
Molecular Targets of Oxidative Stress: Focus on Nuclear Factor Erythroid 2–Related Factor 2 Function in Leukemia and Other Cancers

 , ,
, , 
Abstract

1. Introduction
2. Nrf2′s Functional Domains and Its Regulation by Keap1
KEAP-1-Independent Regulation of Nrf2
3. Regulation of Metabolism by Nrf2
3.1. Glycolytic Pathway
3.2. Pentose Phosphate Pathway
3.3. Purine Biosynthesis Pathways
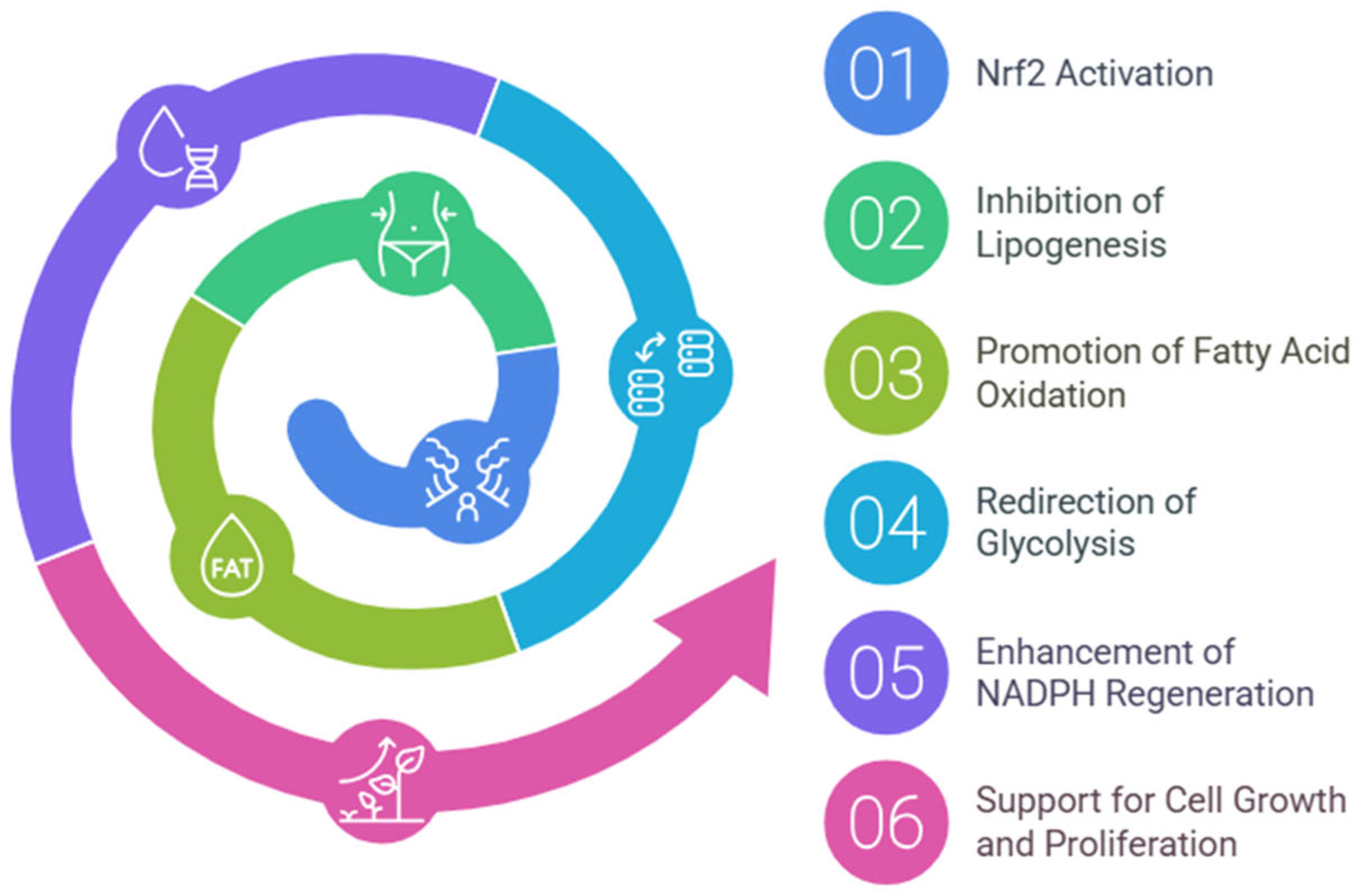
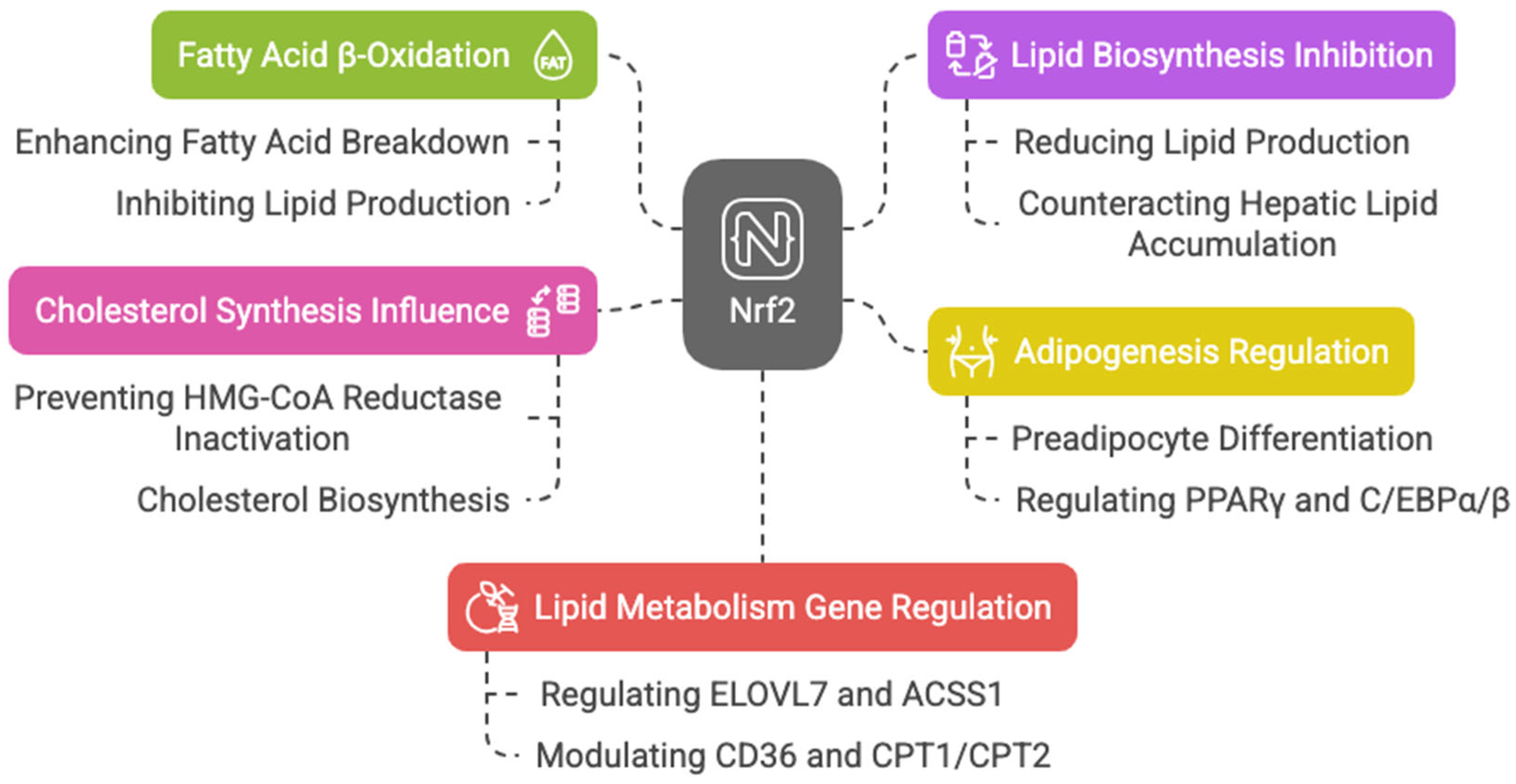
3.4. Lipid Metabolism
3.5. Amino Acid Metabolism
4. Nrf2-Regulated Mitochondrial Functions
5. Nrf2 Dysregulation in Cancer
5.1. Somatic Mutations in Nrf2-Keap1 System in Cancers
5.2. Metabolic Dysregulation in Cancer
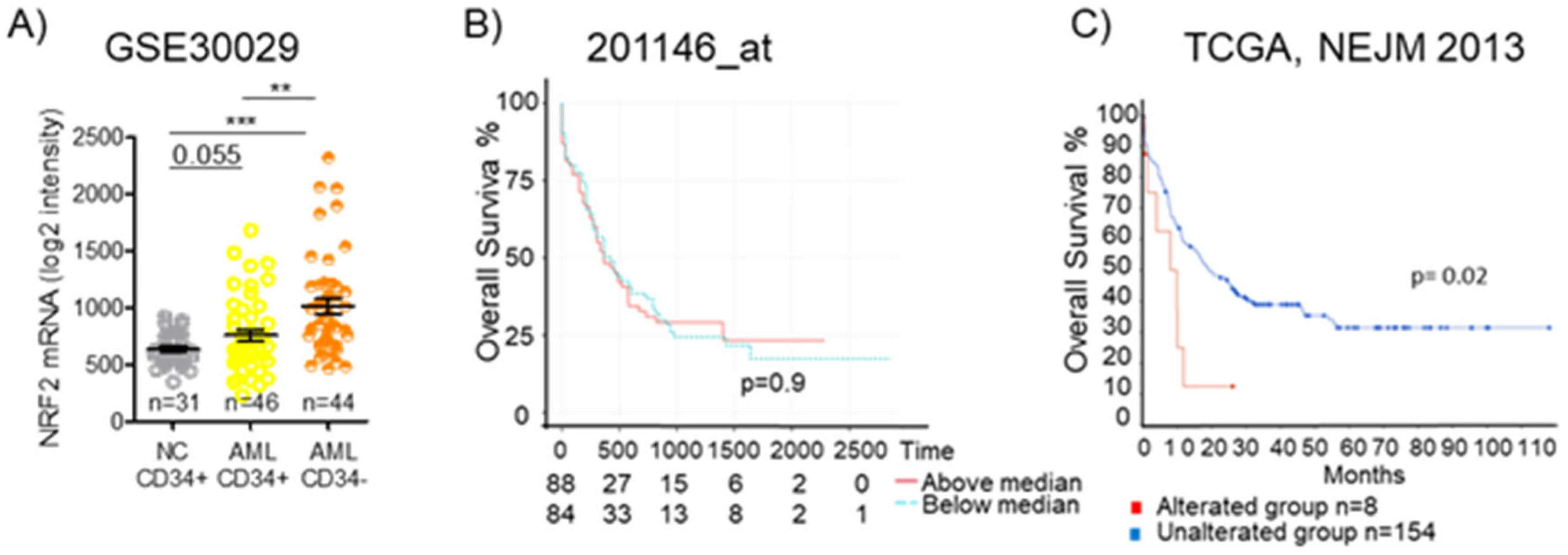
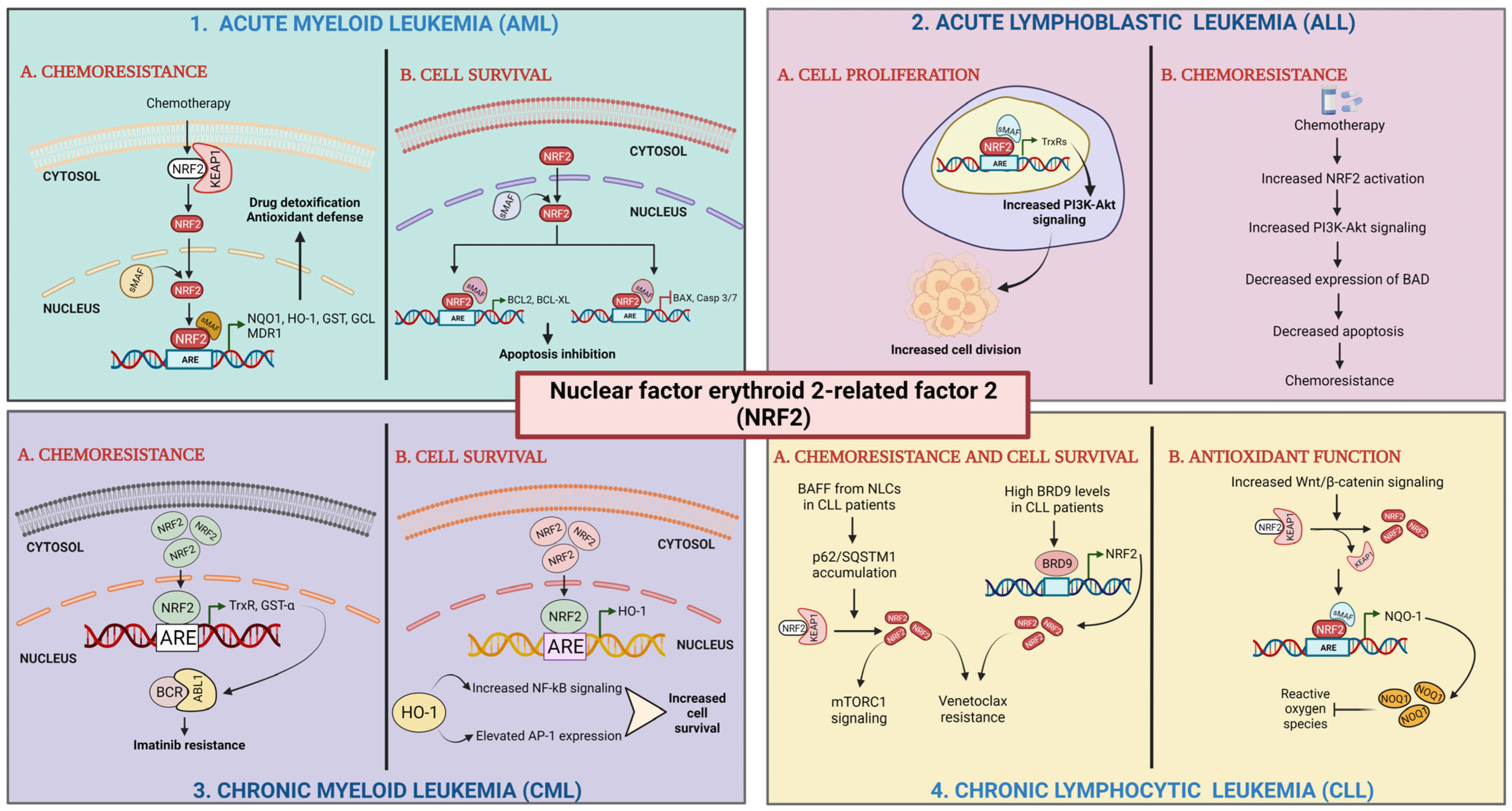
5.3. Nrf2 Regulatory Networks in Leukemia
6. Nrf2 and Cancer Treatment Resistance
Role of Nrf2 in Ferroptosis-Mediated Drug Resistance
7. Nrf2 Inhibitors
8. Nrf2 in Cancer: Opportunities and Problems
8.1. Strengths of Nrf2 as a Target in Cancer
- Protection against oxidative stress: Nrf2 protects cancer cells from the high levels of oxidative stress that result from rapid cell proliferation and altered metabolism. By upregulating antioxidant genes (e.g., HO-1, NQO1, and GCLC), Nrf2 helps maintain cellular redox homeostasis, promoting cancer cell survival in tumors with high oxidative stress. In early-stage cancers, Nrf2 can contribute to DNA repair and resistance to apoptosis, thus supporting tumor growth and favoring the emergence of clones resistant to therapies [202].
- Chemoresistance: Nrf2 activation leads to the expression of drug-metabolizing enzymes (e.g., GST and CYP450), which detoxify chemotherapeutic agents, thus reducing the effectiveness of chemotherapy [204]. In cancers such as non-small-cell lung cancer (NSCLC) and ovarian cancer, elevated Nrf2 expression is linked to poor responses to chemotherapy drugs like cisplatin and paclitaxel [7].
