
Breast Cancer: Mitochondria-Centered Metabolic Alterations in Tumor and Associated Adipose Tissue

 , , , , and
, , , , and
Abstract
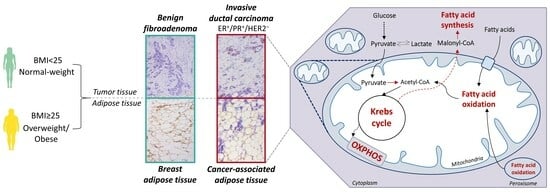
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Sample Collection
2.2. Western Blotting
2.3. Mitochondrial DNA Copy Number
2.4. Light and Electron Microscopy
2.5. Statistical Analyses
3. Results
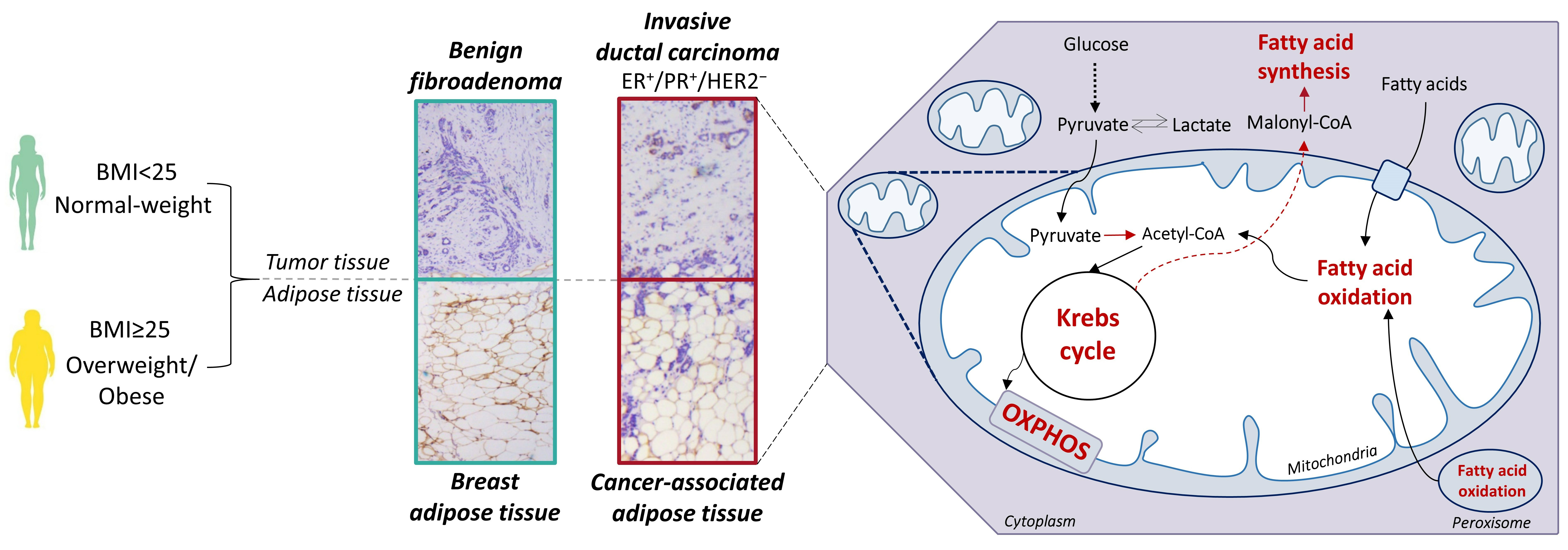
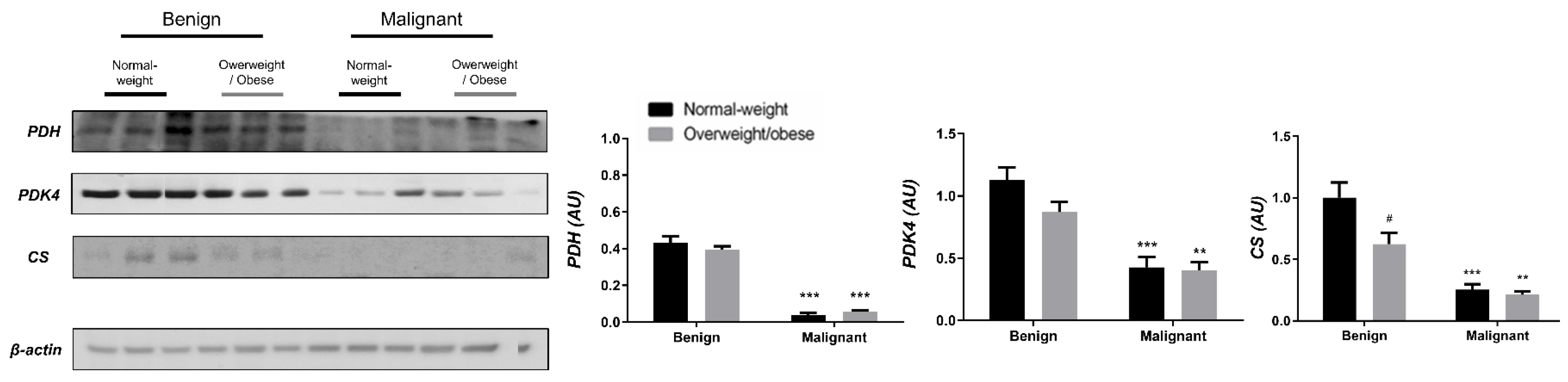
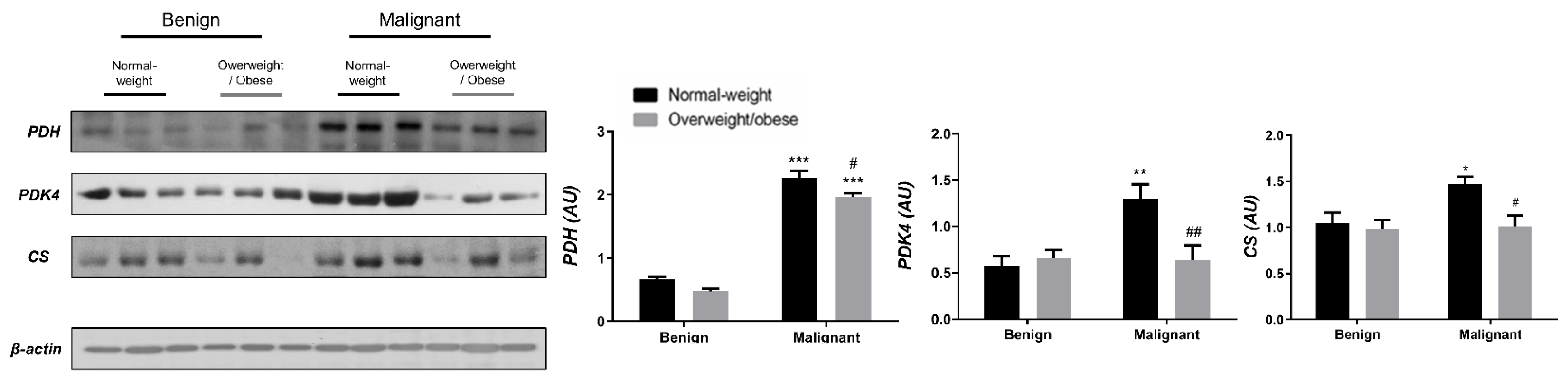
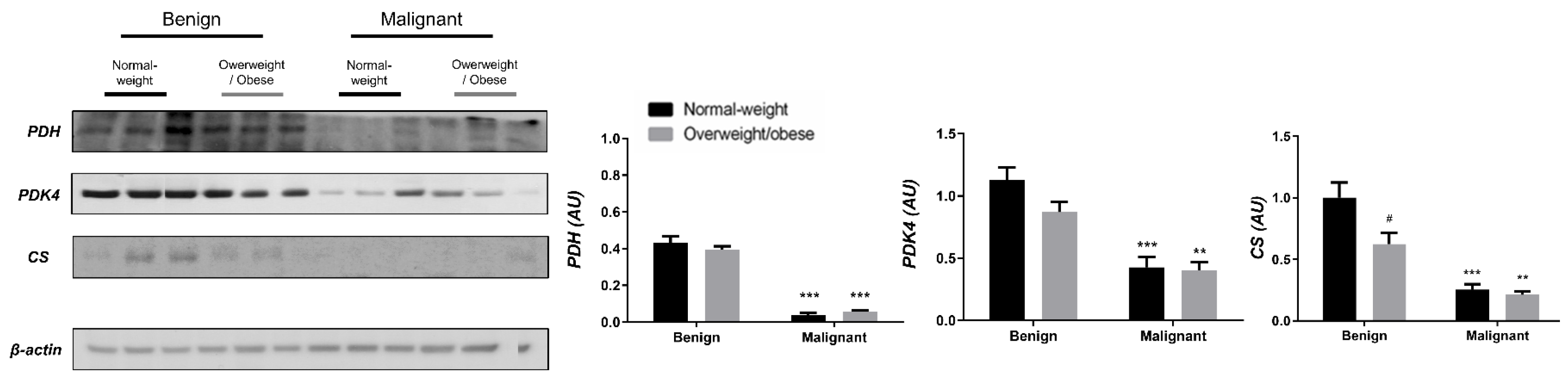
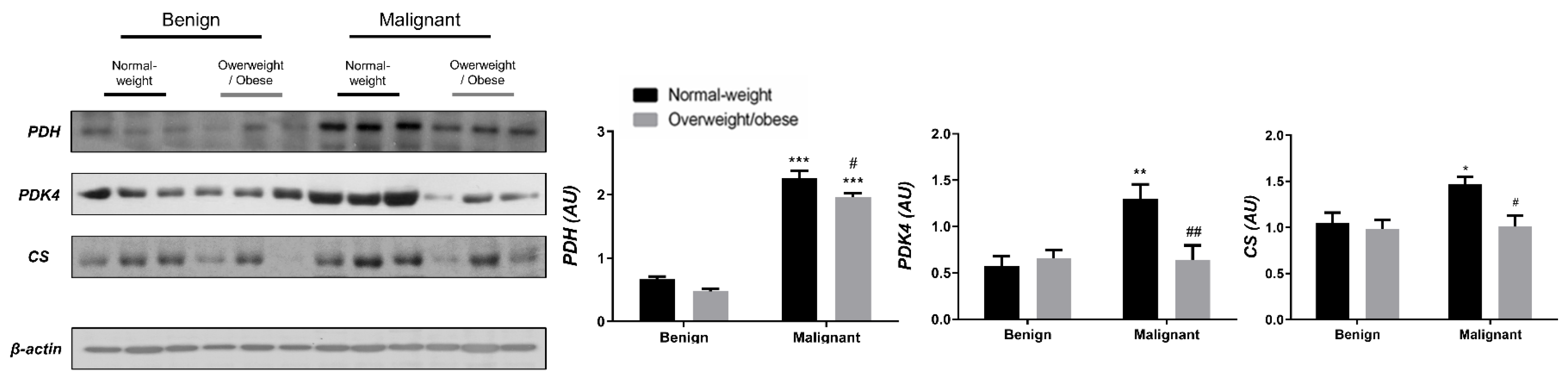
3.1. Contrasting Profiles of Major Metabolic Pathways in Breast Cancer Tissue and CAAT
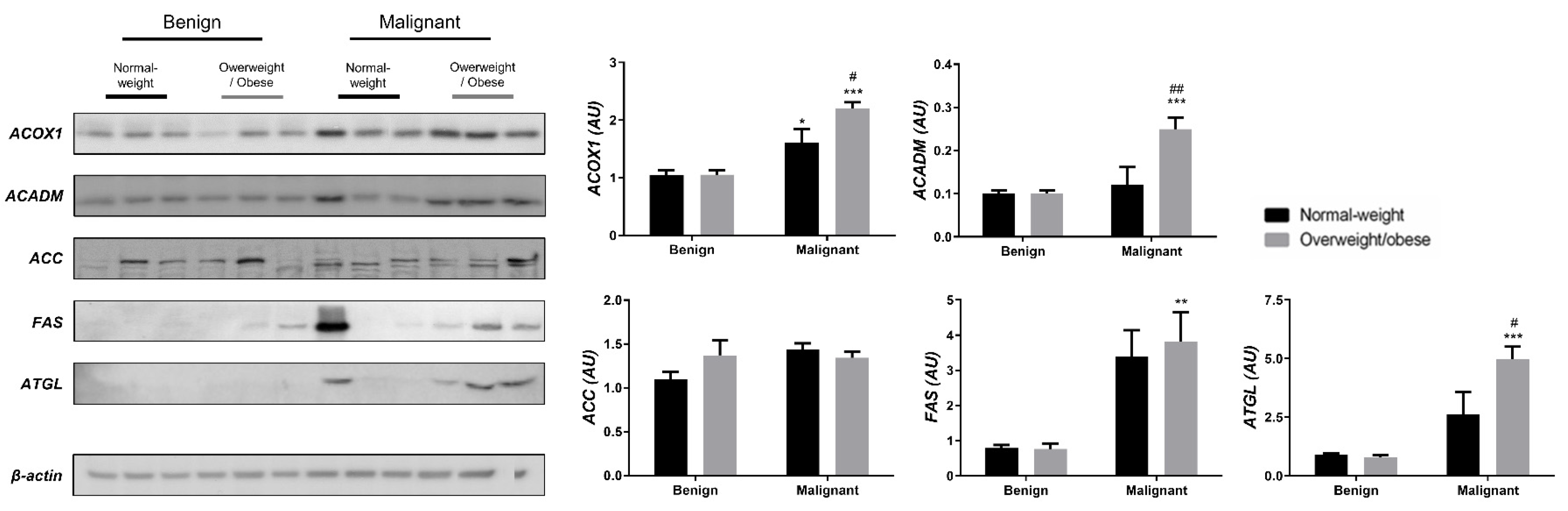
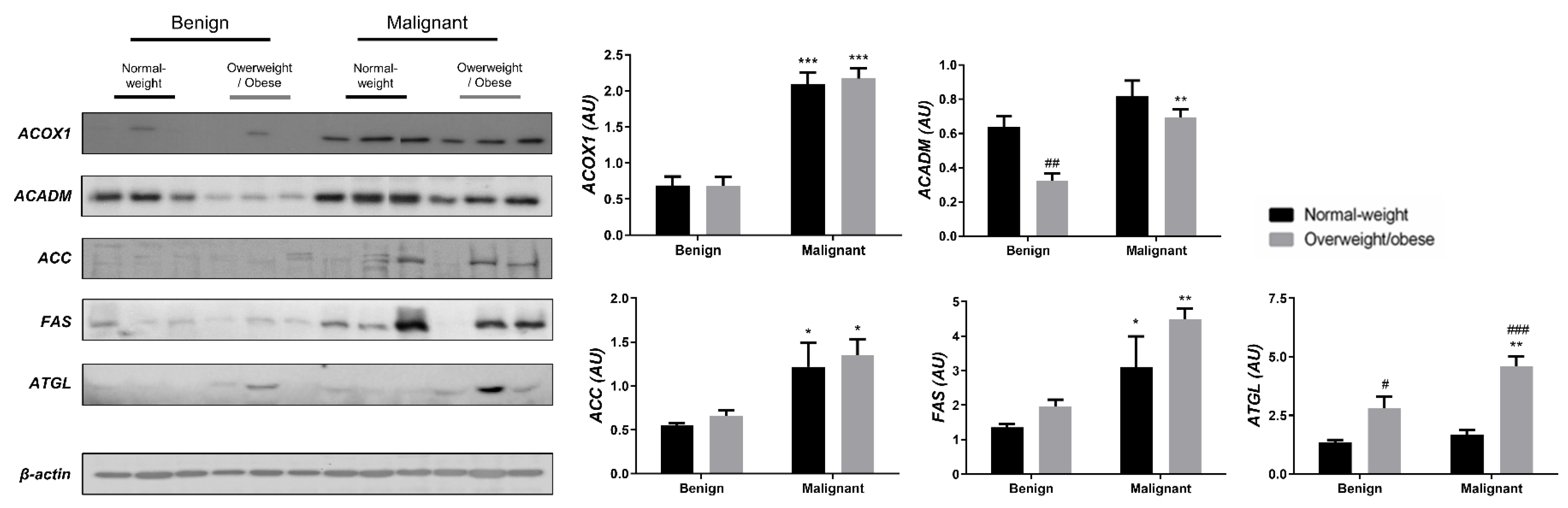
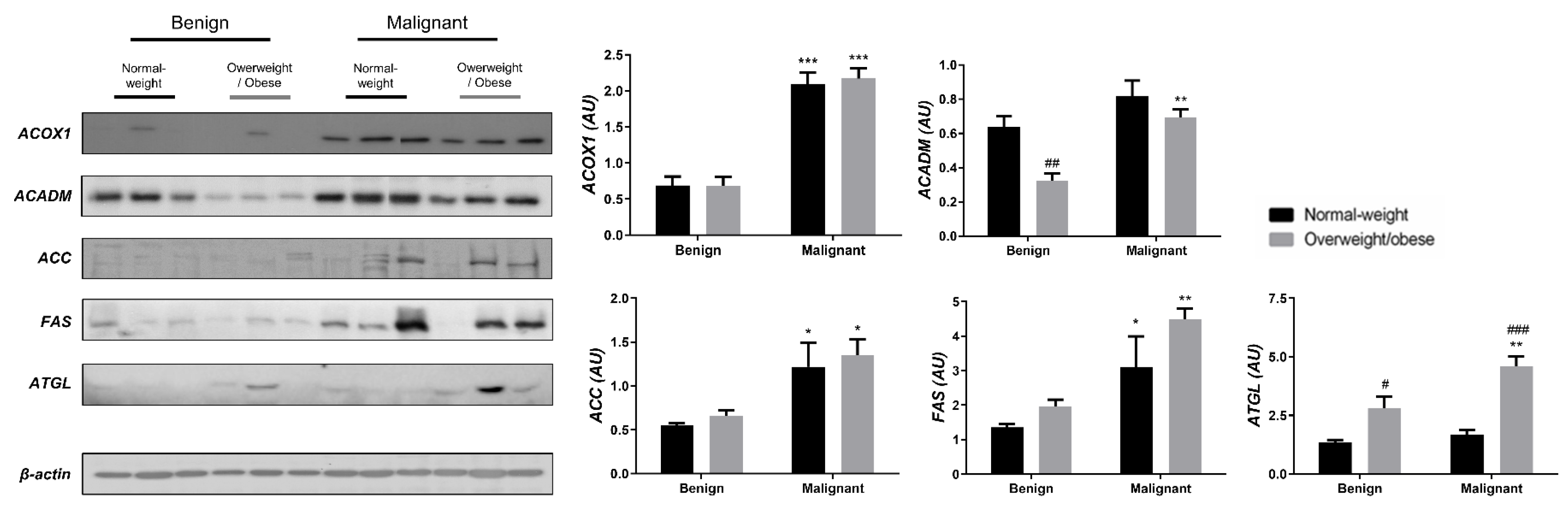
3.2. Mitochondrial Metabolism in Breast Cancer Is Maintained on the Grounds of Lipid Metabolism
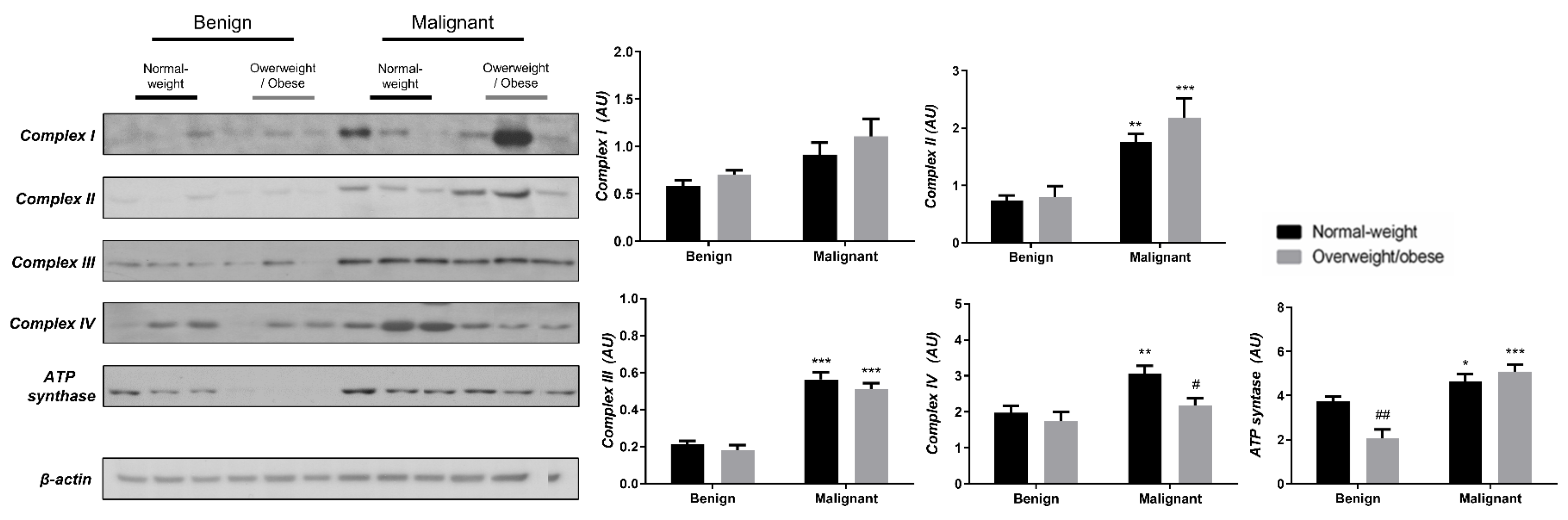
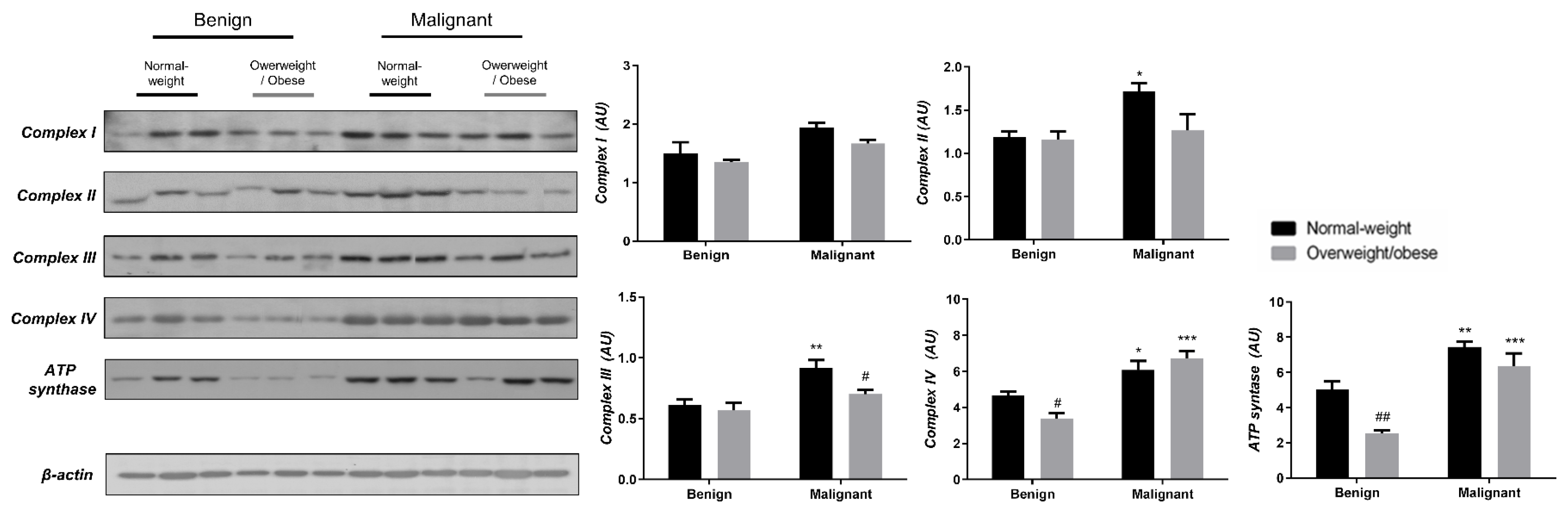
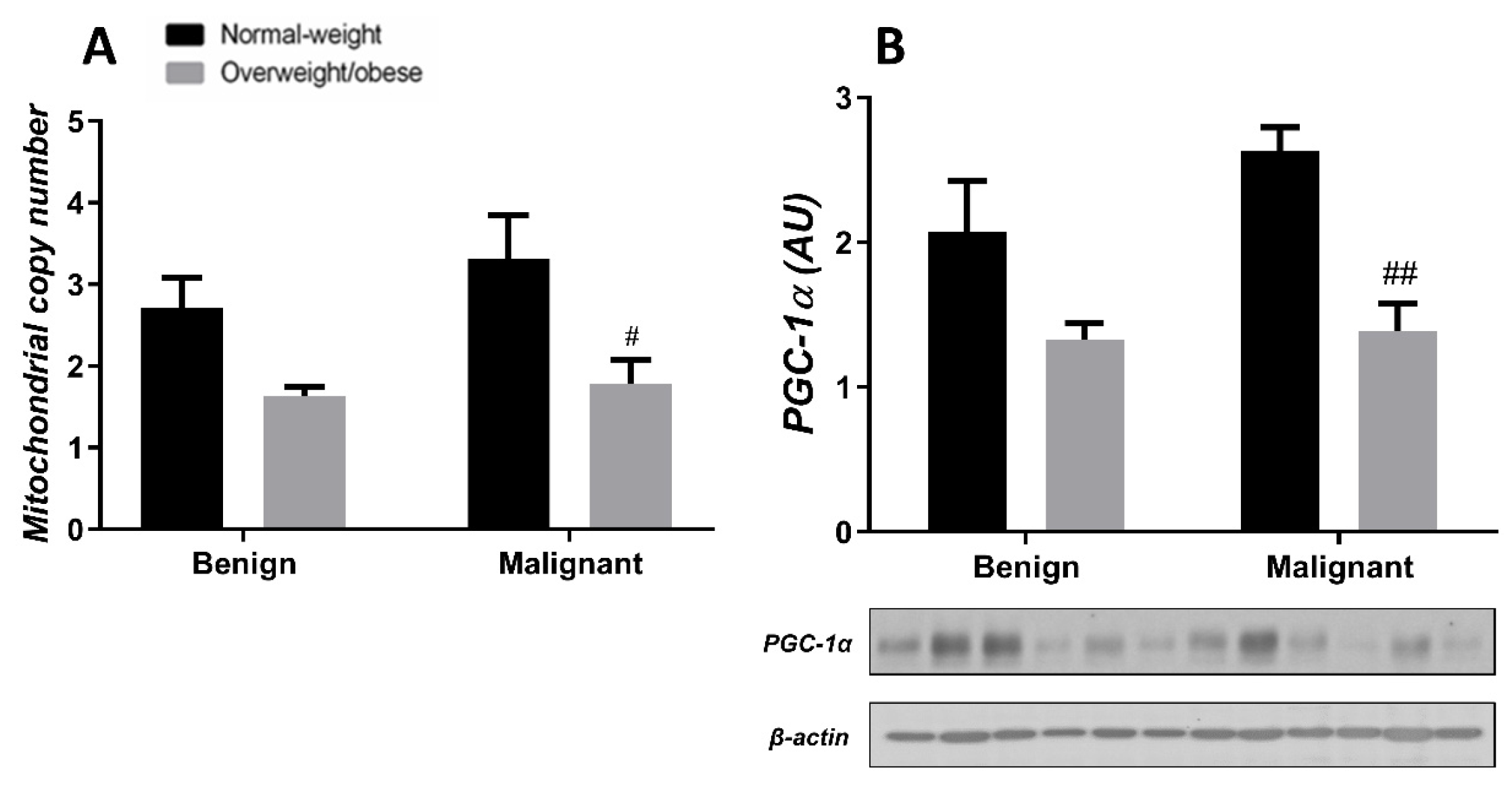
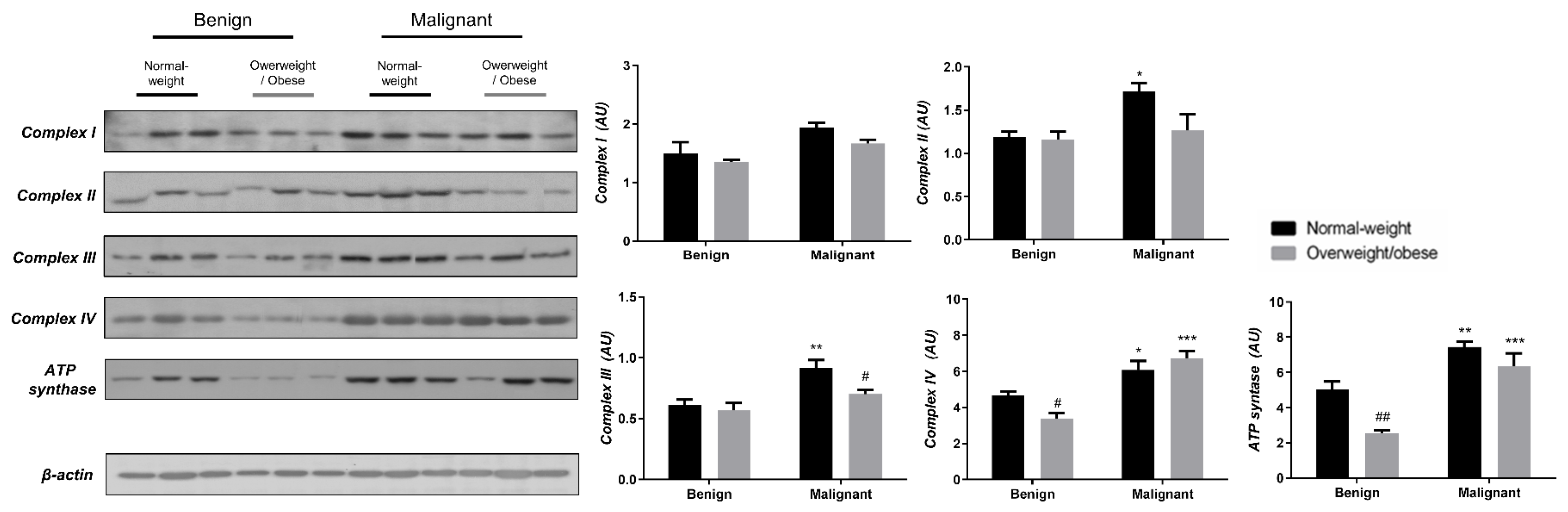
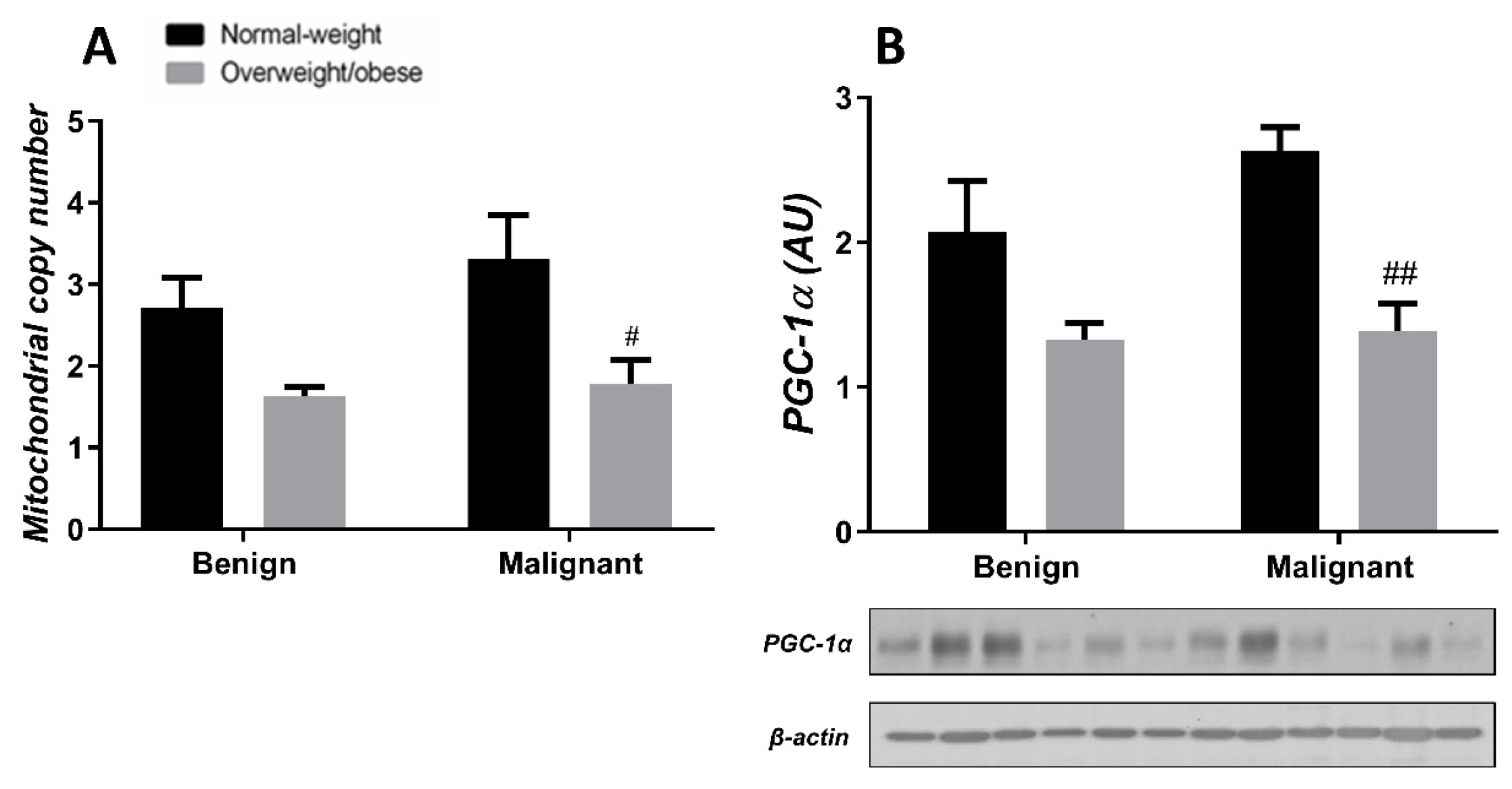
3.3. Obesity-Affected Oxidative Phosphorylation in Mitochondria Is a Major Constituent of Breast Cancer Progression
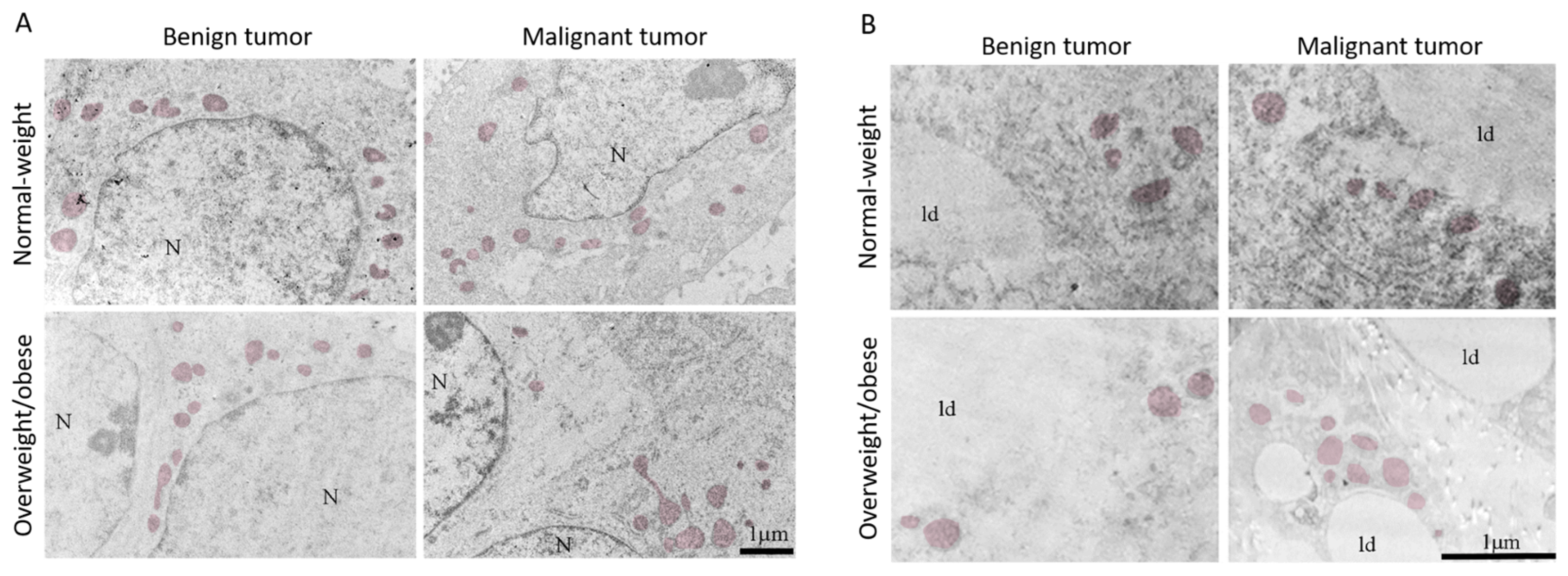
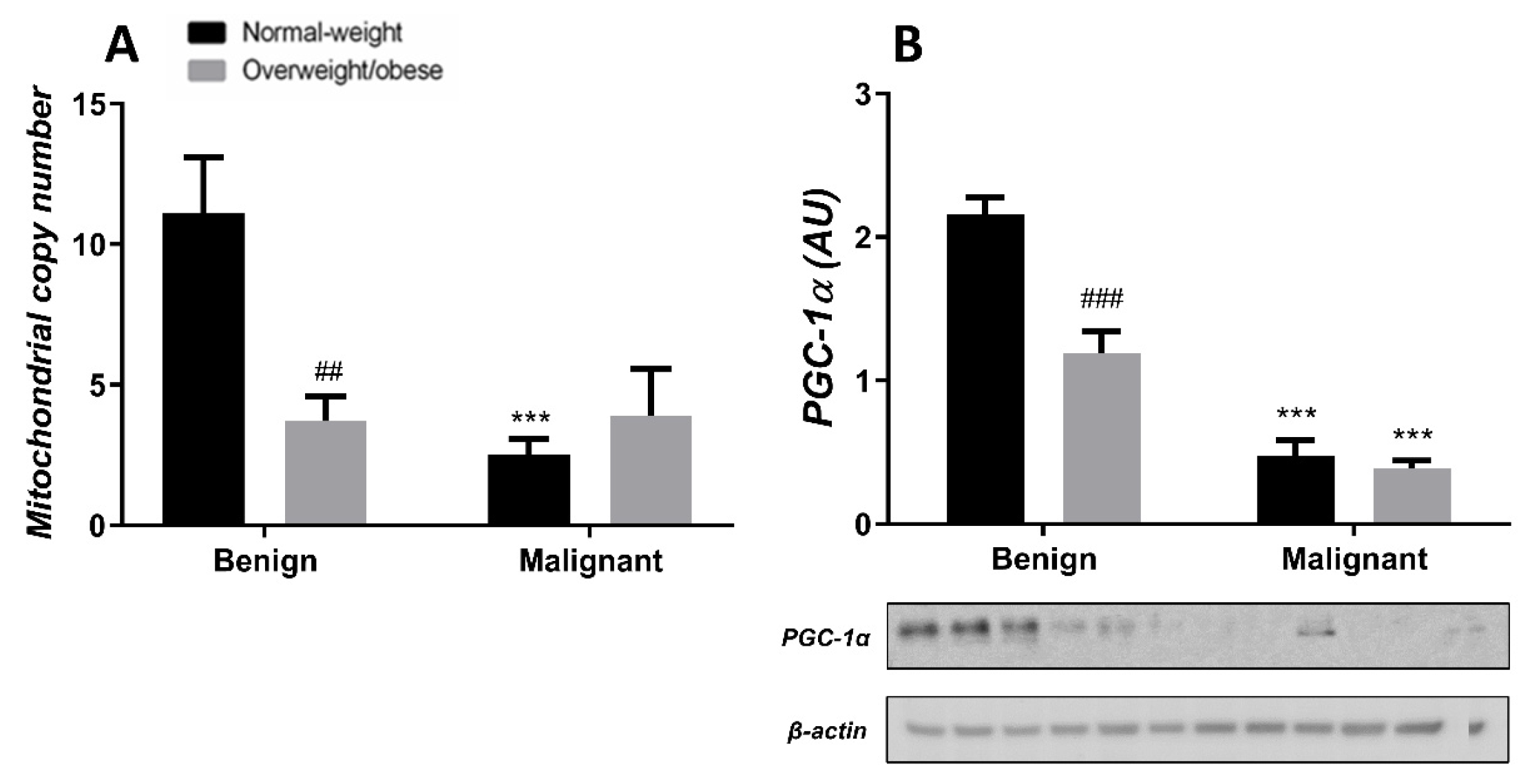
3.4. Mitochondrial Dynamics in Breast Tumor Tissue and CAAT
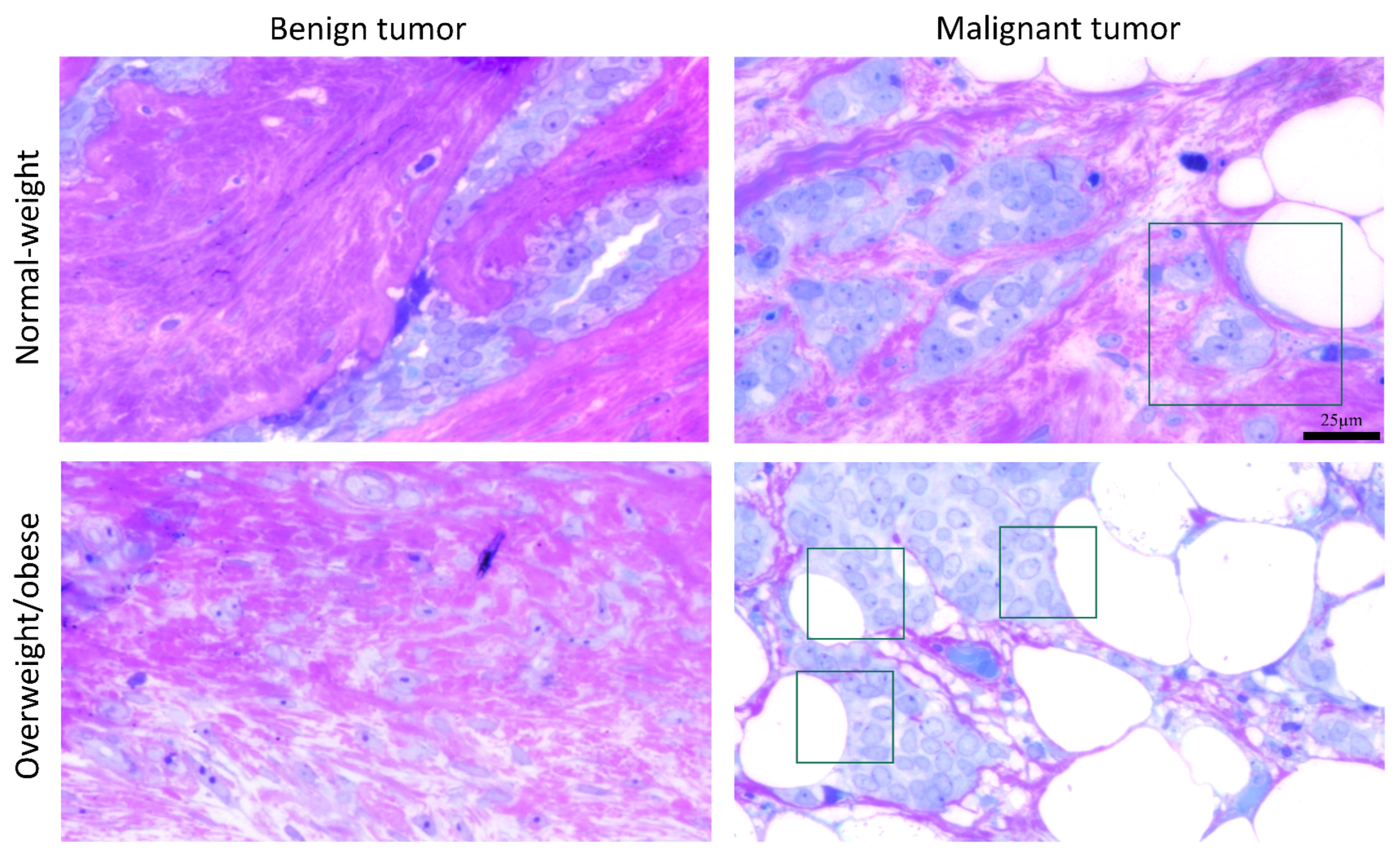
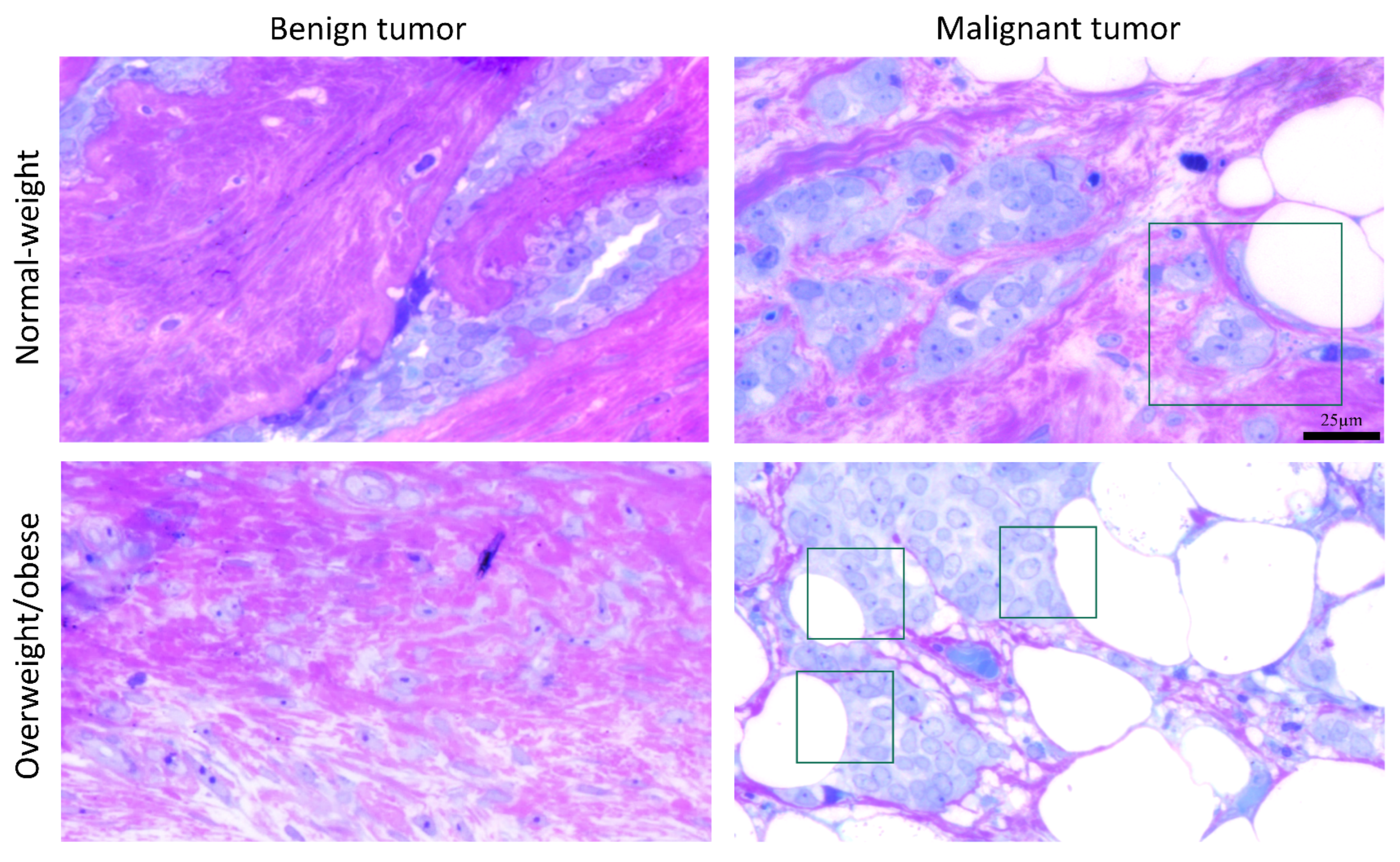
3.5. Microscopy Revealed Tight Connection between Breast Cancer Cells and Associated Adipocytes and Preservation of Mitochondrial Structure in Malignancy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TME | tumor microenvironment |
| CAAT | cancer-associated adipose tissue |
| mtDNA | mitochondrial DNA |
| MCN | mitochondrial DNA copy number |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator-1 alpha |
| BMI | body mass index |
| CS | citrate synthase |
| PDH | pyruvate dehydrogenase |
| PDK4 | pyruvate dehydrogenase kinase 4 |
| ACADM | acyl-CoA dehydrogenase medium chain |
| ACOX1 | acyl-CoA oxidase 1 |
