


Cellular Mechanisms Mediating Exercise-Induced Protection against Cardiotoxic Anthracycline Cancer Therapy
{kind=link}
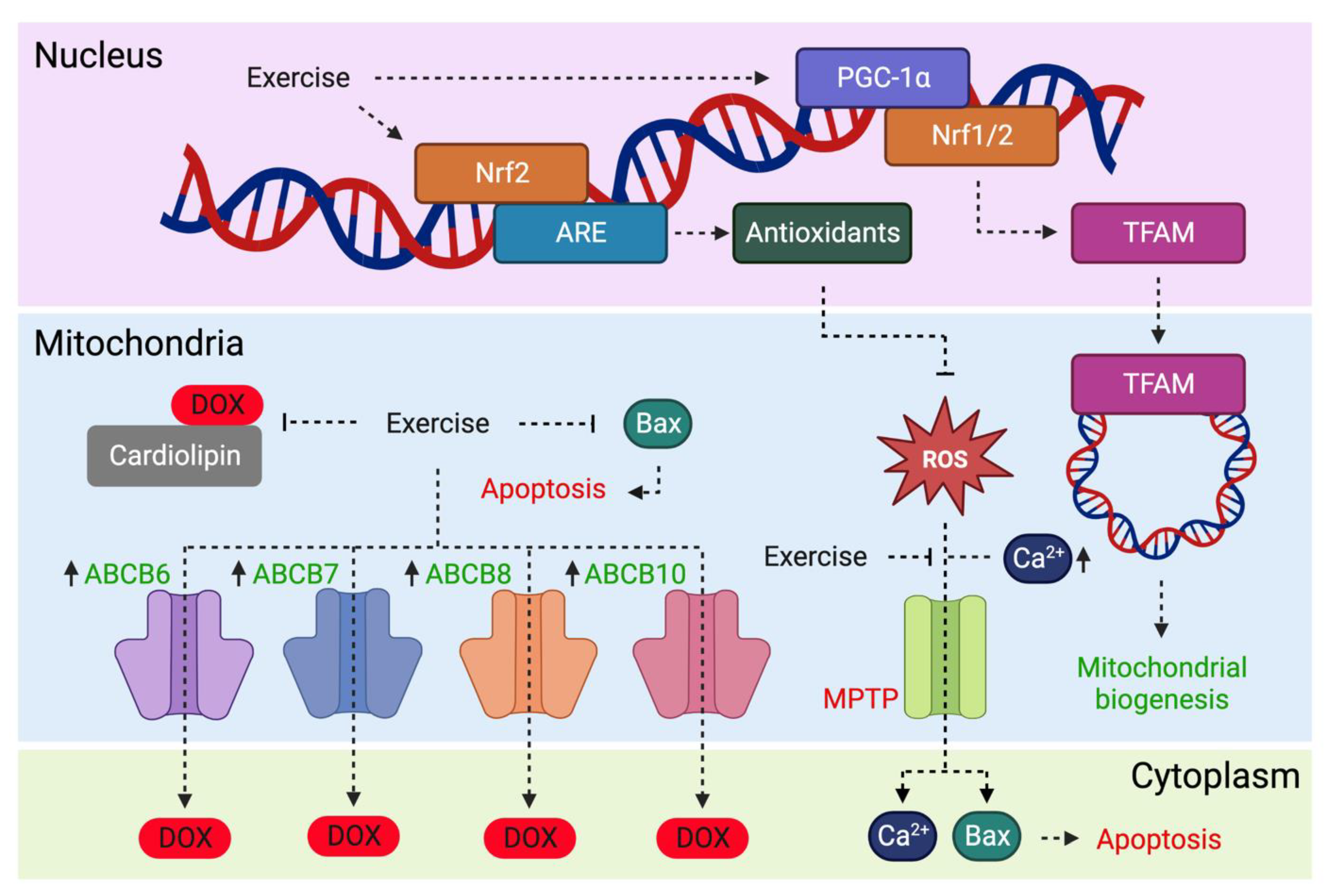{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxidative Stress
2.1. Exercise Prevents Oxidation of Biological Molecules
2.2. Exercise Upregulates Antioxidants
2.3. Exercise Maintains Cellular Protein Homeostasis
3. Mitochondrial Adaptations
3.1. Exercise Preserves Mitochondrial Structure
3.2. Exercise Decreases Susceptibility to MPTP Toxicity
3.3. Exercise Induces Mitochondrial Biogenesis
3.4. Exercise Reduces Mitochondrial Accumulation of DOX
3.5. Exercise Prevents Mitochondrial-Mediated Apoptosis
4. Cardiac Adaptations
4.1. Exercise Preserves Cardiomyocyte Ultrastructure
4.2. Exercise Prevents MHC Isoform Shifts
4.3. Exercise Alleviates Fibrosis
4.4. Exercise Preserves Cardiac Size
4.5. Exercise Preserves SERCA2A Activity
5. Vascular Adaptations
5.1. Exercise Preserves Smooth Muscle Function
5.2. Exercise Preserves Endothelial Function
6. Exerkines
Exercise Releases Cardioprotective Factors
7. Conclusions & Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Nitiss, J.L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Arnaiz, C.; Busto, N.; Leal, J.M.; García, B. New Insights into the Mechanism of the DNA/Doxorubicin Interaction. J. Phys. Chem. B 2014, 118, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Arango, M.G.; Dasari, H.; Calle, M.A.; Adjei, E.; Mesa, J.R.; Scott, C.G.; Thompson, C.A.; Cerhan, J.R.; Haddad, T.C.; et al. Association of Anthracycline with Heart Failure in Patients Treated for Breast Cancer or Lymphoma, 1985–2010. JAMA Netw. Open 2023, 6, e2254669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the Molecular Basis of Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Atwal, M.; Swan, R.L.; Rowe, C.; Lee, K.C.; Lee, D.C.; Armstrong, L.; Cowell, I.G.; Austin, C.A. Intercalating TOP2 Poisons Attenuate Topoisomerase Action at Higher Concentrations. Mol. Pharmacol. 2019, 96, 475–484. [Google Scholar] [CrossRef]
- Min, K.; Kwon, O.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.; Hwang, M.; Szeto, H.H.; Kavazis, A.N.; et al. Increased Mitochondrial Emission of Reactive Oxygen Species and Calpain Activation Are Required for Doxorubicin-induced Cardiac and Skeletal Muscle Myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Mulrooney, D.A.; Yeazel, M.W.; Kawashima, T.; Mertens, A.C.; Mitby, P.; Stovall, M.; Donaldson, S.S.; Green, D.M.; Sklar, C.A.; Robison, L.L.; et al. Cardiac Outcomes in a Cohort of Adult Survivors of Childhood and Adolescent Cancer: Retrospective Analysis of the Childhood Cancer Survivor Study Cohort. BMJ 2009, 339, b4606. [Google Scholar] [CrossRef]