- Support for metastasis: Nrf2 plays a role in the epithelial–mesenchymal transition (EMT), a process that allows cancer cells to acquire migratory and invasive properties. In cancers like breast, pancreatic, and prostate cancer, Nrf2 promotes metastasis by enhancing the invasive potential of tumor cells [205].
- Nrf2 in late-stage cancers: The overexpression of Nrf2 in late-stage cancers (e.g., lung, colon, and liver cancers) is associated with aggressive tumor behavior, as Nrf2 helps cancer cells resist oxidative stress and chemotherapeutic agents. In these cases, Nrf2 inhibitors could reduce tumor progression and enhance the effectiveness of chemotherapy [207,208].
- Cancer stem cells (CSCs): Nrf2 is often involved in maintaining the stem-like properties of cancer stem cells, which are resistant to chemotherapy and contribute to tumor relapse. In cancers like breast, lung, and pancreatic cancers, Nrf2 activation may promote CSC survival and self-renewal, contributing to chemoresistance [205].
- Compromised antioxidant defense in certain tumors: In certain cancers, such as APL, where the Nrf2 half-life is reduced [121] or in glioblastoma and colorectal cancer, where mutations in the Nrf2 pathway impair its antioxidant defense function, these tumors become more vulnerable to oxidative damage [209]. Consequently, they may be more responsive to therapies that induce the generation of ROS, such as radiation or chemotherapy.
8.2. Problems of Nrf2 as a Target in Cancer
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| ROS | Reactive oxygen species |
| PPP | Pentose phosphate pathway |
| ARE | Antioxidant response elements |
| bZIP | Basic leucine zipper |
| Keap1 | Kelch-like ECH-associated protein 1 |
| mTORC1 | Mammalian target for Rapamycin |
| p21 | Cyclin dependent kinase inhibitor1 |
| NEDD8 | Neural precursor cell-expressed developmentally downregulated protein 8 |
| MLL | Mixed-Lineage Leukemia |
| H3K4me3 | H3K4 trimethylation |
| miRNAs | MicroRNAs |
| UTR | Untranslated region |
| EpRE | Electrophile response element |
| FAO | Fatty acids oxidation |
| HK1/2 | Hexokinase 1 and 2 |
| GPI1 | Glucose phosphate isomerase 1 |
| PFK2 | 6-phosphofructo-2-kinase |
| ALDA | Fructose-bisphosphate aldolase A |
| ENO1 | Enolase 1 |
| PK | Pyruvate kinase |
| PPP | Pentose phosphate pathway |
| GBE1 | 1,4-alpha-glucan branching enzyme 1 |
| GAA | Glucosidase alpha |
| PGM | Phosphoglucomutase |
| G1P | Glucose-1-phosphate |
| G6P | Glucose-6-phosphate |
| G6PD | Glucose-6-phosphate dehydrogenase |
| HO-1 | Heme oxygenase 1 |
| TALDO1 | Transaldolase 1 |
| TKT | Transketolase |
| PPAT | Phosphoribosyl pyrophosphate amidotransferase |
| MTHFD2 | Methylenetetrahydrofolate dehydrogenase 2 |
| IDH1 | Isocitrate dehydrogenase 1 |
| ME1 | Malic enzyme 1 |
| PRPP | Phosphoribosyl pyrophosphate |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-CoA |
| AML | Acute myeloid leukemia |
| PUFAs | Polyunsaturated fatty acids |
| α-KG | α-ketoglutarate |
| PGD | Phosphogluconate dehydrogenase |
| HHT | Homoharringtonine |
| HSC | Hematopoietic stem cells |
| GMP | Granulocyte-macrophage progenitors |
| TCGA | The Cancer Genome Atlas |
| OSm | Medium overall survival |
| MMR | Mismatch repair |
| GP | Glutathione peroxidase |
| GST | Glutathione S-transferase |
| GCL | γ-glutamyl-cysteine ligase |
| CA | Carnosic acid |
| AP-1 | Activator protein-1 |
| 1,25D | 1α,25-dihydroxyvitamin D3 |
| APL | Acute promyelocytic leukemia |
| ATO | Arsenic trioxide |
| MRP1 | Multidrug resistance protein 1 |
| 4-HNE | 4-Hydroxynonenal |
| ALL | Acute lymphoblastic leukemia |
| PAQR3 | Progestin and AdipoQ Receptor family member 3 |
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular signal kinase |
| PI3K | Phosphatidylinositol-3-kinase |
| AKT | Protein kinase B |
| UGT | UDP-glucuronosyl transferases |
| ALDH1A1 | Aldehyde dehydrogenase 1 |
| RXR-α | Retinoid-X-receptor α |
| RAR-α | Retinoic acid receptor α |
References
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Zhang, D.D. Thirty years of NRF2: Advances and therapeutic challenges. Nat. Rev. Drug Discov. 2025, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Patinen, T.; Jawahar Deen, A.; Pitkänen, S.; Härkönen, J.; Kansanen, E.; Küblbeck, J.; Levonen, A.L. The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biol. 2023, 63, 102726. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Inhibition of the NRF2/KEAP1 Axis: A Promising Therapeutic Strategy to Alter Redox Balance of Cancer Cells. Antioxid. Redox Signal. 2021, 34, 1428–1483. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- McCord, J.M.; Gao, B.; Hybertson, B.M. The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review. Antioxidants 2023, 12, 366. [Google Scholar] [CrossRef]
- Camiña, N.; Penning, T.M. Genetic and epigenetic regulation of the NRF2-KEAP1 pathway in human lung cancer. Br. J. Cancer 2022, 126, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB Protein Is an Adaptor That Bridges Nrf2 to a Cul3-Based E3 Ligase: Oxidative Stress Sensing by a Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase to Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef]
- Shakya, A.; Liu, P.; Godek, J.; McKee, N.W.; Dodson, M.; Anandhan, A.; Ooi, A.; Garcia, J.G.N.; Costa, M.; Chapman, E.; et al. The NRF2-p97-NRF2 negative feedback loop. Redox Biol. 2023, 65, 102839. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological Significance of Reactive Cysteine Residues of Keap1 in Determining Nrf2 Activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.s.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Sun, Z.; Chen, W.; Zhang, D.D. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell Cycle 2009, 8, 3255. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3β-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275. [Google Scholar] [CrossRef]
- Lo, S.-C.; Hannink, M. CAND1-Mediated Substrate Adaptor Recycling Is Required for Efficient Repression of Nrf2 by Keap1. Mol. Cell. Biol. 2006, 26, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Ah Kang, K.; Piao, M.J.; Ryu, Y.S.; Kang, H.K.; Chang, W.Y.; Keum, Y.S.; Hyun, J.W. Interaction of DNA demethylase and histone methyltransferase upregulates Nrf2 in 5-fluorouracil-resistant colon cancer cells. Oncotarget 2016, 7, 40594–40620. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Mol. Cell. Biol. 2009, 29, 2658. [Google Scholar] [CrossRef]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of Novel microRNAs in Post-Transcriptional Control of Nrf2 Expression and Redox Homeostasis in Neuronal, SH-SY5Y Cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef]
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef]
- Chu, X.; Zhong, L.; Dan, W.; Wang, X.; Zhang, Z.; Liu, Z.; Lu, Y.; Shao, X.; Zhou, Z.; Chen, S.; et al. DNMT3A R882H mutation drives daunorubicin resistance in acute myeloid leukemia via regulating NRF2/NQO1 pathway. Cell Commun. Signal. CCS 2022, 20, 168. [Google Scholar] [CrossRef]