| ACC | acetyl-CoA carboxylase |
| FAS | fatty acid synthase |
| ATGL | adipose triglyceride lipase |
| ETC | electron transport chain |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Muller, C. Tumour-Surrounding Adipocytes Are Active Players in Breast Cancer Progression. Ann. Endocrinol. 2013, 74, 108–110. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.N.; Geneste, A.; Fallone, F.; Li, X.; Dumontet, C.; Muller, C. The Fat and the Bad: Mature Adipocytes, Key Actors in Tumor Progression and Resistance. Oncotarget 2017, 8, 57622–57641. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a Metabolic Disease: Implications for Novel Therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Kalezic, A.; Udicki, M.; Galic, B.S.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B. Lactate Metabolism in Breast Cancer Microenvironment: Contribution Focused on Associated Adipose Tissue and Obesity. Int. J. Mol. Sci. 2020, 21, 9676. [Google Scholar] [CrossRef]
- Kalezic, A.; Udicki, M.; Srdic Galic, B.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B.; García-Otín, L.; Sancho, P. Tissue-Specific Warburg Effect in Breast Cancer and Cancer-Associated Adipose Tissue-Relationship between AMPK and Glycolysis. Cancers 2021, 13, 2731. [Google Scholar] [CrossRef]
- D’Esposito, V.; Ambrosio, M.R.; Giuliano, M.; Cabaro, S.; Miele, C.; Beguinot, F.; Formisano, P. Mammary Adipose Tissue Control of Breast Cancer Progression: Impact of Obesity and Diabetes. Front. Oncol. 2020, 10, 1554. [Google Scholar] [CrossRef]
- Hoy, A.J.; Balaban, S.; Saunders, D.N. Adipocyte–Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol. Med. 2017, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating Cancer Metabolic Plasticity by Coupling Gene Regulation with Metabolic Pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte Lipolysis Links Obesity to Breast Cancer Growth: Adipocyte-Derived Fatty Acids Drive Breast Cancer Cell Proliferation and Migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Kalezic, A.; Udicki, M.; Srdic Galic, B.