- Vo, J.B.; Ramin, C.; Barac, A.; de Gonzalez, A.B.; Veiga, L. Trends in Heart Disease Mortality among Breast Cancer Survivors in the US, 1975–2017. Breast Cancer Res. Treat. 2022, 192, 611–622. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Franco, V.I.; Miller, T.L.; Colan, S.D.; Sallan, S.E. Cardiovascular Disease in Adult Survivors of Childhood Cancer. Annu. Rev. Med. 2015, 66, 161–176. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef] [PubMed]
- Lewinter, C.; Nielsen, T.H.; Edfors, L.R.; Linde, C.; Bland, J.M.; LeWinter, M.; Cleland, J.G.F.; Køber, L.; Braunschweig, F.; Mansson-Broberg, A. A Systematic Review and Meta-Analysis of Beta-Blockers and Renin–Angiotensin System Inhibitors for Preventing Left Ventricular Dysfunction Due to Anthracyclines or Trastuzumab in Patients with Breast Cancer. Eur. Heart J. 2021, 43, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.L.; Hasinoff, B.B. The One-Ring Open Hydrolysis Product Intermediates of the Cardioprotective Agent ICRF-187 (Dexrazoxane) Displace Iron from Iron-Anthracycline Complexes. Agents Actions 1993, 40, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Yan, T.; Jendrny, C.; Nemecek, A.; Vincetic, M.; Gödtel-Armbrust, U.; Wojnowski, L. Dexrazoxane May Prevent Doxorubicin-Induced DNA Damage via Depleting Both Topoisomerase II Isoforms. BMC Cancer 2014, 14, 842. [Google Scholar] [CrossRef]
- Jirkovský, E.; Jirkovská, A.; Bavlovič-Piskáčková, H.; Skalická, V.; Pokorná, Z.; Karabanovich, G.; Kollárová-Brázdová, P.; Kubeš, J.; Lenčová-Popelová, O.; Mazurová, Y.; et al. Clinically Translatable Prevention of Anthracycline Cardiotoxicity by Dexrazoxane Is Mediated by Topoisomerase II Beta and Not Metal Chelation. Circ. Heart Fail. 2021, 14, e008209. [Google Scholar] [CrossRef]
- Reichardt, P.; Tabone, M.-D.; Mora, J.; Morland, B.; Jones, R.L. Riskbenefit of Dexrazoxane for Preventing Anthracycline-Related Cardiotoxicity: Re-Evaluating the European Labeling. Future Oncol. 2018, 14, 2663–2676. [Google Scholar] [CrossRef]
- Naaktgeboren, W.R.; Binyam, D.; Stuiver, M.M.; Aaronson, N.K.; Teske, A.J.; van Harten, W.H.; Groen, W.G.; May, A.M. Efficacy of Physical Exercise to Offset Anthracycline-Induced Cardiotoxicity: A Systematic Review and Meta-Analysis of Clinical and Preclinical Studies. J. Am. Heart Assoc. 2021, 10, e021580. [Google Scholar] [CrossRef]
- Ghignatti, P.V.d.C.; Nogueira, L.J.; Lehnen, A.M.; Leguisamo, N.M. Cardioprotective Effects of Exercise Training on Doxorubicin-Induced Cardiomyopathy: A Systematic Review with Meta-Analysis of Preclinical Studies. Sci. Rep. 2021, 11, 6330. [Google Scholar] [CrossRef]
- Wonders, K.Y.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Acute Exercise Protects against Doxorubicin Cardiotoxicity. Integr. Cancer Ther. 2008, 7, 147–154. [Google Scholar] [CrossRef]