- Díaz, M.; Valdés-Baizabal, C.; de Pablo, D.P.; Marin, R. Age-Dependent Changes in Nrf2/Keap1 and Target Antioxidant Protein Expression Correlate to Lipoxidative Adducts, and Are Modulated by Dietary N-3 LCPUFA in the Hippocampus of Mice. Antioxidants 2024, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, A.; Okamura, Y.; Aoki, Y.; Ishida, N.; Kumada, K.; Minegishi, N.; Katsuoka, F.; Kinoshita, K.; Yamamoto, M. Identification of Dominant Transcripts in Oxidative Stress Response by a Full-Length Transcriptome Analysis. Mol. Cell Biol. 2021, 41, e00472-20. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.P.; Chorley, B.; Hiemstra, S.; Wink, S.; Wang, X.; Bell, D.A.; van de Water, B.; Corton, J.C. Mining a human transcriptome database for chemical modulators of NRF2. PLoS ONE 2020, 15, e0239367. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Wu, K.C.; Klaassen, C.D. Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS ONE 2013, 8, e59122. [Google Scholar] [CrossRef]
- Kulkarni, S.R.; Donepudi, A.C.; Xu, J.; Wei, W.; Cheng, Q.C.; Driscoll, M.V.; Johnson, D.A.; Johnson, J.A.; Li, X.; Slitt, A.L. Fasting induces nuclear factor E2-related factor 2 and ATP-binding Cassette transporters via protein kinase A and Sirtuin-1 in mouse and human. Antioxid. Redox Signal. 2014, 20, 15–30. [Google Scholar] [CrossRef]
- Bauer, M.; Hamm, A.C.; Bonaus, M.; Jacob, A.; Jaekel, J.; Schorle, H.; Pankratz, M.J.; Katzenberger, J.D. Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol. Genom. 2004, 17, 230–244. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, L.M.; Xie, J.Y.; Han, H.; Zhu, B.F.; Wang, L.J.; Wang, W.J. High Expression of PKM2 Was Associated with the Poor Prognosis of Acute Leukemia. Cancer Manag. Res. 2021, 13, 7851–7858. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Yang, Z.; Tang, Y.; Tao, Y.; Zhan, Q.; Lei, L.; Jing, Y.; Jiang, X.; Jin, H.; et al. Glycolytic Enzyme PKM2 Mediates Autophagic Activation to Promote Cell Survival in NPM1-Mutated Leukemia. Int. J. Biol. Sci. 2019, 15, 882–894. [Google Scholar] [CrossRef]
- Fu, J.; Xiong, Z.; Huang, C.; Li, J.; Yang, W.; Han, Y.; Paiboonrungruan, C.; Major, M.B.; Chen, K.N.; Kang, X.; et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019, 294, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Gu, J.; Jiang, H.; Ahmed, A.; Zhang, Z.; Gu, Y. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to the development of pulmonary arterial hypertension. J. Mol. Cell Cardiol. 2016, 91, 179–187. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Chang, C.W.; Chen, Y.S.; Tsay, Y.G.; Han, C.L.; Chen, Y.J.; Yang, C.C.; Hung, K.F.; Lin, C.H.; Huang, T.Y.; Kao, S.Y.; et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 2018, 9, 194. [Google Scholar] [CrossRef]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Ohl, K.; Fragoulis, A.; Klemm, P.; Baumeister, J.; Klock, W.; Verjans, E.; Böll, S.; Möllmann, J.; Lehrke, M.; Costa, I.; et al. Nrf2 Is a Central Regulator of Metabolic Reprogramming of Myeloid-Derived Suppressor Cells in Steady State and Sepsis. Front. Immunol 2018, 9, 1552. [Google Scholar] [CrossRef]
- Xu, I.M.; Lai, R.K.; Lin, S.H.; Tse, A.P.; Chiu, D.K.; Koh, H.Y.; Law, C.T.; Wong, C.M.; Cai, Z.; Wong, C.C.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Datta, S.; Wang, L.; Liu, T.; Flores-Bellver, M.; Sachdeva, M.; Sinha, D.; Handa, J.T. Nrf2 deficiency decreases NADPH from impaired IDH shuttle and pentose phosphate pathway in retinal pigmented epithelial cells to magnify oxidative stress-induced mitochondrial dysfunction. Aging Cell 2021, 20, e13444. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Tan, V.W.T.; Salmi, T.M.; Karamalakis, A.P.; Gillespie, A.; Ong, A.J.S.; Balic, J.J.; Chan, Y.C.; Bladen, C.E.; Brown, K.K.; Dawson, M.A.; et al. SLAM-ITseq identifies that Nrf2 induces liver regeneration through the pentose phosphate pathway. Dev. Cell 2024, 59, 898–910.e896. [Google Scholar] [CrossRef] [PubMed]
- Onoki, T.; Izumi, Y.; Takahashi, M.; Murakami, S.; Matsumaru, D.; Ohta, N.; Wati, S.M.; Hatanaka, N.; Katsuoka, F.; Okutsu, M.; et al. Skeletal muscle-specific Keap1 disruption modulates fatty acid utilization and enhances exercise capacity in female mice. Redox Biol. 2021, 43, 101966. [Google Scholar] [CrossRef]
- Madsen, M.S.; Siersbæk, R.; Boergesen, M.; Nielsen, R.; Mandrup, S. Peroxisome proliferator-activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading. Mol. Cell Biol. 2014, 34, 939–954. [Google Scholar] [CrossRef]
- Kim, B.R.; Lee, G.Y.; Yu, H.; Maeng, H.J.; Oh, T.J.; Kim, K.M.; Moon, J.H.; Lim, S.; Jang, H.C.; Choi, S.H. Suppression of Nrf2 attenuates adipogenesis and decreases FGF21 expression through PPAR gamma in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2018, 497, 1149–1153. [Google Scholar] [CrossRef]
- Kuri-Harcuch, W.; Velez-delValle, C.; Vazquez-Sandoval, A.; Hernández-Mosqueira, C.; Fernandez-Sanchez, V. A cellular perspective of adipogenesis transcriptional regulation. J. Cell Physiol. 2019, 234, 1111–1129. [Google Scholar] [CrossRef]
- Hou, Y.; Xue, P.; Woods, C.G.; Wang, X.; Fu, J.; Yarborough, K.; Qu, W.; Zhang, Q.; Andersen, M.E.; Pi, J. Association between arsenic suppression of adipogenesis and induction of CHOP10 via the endoplasmic reticulum stress response. Environ. Health Perspect. 2013, 121, 237–243. [Google Scholar] [CrossRef]
- Gao, T.; Lai, M.; Zhu, X.; Ren, S.; Yin, Y.; Wang, Z.; Liu, Z.; Zuo, Z.; Hou, Y.; Pi, J.; et al. Rifampicin impairs adipogenesis by suppressing NRF2-ARE activity in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2021, 413, 115393. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Xue, P.; Bai, Y.; Liu, D.; Woods, C.G.; Yarborough, K.; Fu, J.; Zhang, Q.; Sun, G.; Collins, S.; et al. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein β during adipogenesis. Free Radic. Biol. Med. 2012, 52, 462–472. [Google Scholar] [CrossRef]
- Habeos, G.I.; Filippopoulou, F.; Habeos, E.E.; Kalaitzopoulou, E.; Skipitari, M.; Papadea, P.; Lagoumintzis, G.; Niarchos, A.; Georgiou, C.D.; Chartoumpekis, D.V. Maternal Calorie Restriction Induces a Transcriptional Cytoprotective Response in Embryonic Liver Partially Dependent on Nrf2. Antioxidants 2022, 11, 2274. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Chen, H.; Zirkin, B. Sirt1 and Nrf2: Regulation of Leydig cell oxidant/antioxidant intracellular environment and steroid formation†. Biol. Reprod. 2021, 105, 1307–1316. [Google Scholar] [CrossRef]
- Picou, F.; Debeissat, C.; Bourgeais, J.; Gallay, N.; Ferrié, E.; Foucault, A.; Ravalet, N.; Maciejewski, A.; Vallet, N.; Ducrocq, E.; et al. n-3 Polyunsaturated fatty acids induce acute myeloid leukemia cell death associated with mitochondrial glycolytic switch and Nrf2 pathway activation. Pharmacol. Res. 2018, 136, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Paek, J.; Lo, J.Y.; Narasimhan, S.D.; Nguyen, T.N.; Glover-Cutter, K.; Robida-Stubbs, S.; Suzuki, T.; Yamamoto, M.; Blackwell, T.K.; Curran, S.P. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012, 16, 526–537. [Google Scholar] [CrossRef]