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B. Redox Profile of Breast Tumor and Associated Adipose Tissue in Premenopausal Women—Interplay between Obesity and Malignancy. Redox Biol. 2021, 41, 101939. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Shi, Y.; Wei, X.; Yang, Y.; Zhou, Y.; Hao, X.; Zhang, N.; Niu, R. Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells. Toxicol. Lett. 2007, 170, 83–93. [Google Scholar] [CrossRef]
- Imanishi, H.; Hattori, K.; Wada, R.; Ishikawa, K.; Fukuda, S.; Takenaga, K.; Nakada, K.; Hayashi, J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011, 6, e23401. [Google Scholar] [CrossRef]
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished Tumorigenic Phenotype after Depletion of Mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198. [Google Scholar]
- Petrović, V.; Korać, A.; Buzadžić, B.; Korać, B. The effects of L-arginine and L-NAME supplementation on redox-regulation and thermogenesis in interscapular brown adipose tissue. J. Exp. Biol. 2005, 208, 4263–4271. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsz, M.T.; Teahan, O.; et al. Pyruvate Dehydrogenase Complex Activity Controls Metabolic and Malignant Phenotype in Cancer Cells. J. Biol. Chem. 2008, 283, 22700–22708. [Google Scholar] [CrossRef]
- Yonashiro, R.; Eguchi, K.; Wake, M.; Takeda, N.; Nakayama, K. Pyruvate Dehydrogenase PDH-E1b Controls Tumor Progression by Altering the Metabolic Status of Cancer Cells. Cancer Res. 2018, 78, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Atas, E.; Oberhuber, M.; Kenner, L. The Implications of PDK1–4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 2020, 10, 583217. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Qiu, Y.; Hansen, M.K.; Zhu, L.; Zhang, V.; Xie, M.; Okamoto, Y.; Mattie, M.D.; Higashiyama, H.; Asano, S.; et al. Adipose Mitochondrial Biogenesis Is Suppressed in Db/Db and High-Fat Diet–Fed Mice and Improved by Rosiglitazone. Diabetes 2007, 56, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Goossens, G.H.; Bizzarri, A.; Venteclef, N.; Essers, Y.; Cleutjens, J.P.; Konings, E.; Jocken, J.W.E.; Čajlaković, M.; Ribitsch, V.; Clément, K.; et al. Increased Adipose Tissue Oxygen Tension in Obese Compared with Lean Men Is Accompanied by Insulin Resistance, Impaired Adipose Tissue Capillarization, and Inflammation. Circulation 2011, 124, 67–76. [Google Scholar] [CrossRef]
- Christe, M.; Hirzel, E.; Lindinger, A.; Kern, B.; von Flüe, M.; Peterli, R.; Peters, T.; Eberle, A.N.; Lindinger, P.W. Obesity Affects Mitochondrial Citrate Synthase in Human Omental Adipose Tissue. ISRN Obes. 2013, 2013, 826027. [Google Scholar] [CrossRef]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes Activate Mitochondrial Fatty Acid Oxidation and Autophagy to Promote Tumor Growth in Colon Cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte Extracellular Vesicles Carry Enzymes and Fatty Acids That Stimulate Mitochondrial Metabolism and Remodeling in Tumor Cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer. Res. 2020, 39, 156. [Google Scholar] [CrossRef]
- Micallef, P.; Wu, Y.; Bauzá-Thorbrügge, M.; Chanclón, B.; Vujičić, M.; Peris, E.; Ek, C.J.; Asterholm, I.W. Adipose Tissue—Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth. Int. J. Mol. Sci. 2021, 22, 11881. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Sanderson, S.M.; Locasale, J.W. Cancer Metabolism. In Abeloff’s Clinical Oncology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 127–138.e4. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-Tumor Activity in the MMTV-Neu Model of HER2+ Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R.; Colomer, R. Inhibition of Tumor-Associated Fatty Acid Synthase Hyperactivity Induces Synergistic Chemosensitization of HER-2/Neu-Overexpressing Human Breast Cancer Cells to Docetaxel (Taxotere). Breast Cancer Res. Treat. 2004, 84, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Mehmi, I.; Papadimitropoulou, A.; Steen, T.V.; Cuyàs, E.; Verdura, S.; Espinoza, I.; Vellon, L.; Atlas, E.; Lupu, R. Fatty Acid Synthase Is a Key Enabler for Endocrine Resistance in Heregulin-Overexpressing Luminal b-like Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7661. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, S.; Lu, J.; Zhang, Z.; Wu, D.; Wu, Z.; Zheng, Y. GLUL Promotes Cell Proliferation in Breast Cancer. J. Cell. Biochem. 2017, 118, 2018–2025. [Google Scholar] [CrossRef]
- Chen, Y.C.; Sahoo, S.; Brien, R.; Jung, S.; Humphries, B.; Lee, W.; Cheng, Y.H.; Zhang, Z.; Luker, K.E.; Wicha, M.S.; et al. Single-Cell RNA-Sequencing of Migratory Breast Cancer Cells: Discovering Genes Associated with Cancer Metastasis. Analyst 2019, 144, 7296–7309. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Kim, H.M.; Koo, J.S. Expression of Lipid Metabolism-Related Proteins in Metastatic Breast Cancer. PLoS ONE 2015, 10, e0137204. [Google Scholar] [CrossRef]
- Moncur, J.T.; Park, J.P.; Memoli, V.A.; Mohandas, T.K.; Kinlaw, W.B. The “‘Spot 14′” Gene Resides on the Telomeric End of the 11q13 Amplicon and Is Expressed in Lipogenic Breast Cancers: Implications for Control of Tumor Metabolism. Proc. Natl. Acad. Sci. USA 1998, 95, 6989–6994. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Marci, A.; Mangoni, A.; Botti, C.; Di Tondo, U. Expression of Fatty Acid Synthase (FAS) as a Predictor of Recurrence in Stage I Breast Carcinoma Patients. Cancer 1996, 77, 474–482. [Google Scholar] [CrossRef]
- Alo, P.L.; Visca, P.; Trombetta, G.; Mangoni, A.; Lentp, L.; Monaco, S.; Bottj, C.; Serpierp, D.E.; Di Tondo, U. Fatty acid synthase (FAS) predictive strength in poorly differentiated early breast carcinomas. Tumori 1999, 85, 35–40. [Google Scholar] [CrossRef]
- Wang, Y.; Kuhajda, F.P.; Li, J.N.; Pizer, E.S.; Han, W.F.; Sokoll, L.J.; Chan, D.W. Fatty Acid Synthase (FAS) Expression in Human Breast Cancer Cell Culture Supernatants and in Breast Cancer Patients. Cancer Lett. 2001, 167, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Thupari, J.N.; Pinn, M.L.; Kuhajda, F.P. Fatty Acid Synthase Inhibition in Human Breast Cancer Cells Leads to Malonyl-CoA-Induced Inhibition of Fatty Acid Oxidation and Cytotoxicity. Biochem. Biophys. Res. Commun. 