biomarkers in patients with type 2 diabetes treated with canagliflozin. J. Clin. Endocrinol. Metab. 2016, 101, 44–51. [Google Scholar] [CrossRef]
- Rosenstock, J.; Frias, J.; Pall, D.; Charbonnel, B.; Pascu, R.; Saur, D.; Darekar, A.; Huyck, S.; Shi, H.; Lauring, B.; et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy [VERTIS MET]. Diabetes Obes. Metab. 2018, 20, 520–529. [Google Scholar] [CrossRef]
- Ljunggren, O.; Bolinder, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sjostrom, C.D.; Sugg, J.; Parikh, S. Dapagliflozin has no effect on markers of bone formation and resorption or bone mineral density in patients with inadequately controlled type 2 diabetes mellitus on metformin. Diabetes Obes. Metab. 2012, 14, 990–999. [Google Scholar] [CrossRef]
- Lee, S.; Yu, M.H.; Hong, N.; Kim, K.J.; Kim, H.K.; Rhee, Y.; Lee, M.; Kim, K.M. Association of sodium-glucose cotransporter 2 inhibitor use with risk of osteoporotic fracture among older women: A nationwide, population-based cohort study. Diabetes Res. Clin. Pract. 2024, 213, 111712. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Guo, A.; Zhang, K.; Jiang, Y.; Liu, H. The effect of empagliflozin [sodium–glucose cotransporter-2 inhibitor] on osteoporosis and glycemic parameters in patients with type 2 diabetes: A quasi-experimental study. BMC Musculoskelet. Disord. 2024, 25, 793. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Yu, Y.; Duan, J.; Bi, S.; Swe, K.N.C.; Xi, Z.; Gao, Y.; Zhou, Y.; Nie, X.; Liu, W. Sodium-glucose cotransporter 2 inhibitors and fracture risk in patients with type 2 diabetes mellitus: A meta-analysis of randomized controlled trials. Ther. Adv. Chronic Dis. 2020, 11, 2040622320961599. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Neal, B.; Perkovic, V.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Fabbrini, E.; Sun, T.; Li, Q.; et al. CANVAS Program Collaborative Group. Canagliflozin for Primary and Secondary Prevention of Cardiovascular Events: Results From the CANVAS Program [Canagliflozin Cardiovascular Assessment Study]. Circulation 2018, 137, 323–334. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neal, B.; Perkovic, V.; Matthews, D.R.; Mahaffey, K.W.; Fulcher, G.; Meininger, G.; Erondu, N.; Desai, M.; Shaw, W.; Vercruysse, F.; et al. Rationale, design and baseline characteristics of the CANagliflozin cardioVascular Assessment Study–Renal (CANVAS-R): A randomized, placebo-controlled trial. Diabetes Obes. Metab. 2017, 19, 387–393. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bellone, F.; Cinquegrani, M.; Nicotera, R.; Carullo, N.; Casarella, A.; Presta, P.; Andreucci, M.; Squadrito, G.; Mandraffino, G.; Prunestì, M.; et al. Role of Vitamin K in Chronic Kidney Disease: A Focus on Bone and Cardiovascular Health. Int. J. Mol. Sci. 2022, 23, 5282. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lupsa, B.C.; Inzucchi, S.E. Use of SGLT2 inhibitors in type 2 diabetes: Weighing the risks and benefits. Diabetologia 2018, 61, 2118–2125. [Google Scholar] [CrossRef]
- Gorboulev, V.; Schurmann, A.; Vallon, V.; Kipp, H.; Jaschke, A.; Klessen, D.; Friedrich, A.; Scherneck, S.; Rieg, T.; Cunard, R.; et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 2012, 61, 187–196. [Google Scholar] [CrossRef]
- Sha, S.; Polidori, D.; Farrell, K.; Ghosh, A.; Natarajan, J.; Vaccaro, N.; Pinheiro, J.; Rothenberg, P.; Plum-Mörschel, L. Pharmacodynamic differences between canagliflozin and dapagliflozin: Results of a randomized, double-blind, crossover study. Diabetes Obes. Metab. 2015, 17, 188–197. [Google Scholar] [CrossRef]
- Mamidi, R.N.; Proctor, J.; De Jonghe, S.; Feyen, B.; Moesen, E.; Vinken, P.; Ma, J.Y.; Bryant, S.; Snook, S.; Louden, C.; et al. Carbohydrate malabsorption mechanism for tumor formation in rats treated with the SGLT2 inhibitor canagliflozin. Chem. Biol. Interact. 2014, 221, 109–118. [Google Scholar] [CrossRef]