- Maruyama, A.; Tsukamoto, S.; Nishikawa, K.; Yoshida, A.; Harada, N.; Motojima, K.; Ishii, T.; Nakane, A.; Yamamoto, M.; Itoh, K. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch. Biochem. Biophys. 2008, 477, 139–145. [Google Scholar] [CrossRef]
- Barroso, E.; Rodríguez-Rodríguez, R.; Zarei, M.; Pizarro-Degado, J.; Planavila, A.; Palomer, X.; Villarroya, F.; Vázquez-Carrera, M. SIRT3 deficiency exacerbates fatty liver by attenuating the HIF1α-LIPIN 1 pathway and increasing CD36 through Nrf2. Cell Commun. Signal. 2020, 18, 147. [Google Scholar] [CrossRef]
- Pang, S.; Lynn, D.A.; Lo, J.Y.; Paek, J.; Curran, S.P. SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation. Nat. Commun. 2014, 5, 5048. [Google Scholar] [CrossRef]
- Gao, L.; Kumar, V.; Vellichirammal, N.N.; Park, S.Y.; Rudebush, T.L.; Yu, L.; Son, W.M.; Pekas, E.J.; Wafi, A.M.; Hong, J.; et al. Functional, proteomic and bioinformatic analyses of Nrf2- and Keap1-null skeletal muscle. J. Physiol. 2020, 598, 5427–5451. [Google Scholar] [CrossRef]
- Ryan, D.G.; Knatko, E.V.; Casey, A.M.; Hukelmann, J.L.; Dayalan Naidu, S.; Brenes, A.J.; Ekkunagul, T.; Baker, C.; Higgins, M.; Tronci, L.; et al. Nrf2 activation reprograms macrophage intermediary metabolism and suppresses the type I interferon response. iScience 2022, 25, 103827. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liao, Q.; Li, D.; Chen, L.; Zhang, H.; Yi, B. Vitamin D receptor alleviates lipid peroxidation in diabetic nephropathy by regulating ACLY/Nrf2/Keap1 pathway. FASEB J. 2024, 38, e70060. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhai, X.; Qiu, Y.; Lu, X.; Jiao, Y. The Nrf2 in Obesity: A Friend or Foe? Antioxidants 2022, 11, 2067. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Zheng, Z.; Hu, S.; Wang, M.; Feng, J.; Mattjus, P.; Zhang, Z.; Zhang, Y. Loss of Nrf1 rather than Nrf2 leads to inflammatory accumulation of lipids and reactive oxygen species in human hepatoma cells, which is alleviated by 2-bromopalmitate. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119644. [Google Scholar] [CrossRef]
- Egbujor, M.C.; Olaniyan, O.T.; Emeruwa, C.N.; Saha, S.; Saso, L.; Tucci, P. An insight into role of amino acids as antioxidants via NRF2 activation. Amino Acids 2024, 56, 23. [Google Scholar] [CrossRef]
- He, F.; Wu, C.; Li, P.; Li, N.; Zhang, D.; Zhu, Q.; Ren, W.; Peng, Y. Functions and Signaling Pathways of Amino Acids in Intestinal Inflammation. Biomed. Res. Int. 2018, 2018, 9171905. [Google Scholar] [CrossRef]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Reisz, J.A.; Zaberezhnyy, V.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 2018, 58, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Ye, J.; Mancuso, A.; Tong, X.; Ward, P.S.; Fan, J.; Rabinowitz, J.D.; Thompson, C.B. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 6904–6909. [Google Scholar] [CrossRef] [PubMed]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef]
- Alam, M.M.; Kishino, A.; Sung, E.; Sekine, H.; Abe, T.; Murakami, S.; Akaike, T.; Motohashi, H. Contribution of NRF2 to sulfur metabolism and mitochondrial activity. Redox Biol. 2023, 60, 102624. [Google Scholar] [CrossRef]
- Zamponi, E.; Zamponi, N.; Coskun, P.; Quassollo, G.; Lorenzo, A.; Cannas, S.A.; Pigino, G.; Chialvo, D.R.; Gardiner, K.; Busciglio, J.; et al. Nrf2 stabilization prevents critical oxidative damage in Down syndrome cells. Aging Cell 2018, 17, e12812. [Google Scholar] [CrossRef]
- Imhoff, B.R.; Hansen, J.M. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem. J. 2009, 424, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic. Biol. Med. 2022, 189, 136–153. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Cho, H.Y.; Miller-DeGraff, L.; Blankenship-Paris, T.; Wang, X.; Bell, D.A.; Lih, F.; Deterding, L.; Panduri, V.; Morgan, D.L.; Yamamoto, M.; et al. Sulforaphane enriched transcriptome of lung mitochondrial energy metabolism and provided pulmonary injury protection via Nrf2 in mice. Toxicol. Appl. Pharmacol. 2019, 364, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, L.; Xu, H.; Zhou, Q.; Tan, B.; Yi, Q.; Yan, L.; Xie, M.; Zhang, Y.; Tian, J.; et al. NRF2 is required for structural and metabolic maturation of human induced pluripotent stem cell-derived ardiomyocytes. Stem Cell Res. Ther. 2021, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef]
- Cvetko, F.; Caldwell, S.T.; Higgins, M.; Suzuki, T.; Yamamoto, M.; Prag, H.A.; Hartley, R.C.; Dinkova-Kostova, A.T.; Murphy, M.P. Nrf2 is activated by disruption of mitochondrial thiol homeostasis but not by enhanced mitochondrial superoxide production. J. Biol. Chem. 2021, 296, 100169. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci. Rep. 2018, 8, 7420. [Google Scholar] [CrossRef]
- O’Mealey, G.B.; Plafker, K.S.; Berry, W.L.; Janknecht, R.; Chan, J.Y.; Plafker, S.M. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J. Cell Sci. 2017, 130, 3467–3480. [Google Scholar] [CrossRef]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [CrossRef]
- Hoang, D.H.; Buettner, R.; Valerio, M.; Ghoda, L.; Zhang, B.; Kuo, Y.H.; Rosen, S.T.; Burnett, J.; Marcucci, G.; Pullarkat, V.; et al. Arsenic Trioxide and Venetoclax Synergize against AML Progenitors by ROS Induction and Inhibition of Nrf2 Activation. Int. J. Mol. Sci. 2022, 23, 6568. [Google Scholar] [CrossRef]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Sparaneo, A.; Muscarella, L.A. NRF2 Regulation by Noncoding RNAs in Cancers: The Present Knowledge and the Way Forward. Cancers 2020, 12, 3621. [Google Scholar] [CrossRef]
- Feng, J.; Read, O.J.; Dinkova-Kostova, A.T. Nrf2 in TIME: The Emerging Role of Nuclear Factor Erythroid 2-Related Factor 2 in the Tumor Immune Microenvironment. Mol. Cells 2023, 46, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.J.; Yoo, H.S.; Shin, S.; Park, Y.J.; Jeon, S.M. Dysregulation of NRF2 in Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Biomol. Ther. 2018, 26, 57–68. [Google Scholar] [CrossRef]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Karim, M.R.; Salam Teeli, A.; Smiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular Mechanisms Underlying Hepatocellular Carcinoma Induction by Aberrant NRF2 Activation-Mediated Transcription Networks: Interaction of NRF2-KEAP1 Controls the Fate of Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef]
- Hast, B.E.; Cloer, E.W.; Goldfarb, D.; Li, H.; Siesser, P.F.; Yan, F.; Walter, V.; Zheng, N.; Hayes, D.N.; Major, M.B. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014, 74, 808–817. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- Budihardjo, I.I.; Walker, D.L.; Svingen, P.A.; Buckwalter, C.A.; Desnoyers, S.; Eckdahl, S.; Shah, G.M.; Poirier, G.G.; Reid, J.M.; Ames, M.M.; et al. 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin. Cancer Res. 1998, 4, 117–130. [Google Scholar]