2001, 285, 217–223. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.; Muniandy, M.; Buzkova, J.; Mardinoglu, A.; Rodríguez, A.; Frühbeck, G.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Kaprio, J.; et al. Mitochondria-Related Transcriptional Signature Is Downregulated in Adipocytes in Obesity: A Study of Young Healthy MZ Twins. Diabetologia 2017, 60, 169–181. [Google Scholar] [CrossRef]
- Yin, X.; Lanza, I.R.; Swain, J.M.; Sarr, M.G.; Nair, K.S.; Jensen, M.D. Adipocyte Mitochondrial Function Is Reduced in Human Obesity Independent of Fat Cell Size. J. Clin. Endocrinol. Metab. 2014, 99, E209–E216. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, J.I.; Takemitsu, M.; Nonaka, I. Recovery of the Missing Tumorigenicity in Mitochondrial DNA-Less HeLa Cells by Introduction of Mitochondrial DNA from Normal Human Cells. Somat. Cell Mol. Genet. 1992, 18, 123–129. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA Variation and Cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Zhu, W.; Qin, W.; Bradley, P.; Wessel, A.; Puckett, C.L.; Sauter, E.R. Mitochondrial DNA Mutations in Breast Cancer Tissue and in Matched Nipple Aspirate Fluid. Carcinogenesis 2005, 26, 145–152. [Google Scholar] [CrossRef]
- Mambo, E.; Chatterjee, A.; Xing, M.; Tallini, G.; Haugen, B.R.; Yeung, S.C.J.; Sukumar, S.; Sidransky, D. Tumor-Specific Changes in MtDNA Content in Human Cancer. Int. J. Cancer 2005, 116, 920–924. [Google Scholar] [CrossRef]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA Mutations and Mitochondrial DNA Depletion in Breast Cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.X.C.; Radpour, R.; Haghighi, M.M.; Kohler, C.; Xia, P.; Hahn, S.; Holzgreve, W.; Zhong, X.Y. Mitochondrial DNA Content in Paired Normal and Cancerous Breast Tissue Samples from Patients with Breast Cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 983–989. [Google Scholar] [CrossRef]