- De Jonghe, S.; Proctor, J.; Vinken, P.; Feyen, B.; Wynant, I.; Marien, D.; Geys, H.; Mamidi, R.N.; Johnson, M.D. Carcinogenicity in rats of the antidiabetic SGLT2 inhibitor canagliflozin. Chem. Biol. Interact. 2014, 224, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, F.; Zhang, Y.; Zhang, J.; Sheng, Y.; Wang, W.; Li, Y. Effect of SGLT2 inhibitors on fractures, BMD, and bone metabolism markers in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Osteoporos. Int. 2023, 34, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, T.; Cheng, Y.; Lu, Y.; Xue, M.; Xu, L.; Liu, X.; Yu, X.; Sun, B.; Chen, L. Effects of SGLT2 inhibitors on fractures and bone mineral density in type 2 diabetes: An updated meta-analysis. Diabetes Metab. Res. Rev. 2019, 35, e3170. [Google Scholar] [CrossRef] [PubMed]

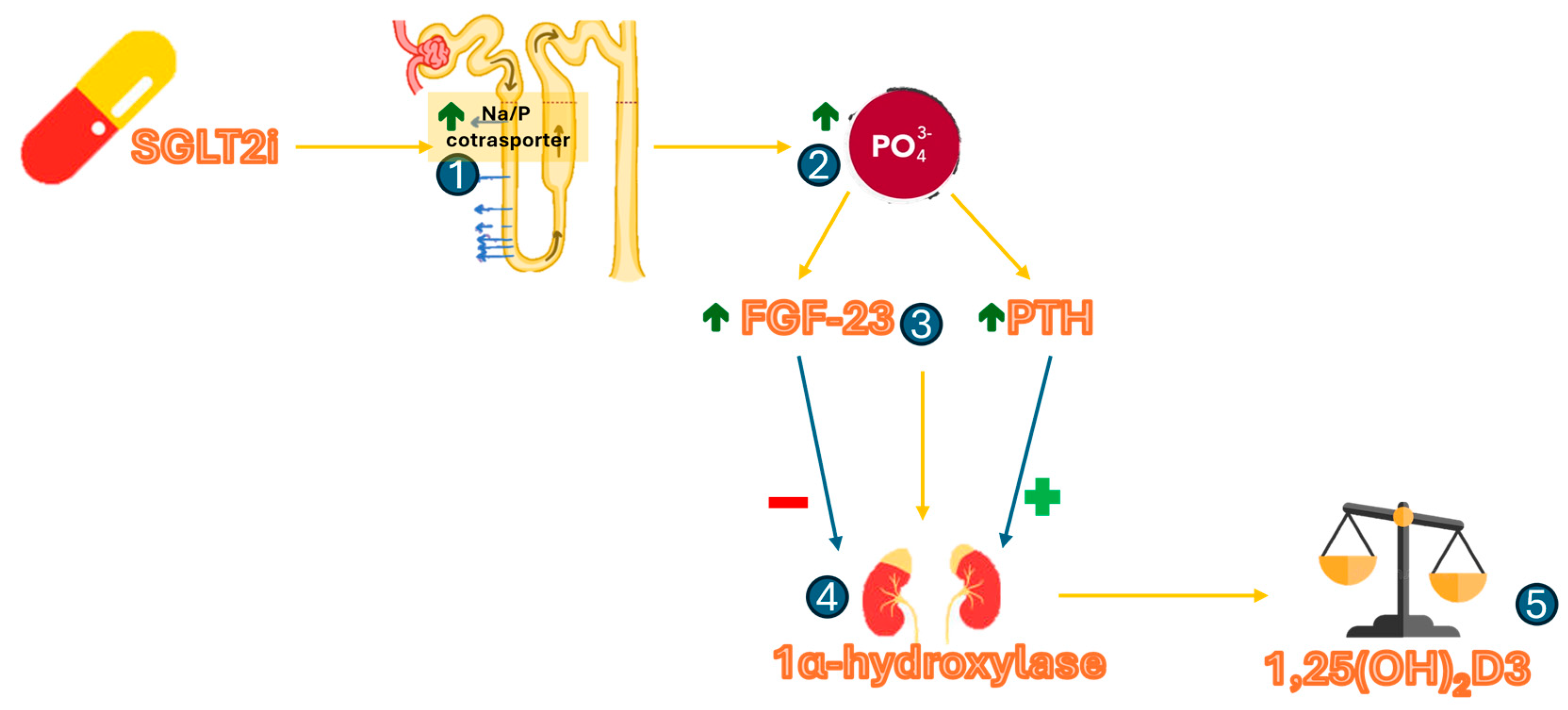
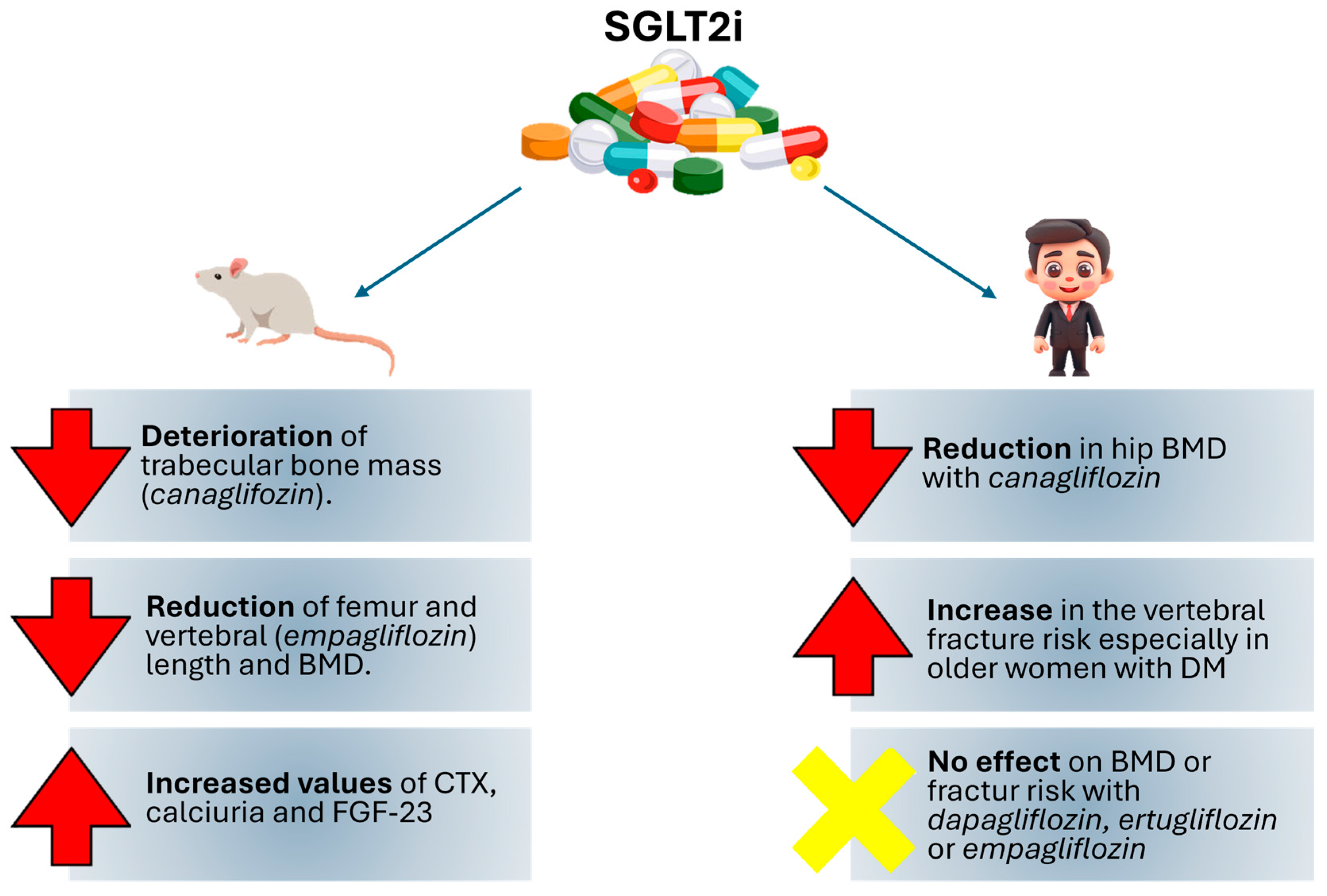

{kind=link}
{kind=link}
{kind=link}
| Methods | Population | Drug | Results | Ref. |
|---|---|---|---|---|
| Randomized study. 26 weeks, double-blind, placebo-controlled period and a 78 weeks, double-blind, placebo-controlled extension. | 716 T2D patients aged 55–80 years | Canagliflozin 100 mg or 300 mg | ↓ Total hip BMD over 104 weeks ↑ Collagen type 1 β-carboxy-telopeptide at week 52 ↓ Estradiol | [38] |
| Double-blind, 26 weeks, multicenter study with ongoing 78 weeks extension | 621 T2D patients | Ertugliflozin 5 or 15 mg | No adverse impact on BMD at week 26 | [39] |
| International, multi-center, randomized, parallel-group, double-blind, placebo-controlled study | 182 T2D patients (women 55–75 years and men 30–75 years) | Dapagliflozin 10 mg | No significant changes from baseline in P1NP, CTX, or BMD | [40] |
| The population-based cohort study data from the National Health Insurance Service of Korea (2013–2020) | 3959 T2D women of age > 65 years | 1333 patients in treatment with dapagliflozin, empagliflozin, ipragliflozin, and ertugliflozin or other drugs | ↑ Risk of vertebral fracture than non-SGLT2i use in elderly women | [41] |