- Zhao, D.; Badur, M.G.; Luebeck, J.; Magaña, J.H.; Birmingham, A.; Sasik, R.; Ahn, C.S.; Ideker, T.; Metallo, C.M.; Mali, P. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol. Cell 2018, 69, 699–708.e697. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, C.; Hou, Z.S.; He, X.D.; Xie, S.; Ni, J.; Qian, C.; Cheng, X.; Jiang, T.; Yang, C.; et al. PI3Kγ maintains the self-renewal of acute myeloid leukemia stem cells by regulating the pentose phosphate pathway. Blood 2024, 143, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Egnell, C.; Ranta, S.; Banerjee, J.; Merker, A.; Niinimäki, R.; Lund, B.; Mogensen, P.R.; Jonsson, Ó.; Vaitkeviciene, G.; Lepik, K.; et al. Impact of body mass index on relapse in children with acute lymphoblastic leukemia treated according to Nordic treatment protocols. Eur. J. Haematol. 2020, 105, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Perea, G.; Domingo, A.; Villamor, N.; Palacios, C.; Juncà, J.; Torres, P.; Llorente, A.; Fernández, C.; Tormo, M.; Queipo de Llano, M.P.; et al. Adverse prognostic impact of CD36 and CD2 expression in adult de novo acute myeloid leukemia patients. Leuk. Res. 2005, 29, 1109–1116. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 52. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef]
- Huang, D.; Zhang, C.; Xiao, M.; Li, X.; Chen, W.; Jiang, Y.; Yuan, Y.; Zhang, Y.; Zou, Y.; Deng, L.; et al. Redox metabolism maintains the leukemogenic capacity and drug resistance of AML cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2210796120. [Google Scholar] [CrossRef]
- Khodakarami, A.; Adibfar, S.; Karpisheh, V.; Abolhasani, S.; Jalali, P.; Mohammadi, H.; Gholizadeh Navashenaq, J.; Hojjat-Farsangi, M.; Jadidi-Niaragh, F. The molecular biology and therapeutic potential of Nrf2 in leukemia. Cancer Cell Int. 2022, 22, 241. [Google Scholar] [CrossRef]
- Banella, C.; Catalano, G.; Travaglini, S.; Divona, M.; Masciarelli, S.; Guerrera, G.; Fazi, F.; Lo Coco, F.; Voso, M.T.; Noguera, N. PML/RARa Interferes with NRF2 Transcriptional Activity Increasing the Sensitivity to Ascorbate of Acute Promyelocytic Leukemia Cells. Cancers 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Pan, C.; Zhang, T.; Ni, M.; Wang, W.; Zhang, S.; Chen, Y.; Wang, J.; Fang, Q. Nrf2 overexpression increases the resistance of acute myeloid leukemia to cytarabine by inhibiting replication factor C4. Cancer Gene Ther. 2022, 29, 1773–1790. [Google Scholar] [CrossRef]
- Bobilev, I.; Novik, V.; Levi, I.; Shpilberg, O.; Levy, J.; Sharoni, Y.; Studzinski, G.P.; Danilenko, M. The Nrf2 transcription factor is a positive regulator of myeloid differentiation of acute myeloid leukemia cells. Cancer Biol. Ther. 2011, 11, 317–329. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef]
- Nishimoto, S.; Suzuki, T.; Koike, S.; Yuan, B.; Takagi, N.; Ogasawara, Y. Nrf2 activation ameliorates cytotoxic effects of arsenic trioxide in acute promyelocytic leukemia cells through increased glutathione levels and arsenic efflux from cells. Toxicol. Appl. Pharmacol. 2016, 305, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Bonovolias, I.D.; Tsiftsoglou, A.S. Hemin counteracts the repression of Bcl-2 and NrF2 genes and the cell killing induced by imatinib in human Bcr-Abl(+) CML cells. Oncol. Res. 2009, 17, 535–547. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, Y.; Pan, F.; Zhu, M.; Yao, L.; Liu, Y.; Feng, J.; Xiong, J.; Chen, X.; Ren, F.; et al. Inhibition of the Nrf2-TrxR Axis Sensitizes the Drug-Resistant Chronic Myelogenous Leukemia Cell Line K562/G01 to Imatinib Treatments. Biomed. Res. Int. 2019, 2019, 6502793. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Chen, W.; Wang, Y.; Hui, L.; Liu, J.; Li, N.; Zhang, L.; Zou, Y.; Wang, F. Nrf-2/Gst-alpha mediated imatinib resistance through rapid 4-HNE clearance. Exp. Cell Res. 2017, 353, 72–78. [Google Scholar] [CrossRef]
- Akin-Bali, D.F.; Aktas, S.H.; Unal, M.A.; Kankilic, T. Identification of novel Nrf2/Keap1 pathway mutations in pediatric acute lymphoblastic leukemia. Pediatr. Hematol. Oncol. 2020, 37, 58–75. [Google Scholar] [CrossRef]
- Jin, L.; Tong, L. PAQR3 inhibits proliferation and aggravates ferroptosis in acute lymphoblastic leukemia through modulation Nrf2 stability. Immun. Inflamm. Dis. 2021, 9, 827–839. [Google Scholar] [CrossRef]
- Jorge, J.; Magalhaes, N.; Alves, R.; Lapa, B.; Goncalves, A.C.; Sarmento-Ribeiro, A.B. Antitumor Effect of Brusatol in Acute Lymphoblastic Leukemia Models Is Triggered by Reactive Oxygen Species Accumulation. Biomedicines 2022, 10, 2207. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Kang, Q.; Pan, C.; Zhang, T.; Feng, C.; Chen, L.; Wei, S.; Wang, J. Nrf2 Overexpression Decreases Vincristine Chemotherapy Sensitivity Through the PI3K-AKT Pathway in Adult B-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2022, 12, 876556. [Google Scholar] [CrossRef]
- Jasek-Gajda, E.; Jurkowska, H.; Jasinska, M.; Lis, G.J. Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants 2020, 9, 633. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.P.; Hayashi, T.; Cottam, H.B.; Jin, G.; Yao, S.; Wu, C.C.; Rosenbach, M.D.; Corr, M.; Schwab, R.B.; Carson, D.A. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 7479–7484. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Ghia, E.M.; Antonucci, L.; Sharma, N.; Rassenti, L.Z.; Xu, J.; Sun, B.; Kipps, T.J.; Karin, M. NF-kappaB-p62-NRF2 survival signaling is associated with high ROR1 expression in chronic lymphocytic leukemia. Cell Death Differ. 2020, 27, 2206–2216. [Google Scholar] [CrossRef]
- Shah, N.M.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J.; Rushworth, S.A. NRF2-driven miR-125B1 and miR-29B1 transcriptional regulation controls a novel anti-apoptotic miRNA regulatory network for AML survival. Cell Death Differ. 2015, 22, 654–664. [Google Scholar] [CrossRef]
- Noguera, N.I.; Pelosi, E.; Angelini, D.F.; Piredda, M.L.; Guerrera, G.; Piras, E.; Battistini, L.; Massai, L.; Berardi, A.; Catalano, G.; et al. High-dose ascorbate and arsenic trioxide selectively kill acute myeloid leukemia and acute promyelocytic leukemia blasts in vitro. Oncotarget 2017, 8, 32550–32565. [Google Scholar] [CrossRef]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife 2017, 6, 18970. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Ruefli-Brasse, A.; Reed, J.C. Therapeutics targeting Bcl-2 in hematological malignancies. Biochem. J. 2017, 474, 3643–3657. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2013, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, 2949–2955. [Google Scholar] [CrossRef]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. Mol. Mech. Mutagen. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Karathedath, S.; Rajamani, B.M.; Aalam, S.M.M.; Abraham, A.; Varatharajan, S.; Krishnamurthy, P.; Mathews, V.; Velayudhan, S.R.; Balasubramanian, P. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS ONE 2017, 12, e0177227. [Google Scholar] [CrossRef]