- Domínguez-de-la-Cruz, E.; Muñoz, M.d.L.; Pérez-Muñoz, A.; García-Hernández, N.; Moctezuma-Meza, C.; Hinojosa-Cruz, J.C. Reduced Mitochondrial DNA Copy Number Is Associated with the Haplogroup, and Some Clinical Features of Breast Cancer in Mexican Patients. Gene 2020, 761, 145047. [Google Scholar] [CrossRef]
- Radzak, S.M.A.; Khair, S.Z.N.M.; Ahmad, F.; Patar, A.; Idris, Z.; Yusoff, A.A.M. Insights Regarding Mitochondrial DNA Copy Number Alterations in Human Cancer (Review). Int. J. Mol. Med. 2022, 50, 104. [Google Scholar] [CrossRef]
- Rai, N.K.; Panjwani, G.; Ghosh, A.K.; Haque, R.; Sharma, L.K. Analysis of Mitochondrial DNA Copy Number Variation in Blood and Tissue Samples of Metastatic Breast Cancer Patients (A Pilot Study). Biochem. Biophys. Rep. 2021, 26, 100931. [Google Scholar] [CrossRef]
- Bai, R.K.; Chang, J.; Yeh, K.T.; Lou, M.A.; Lu, J.F.; Tan, D.J.; Liu, H.; Wong, L.J.C. Mitochondrial DNA Content Varies with Pathological Characteristics of Breast Cancer. J. Oncol. 2011, 2011, 496189. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Srinivasan, S.; Raman, P.; Jiang, Y.; Kaufman, B.A.; Taylor, D.; Dong, D.; Chakrabarti, R.; Picard, M.; Carstens, R.P.; et al. Aggressive Triple Negative Breast Cancers Have Unique Molecular Signature on the Basis of Mitochondrial Genetic and Functional Defects. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Srinivasan, S.; Ruthel, G.; Kashina, A.K.; Carstens, R.P.; Mendoza, A.; Khanna, C.; Van Winkle, T.; Avadhani, N.G. Mitochondrial Retrograde Signaling Induces Epithelial–Mesenchymal Transition and Generates Breast Cancer Stem Cells. Oncogene 2014, 33, 5238. [Google Scholar] [CrossRef]
- Watkins, G.; Douglas-Jones, A.; Mansel, R.E.; Jiang, W.G. The Localisation and Reduction of Nuclear Staining of PPARγ and PGC-1 in Human Breast Cancer. Oncol. Rep. 2004, 12, 483–488. [Google Scholar] [CrossRef]
- Lebleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e5. [Google Scholar] [CrossRef] [PubMed]