| Quasi-experimental study | 100 T2D patients with osteoporosis | Empagliflozin or placebo | ↑ Bone mineral density, ↑phosphorus and calcium metabolism ↓ Incidence of fracture | [42] |
| Multicenter, prospective, randomized, double-blind, placebo-controlled trial | 10142 T2D patients | Canagliflozin or placebo | ↑ Risk of low-trauma fractures and all fractures in the canagliflozin group | [44] |
| Multicenter, prospective, randomized, double-blind, placebo-controlled trial | 5812 T2D patients | Canagliflozin 100 or 300 mg | No difference in risk factors | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuttone, A.; Xourafa, A.; Morace, C.; Cannavò, V.; Bueti, F.M.; Mandraffino, G.; Squadrito, G.; Basile, G.; Gaudio, A.; Catalano, A.; et al. Effects of Sodium–Glucose Cotransporter 2 Inhibitors on Calcium Homeostasis: Where We Stand Now. Cells 2025, 14, 724. https://doi.org/10.3390/cells14100724
Cuttone A, Xourafa A, Morace C, Cannavò V, Bueti FM, Mandraffino G, Squadrito G, Basile G, Gaudio A, Catalano A, et al. Effects of Sodium–Glucose Cotransporter 2 Inhibitors on Calcium Homeostasis: Where We Stand Now. Cells. 2025; 14(10):724. https://doi.org/10.3390/cells14100724
Chicago/Turabian StyleCuttone, Alessandro, Anastasia Xourafa, Carmela Morace, Vittorio Cannavò, Francesca Maria Bueti, Giuseppe Mandraffino, Giovanni Squadrito, Giorgio Basile, Agostino Gaudio, Antonino Catalano, and et al. 2025. "Effects of Sodium–Glucose Cotransporter 2 Inhibitors on Calcium Homeostasis: Where We Stand Now" Cells 14, no. 10: 724. https://doi.org/10.3390/cells14100724
APA StyleCuttone, A., Xourafa, A., Morace, C., Cannavò, V., Bueti, F. M., Mandraffino, G., Squadrito, G., Basile, G., Gaudio, A., Catalano, A., Russo, G. T., & Bellone, F. (2025). Effects of Sodium–Glucose Cotransporter 2 Inhibitors on Calcium Homeostasis: Where We Stand Now. Cells, 14(10), 724. https://doi.org/10.3390/cells14100724