- Naumann, F.V.; Sweep, F.C.G.J.; Adema, G.J.; Peeters, W.J.M.; Martens, J.W.M.; Bussink, J.; Span, P.N. Tamoxifen induces radioresistance through NRF2-mediated metabolic reprogramming in breast cancer. Cancer Metab. 2023, 11, 3. [Google Scholar] [CrossRef]
- Ho, C.J.; Gorski, S.M. Molecular Mechanisms Underlying Autophagy-Mediated Treatment Resistance in Cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Choudhury, D.; Das, A.; Mukherjee, D.D.; Dasgupta, M.; Bandopadhyay, S.; Chakrabarti, G. Autophagy inhibition with chloroquine reverts paclitaxel resistance and attenuates metastatic potential in human nonsmall lung adenocarcinoma A549 cells via ROS mediated modulation of β-catenin pathway. Apoptosis 2019, 24, 414–433. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Luo, M.; Bao, L.; Xue, Y.; Zhu, M.; Kumar, A.; Xing, C.; Wang, J.E.; Wang, Y.; Luo, W. ZMYND8 protects breast cancer stem cells against oxidative stress and ferroptosis through activation of NRF2. J. Clin. Investig. 2024, 134, e171166. [Google Scholar] [CrossRef] [PubMed]
- Yeldag, G.; Rice, A.; Hernández, A.d.R. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef]
- Bocci, F.; Tripathi, S.C.; Vilchez Mercedes, S.A.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. 2019, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hong, X.; Ma, N.; Wei, Z.; Ci, X.; Zhang, S. The promoting effect and mechanism of Nrf2 on cell metastasis in cervical cancer. J. Transl. Med. 2023, 21, 433. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, A.; Schenkel, J.; Schröder, N.; Brandt, E.F.; Weiand, M.; Neu, T.; Ramadori, P.; Caspers, T.; Kant, S.; Pufe, T.; et al. Nrf2 induces malignant transformation of hepatic progenitor cells by inducing β-catenin expression. Redox Biol. 2022, 57, 102453. [Google Scholar] [CrossRef]
- Hamada, S.; Matsumoto, R.; Masamune, A. HIF-1 and NRF2; Key Molecules for Malignant Phenotypes of Pancreatic Cancer. Cancers 2022, 14, 411. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, K.; Matsuno, Y.; Yoshida, K.; Sherpa, M.; Nakajima, M.; Matsuyama, M.; Kiwamoto, T.; Morishima, Y.; Ishii, Y.; Hizawa, N. ROS-Nrf2 pathway mediates the development of TGF-β1-induced epithelial-mesenchymal transition through the activation of Notch signaling. Eur. J. Cell Biol. 2021, 100, 151181. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Z.; Peng, H. Molecular Mechanisms of Ferroptosis and Its Roles in Hematologic Malignancies. Front. Oncol. 2021, 11, 743006. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Weber, S.; Parmon, A.; Kurrle, N.; Schnütgen, F.; Serve, H. The Clinical Significance of Iron Overload and Iron Metabolism in Myelodysplastic Syndrome and Acute Myeloid Leukemia. Front. Immunol. 2020, 11, 627662. [Google Scholar] [CrossRef]
- Hagag, A.A.; Badraia, I.M.; Abdelmageed, M.M.; Hablas, N.M.; Hazzaa, S.M.E.; Nosair, N.A. Prognostic Value of Transferrin Receptor-1 (CD71) Expression in Acute Lymphoblastic Leukemia. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Bordini, J.; Galvan, S.; Ponzoni, M.; Bertilaccio, M.T.; Chesi, M.; Bergsagel, P.L.; Camaschella, C.; Campanella, A. Induction of iron excess restricts malignant plasma cells expansion and potentiates bortezomib effect in models of multiple myeloma. Leukemia 2017, 31, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.; Huang, K.; Lin, G. Triptolide promotes ferroptosis by suppressing Nrf2 to overcome leukemia cell resistance to doxorubicin. Mol. Med. Rep. 2023, 27, 17. [Google Scholar] [CrossRef]
- Emmanuel, N.; Li, H.; Chen, J.; Zhang, Y. FSP1, a novel KEAP1/NRF2 target gene regulating ferroptosis and radioresistance in lung cancers. Oncotarget 2022, 13, 1136–1139. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Cahuzac, K.M.; Lubin, A.; Bosch, K.; Stokes, N.; Shoenfeld, S.M.; Zhou, R.; Lemon, H.; Asara, J.; Parsons, R.E. AKT activation because of PTEN loss upregulates xCT via GSK3beta/NRF2, leading to inhibition of ferroptosis in PTEN-mutant tumor cells. Cell Rep. 2023, 42, 112536. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhong, S.; Qiu, K.; Chen, X.; Wu, W.; Zheng, J.; Liu, Y.; Wu, H.; Fan, S.; Nie, D.; et al. Targeting NRF2 uncovered an intrinsic susceptibility of acute myeloid leukemia cells to ferroptosis. Exp. Hematol. Oncol. 2023, 12, 47. [Google Scholar] [CrossRef]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- Rai, A.; Patwardhan, R.S.; Jayakumar, S.; Pachpatil, P.; Das, D.; Panigrahi, G.C.; Gota, V.; Patwardhan, S.; Sandur, S.K. Clobetasol propionate, a Nrf-2 inhibitor, sensitizes human lung cancer cells to radiation-induced killing via mitochondrial ROS-dependent ferroptosis. Acta Pharmacol. Sin. 2024, 45, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- de Souza, I.; Monteiro, L.K.S.; Guedes, C.B.; Silva, M.M.; Andrade-Tomaz, M.; Contieri, B.; Latancia, M.T.; Mendes, D.; Porchia, B.F.M.M.; Lazarini, M.; et al. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation. Cell Death Dis. 2022, 13, 591. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.M.; Ke, Z.P.; Wang, J.N.; Yang, J.Y.; Chen, S.Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis 2013, 34, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef]
- Lee, J.; Kang, J.S.; Nam, L.B.; Yoo, O.K.; Keum, Y.S. Suppression of NRF2/ARE by convallatoxin sensitises A549 cells to 5-FU-mediated apoptosis. Free Radic. Res. 2018, 52, 1416–1423. [Google Scholar] [CrossRef]
- Choi, E.J.; Jung, B.J.; Lee, S.H.; Yoo, H.S.; Shin, E.A.; Ko, H.J.; Chang, S.; Kim, S.Y.; Jeon, S.M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Manna, A.; De Sarkar, S.; De, S.; Bauri, A.K.; Chattopadhyay, S.; Chatterjee, M. The variable chemotherapeutic response of Malabaricone-A in leukemic and solid tumor cell lines depends on the degree of redox imbalance. Phytomedicine 2015, 22, 713–723. [Google Scholar] [CrossRef]
- Bollong, M.J.; Yun, H.; Sherwood, L.; Woods, A.K.; Lairson, L.L.; Schultz, P.G. A Small Molecule Inhibits Deregulated NRF2 Transcriptional Activity in Cancer. ACS Chem. Biol. 2015, 10, 2193–2198. [Google Scholar] [CrossRef]
- Ki, S.H.; Cho, I.J.; Choi, D.W.; Kim, S.G. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: Role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell Biol. 2005, 25, 4150–4165. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Zhang, F.; Sun, Z.; Zhou, W.; Li, Z.Y.; You, Q.D.; Guo, Q.L.; Hu, R. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response. Mol. Carcinog. 2013, 52, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef]
- Limonciel, A.; Jennings, P. A review of the evidence that ochratoxin A is an Nrf2 inhibitor: Implications for nephrotoxicity and renal carcinogenicity. Toxins 2014, 6, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Su, L.; Ye, Q.; Zhang, S.; Kung, H.; Jiang, F.; Jiang, G.; Miao, J.; Zhao, B. Discovery of a novel Nrf2 inhibitor that induces apoptosis of human acute myeloid leukemia cells. Oncotarget 2017, 8, 7625–7636. [Google Scholar] [CrossRef]