- Kaaman, M.; Sparks, L.M.; Van Harmelen, V.; Smith, S.R.; Sjölin, E.; Dahlman, I.; Arner, P. Strong Association between Mitochondrial DNA Copy Number and Lipogenesis in Human White Adipose Tissue. Diabetologia 2007, 50, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, S.; Mepani, R.J.; Laznik, D.; Ye, L.; Jurczak, M.J.; Jornayvaz, F.R.; Estall, J.L.; Bhowmick, D.C.; Shulman, G.I.; Spiegelman, B.M. Development of Insulin Resistance in Mice Lacking PGC-1α in Adipose Tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 9635–9640. [Google Scholar] [CrossRef]
- Semple, R.K.; Crowley, V.C.; Sewter, C.P.; Laudes, M.; Christodoulides, C.; Considine, R.V.; Vidal-Puig, A.; O’Rahilly, S. Expression of the Thermogenic Nuclear Hormone Receptor Coactivator PGC-1α Is Reduced in the Adipose Tissue of Morbidly Obese Subjects. Int. J. Obes. 2004, 28, 176–179. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakic, T.; Kalezic, A.; Drvendzija, Z.; Udicki, M.; Ivkovic Kapicl, T.; Srdic Galic, B.; Korac, A.; Jankovic, A.; Korac, B. Breast Cancer: Mitochondria-Centered Metabolic Alterations in Tumor and Associated Adipose Tissue. Cells 2024, 13, 155. https://doi.org/10.3390/cells13020155
Zakic T, Kalezic A, Drvendzija Z, Udicki M, Ivkovic Kapicl T, Srdic Galic B, Korac A, Jankovic A, Korac B. Breast Cancer: Mitochondria-Centered Metabolic Alterations in Tumor and Associated Adipose Tissue. Cells. 2024; 13(2):155. https://doi.org/10.3390/cells13020155
Chicago/Turabian StyleZakic, Tamara, Andjelika Kalezic, Zorka Drvendzija, Mirjana Udicki, Tatjana Ivkovic Kapicl, Biljana Srdic Galic, Aleksandra Korac, Aleksandra Jankovic, and Bato Korac. 2024. "Breast Cancer: Mitochondria-Centered Metabolic Alterations in Tumor and Associated Adipose Tissue" Cells 13, no. 2: 155. https://doi.org/10.3390/cells13020155
APA StyleZakic, T., Kalezic, A., Drvendzija, Z., Udicki, M., Ivkovic Kapicl, T., Srdic Galic, B., Korac, A., Jankovic, A., & Korac, B. (2024). Breast Cancer: Mitochondria-Centered Metabolic Alterations in Tumor and Associated Adipose Tissue. Cells, 13(2), 155. https://doi.org/10.3390/cells13020155