- Tarumoto, T.; Nagai, T.; Ohmine, K.; Miyoshi, T.; Nakamura, M.; Kondo, T.; Mitsugi, K.; Nakano, S.; Muroi, K.; Komatsu, N.; et al. Ascorbic acid restores sensitivity to imatinib via suppression of Nrf2-dependent gene expression in the imatinib-resistant cell line. Exp. Hematol. 2004, 32, 375–381. [Google Scholar] [CrossRef]
- Jayakumar, S.; Patwardhan, R.S.; Pal, D.; Sharma, D.; Sandur, S.K. Dimethoxycurcumin, a metabolically stable analogue of curcumin enhances the radiosensitivity of cancer cells: Possible involvement of ROS and thioredoxin reductase. Biochem. Biophys. Res. Commun. 2016, 478, 446–454. [Google Scholar] [CrossRef]
- Jayakumar, S.; Patwardhan, R.S.; Pal, D.; Singh, B.; Sharma, D.; Kutala, V.K.; Sandur, S.K. Mitochondrial targeted curcumin exhibits anticancer effects through disruption of mitochondrial redox and modulation of TrxR2 activity. Free Radic. Biol. Med. 2017, 113, 530–538. [Google Scholar] [CrossRef]
- Modi, R.; McKee, N.; Zhang, N.; Alwali, A.; Nelson, S.; Lohar, A.; Ostafe, R.; Zhang, D.D.; Parkinson, E.I. Stapled Peptides as Direct Inhibitors of Nrf2-sMAF Transcription Factors. J. Med. Chem. 2023, 66, 6184–6192. [Google Scholar] [CrossRef]
- Simov, V.; Altman, M.D.; Bianchi, E.; DelRizzo, S.; DiNunzio, E.N.; Feng, G.; Goldenblatt, P.; Ingenito, R.; Johnson, S.A.; Mansueto, M.S.; et al. Discovery and characterization of novel peptide inhibitors of the NRF2/MAFG/DNA ternary complex for the treatment of cancer. Eur. J. Med. Chem. 2021, 224, 113686. [Google Scholar] [CrossRef]
- Ji, J.; Ma, S.; Zhu, Y.; Zhao, J.; Tong, Y.; You, Q.; Jiang, Z. ARE-PROTACs Enable Co-degradation of an Nrf2-MafG Heterodimer. J. Med. Chem. 2023, 66, 6070–6081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. The Nrf2-Keap1-ARE signaling pathway: The regulation and dual function of Nrf2 in cancer. Antioxid. Redox Signal. 2010, 13, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Aboulkassim, T.; Tian, X.; Liu, Q.; Qiu, D.; Hancock, M.; Wu, J.H.; Batist, G. A NRF2 inhibitor selectively sensitizes KEAP1 mutant tumor cells to cisplatin and gefitinib by restoring NRF2-inhibitory function of KEAP1 mutants. Cell Rep. 2023, 42, 113104. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded Nrf2 activation on phase-I and -II drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006. [Google Scholar] [CrossRef]
- Hallis, S.P.; Kim, J.M.; Kwak, M.K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 2023, 46, 153–164. [Google Scholar] [CrossRef]
- Baiskhanova, D.; Schäfer, H. The Role of Nrf2 in the Regulation of Mitochondrial Function and Ferroptosis in Pancreatic Cancer. Antioxidants 2024, 13, 696. [Google Scholar] [CrossRef]
- Bae, T.; Hallis, S.P.; Kwak, M.K. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp. Mol. Med. 2024, 56, 501–514. [Google Scholar] [CrossRef]
- Lin, L.; Wu, Q.; Lu, F.; Lei, J.; Zhou, Y.; Liu, Y.; Zhu, N.; Yu, Y.; Ning, Z.; She, T.; et al. Nrf2 signaling pathway: Current status and potential therapeutic targetable role in human cancers. Front. Oncol. 2023, 13, 1184079. [Google Scholar] [CrossRef]
- Hu, M.; Yuan, L.; Zhu, J. The Dual Role of NRF2 in Colorectal Cancer: Targeting NRF2 as a Potential Therapeutic Approach. J. Inflamm. Res. 2024, 17, 5985–6004. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Leukemia | Nrf2 Stimulation (↑)/Inhibition (↓) Activity | References |
|---|---|---|
| Acute myeloid leukemia (AML) | Arsenic ↑ Nrf2 translocation to nucleus; ↓ ROS production; ↑ expression of antioxidant enzymes | [100] |
| ↑ Nrf2 expression; ↑ resistance to chemotherapy | [122] | |
| Vitamin D activates Nrf2; ↑ myeloid differentiation | [123] | |
| NF-κB ↑ Nrf2; ↑ increases oncogenic cell proliferation and survival; ↑ chemoresistance | [124] | |
| Acute promyelocytic leukemia (APL) | ↓ Nrf2 and its target genes led to ↑ sensitivity to oxidative stress therapy, such as ascorbic acid | [121,138] |
| Acute Lymphocytic Leukemia (ALL) | Mutations in Nrf2/Keap1 pathways (73% of pediatric ALL patients) | [129] |
| Elevated Nrf2 expression ↓ PAQR3;↑ ALL progression | [130] | |
| Nrf2 signaling inhibition by brusatol ↑ ROS and O2− and apoptosis of ALL cells | [131] | |
| Nrf2 overexpression ↑ PI3-AKT signaling; ↓ BAD response to chemotherapy | [132] | |
| Chronic myeloid leukemia (CML) | Nrf2 targets HO-1 and NQO1↓ apoptosis; ↑ resistance to imatinib | [126] |
| ↓ Nrf2; ↑ ROS and imatinib induced apoptosis | [127] | |
| Inhibitors of Nrf2 ↓ GST-α; ↑ 4HNE and sensitivity toward imatinib | [128] | |
| Chronic lymphocytic leukemia (CLL) | ↑ Nrf2 signaling in CLL; electrophilic and antioxidant compounds;↓ Nrf2 signaling; ↑ CLL-selective cytotoxicity | [135] |
| Cross talk between NF-κB signaling and Nrf2 via p62/mTORC1; ↑ NQO1 and ROR1; ↓ response to ROS-inducing therapy | [136] |
| Nrf2 Inhibitors | Mechanism of Action | Reference |
|---|---|---|
| ML385 | Inhibition of transactivation of Nrf2 | [178] |
| malabaricone-A | Inhibition of transactivation of Nrf2 | [188] |
| AEM1 | Inhibition of transactivation of Nrf2 | [189] |
| brusatol | Inhibition of protein synthesis | [181] |
| clobetasol propionate | Ligand of glucocorticoid receptor | [187] |
| dexamethasone | Ligand of glucocorticoid receptor | [190] |
| bexarotene | Agonists of the retinoic acid receptor-α | [180] |
| all-trans retinoic acid | Agonists of the retinoic acid receptor-α | [191] |
| wogonin | Impacting the stability of Nrf2 transcript | [192] |
| luteolin | Impacting the stability of Nrf2 transcript | [193] |
| ochratoxin A | Prevents nuclear translocation | [194] |
| trigonelline | Prevents nuclear translocation | [183] |
| halofuginone | Decreased Nrf2 protein | [182] |
| pyrazolyl hydroxamic acid | Pyrazolyl hydroxamic acid | [195] |
| ascorbic acid | Reduced Nrf2/ARE complex | [196] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, S.K.; Jayakumar, S.; Espina Barroso, E.; Jha, A.; Catalano, G.; Sandur, S.K.; Noguera, N.I. Molecular Targets of Oxidative Stress: Focus on Nuclear Factor Erythroid 2–Related Factor 2 Function in Leukemia and Other Cancers. Cells 2025, 14, 713. https://doi.org/10.3390/cells14100713
Hasan SK, Jayakumar S, Espina Barroso E, Jha A, Catalano G, Sandur SK, Noguera NI. Molecular Targets of Oxidative Stress: Focus on Nuclear Factor Erythroid 2–Related Factor 2 Function in Leukemia and Other Cancers. Cells. 2025; 14(10):713. https://doi.org/10.3390/cells14100713
Chicago/Turabian StyleHasan, Syed K., Sundarraj Jayakumar, Eliezer Espina Barroso, Anup Jha, Gianfranco Catalano, Santosh K. Sandur, and Nelida I. Noguera. 2025. "Molecular Targets of Oxidative Stress: Focus on Nuclear Factor Erythroid 2–Related Factor 2 Function in Leukemia and Other Cancers" Cells 14, no. 10: 713. https://doi.org/10.3390/cells14100713
APA StyleHasan, S. K., Jayakumar, S., Espina Barroso, E., Jha, A., Catalano, G., Sandur, S. K., & Noguera, N. I. (2025). Molecular Targets of Oxidative Stress: Focus on Nuclear Factor Erythroid 2–Related Factor 2 Function in Leukemia and Other Cancers. Cells, 14(10), 713. https://doi.org/10.3390/cells14100713