- Lien, C.-Y.; Jensen, B.T.; Hydock, D.S.; Hayward, R. Short-Term Exercise Training Attenuates Acute Doxorubicin Cardiotoxicity. J. Physiol. Biochem. 2015, 71, 669–678. [Google Scholar] [CrossRef]
- Howden, E.J.; Bigaran, A.; Beaudry, R.; Fraser, S.; Selig, S.; Foulkes, S.; Antill, Y.; Nightingale, S.; Loi, S.; Haykowsky, M.J.; et al. Exercise as a Diagnostic and Therapeutic Tool for the Prevention of Cardiovascular Dysfunction in Breast Cancer Patients. Eur. J. Prev. Cardiol. 2018, 26, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Hornsby, W.E.; Douglas, P.S.; West, M.J.; Kenjale, A.A.; Lane, A.R.; Schwitzer, E.R.; Ray, K.A.; Herndon, J.E.; Coan, A.; Gutierrez, A.; et al. Safety and Efficacy of Aerobic Training in Operable Breast Cancer Patients Receiving Neoadjuvant Chemotherapy: A Phase II Randomized Trial. Acta Oncol. 2014, 53, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, S.J.; Howden, E.J.; Haykowsky, M.J.; Antill, Y.; Salim, A.; Nightingale, S.S.; Loi, S.; Claus, P.; Janssens, K.; Mitchell, A.M.; et al. Exercise for the Prevention of Anthracycline-Induced Functional Disability and Cardiac Dysfunction: The BReast Cancer Randomized EXercise InTervention (BREXIT) Study. Circulation 2022, 147, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox Cycling of Anthracyclines by Cardiac Mitochondria. I. Anthracycline Radical Formation by NADH Dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef] [PubMed]
- Pawłowska, J.; Tarasiuk, J.; Wolf, C.R.; Paine, M.J.I.; Borowski, E. Differential Ability of Cytostatics from Anthraquinone Group to Generate Free Radicals in Three Enzymatic Systems: NADH Dehydrogenase, NADPH Cytochrome P450 Reductase, and Xanthine Oxidase. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2003, 13, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Garner, A.P.; Paine, M.J.; Rodriguez-Crespo, I.; Chinje, E.C.; Montellano, P.O.D.; Stratford, I.J.; Tew, D.G.; Wolf, C.R. Nitric Oxide Synthases Catalyze the Activation of Redox Cycling and Bioreductive Anticancer Agents. Cancer Res. 1999, 59, 1929–1934. [Google Scholar]
- Geest, B.D.; Mishra, M. Doxorubicin-Induced Cardiomyopathy: TERT Gets to the Heart of the Matter. Mol. Ther. 2021, 29, 1363–1365. [Google Scholar] [CrossRef]
- Myers, C. The Role of Iron in Doxorubicin-Induced Cardiomyopathy. Semin. Oncol. 1998, 25 (Suppl. S10), 10–14. [Google Scholar]
- Radi, R. Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Marques-Aleixo, I.; Santos-Alves, E.; Mariani, D.; Rizo-Roca, D.; Padrão, A.I.; Rocha-Rodrigues, S.; Viscor, G.; Torrella, J.R.; Ferreira, R.; Oliveira, P.J.; et al. Physical Exercise Prior and during Treatment Reduces Sub-Chronic Doxorubicin-Induced Mitochondrial Toxicity and Oxidative Stress. Mitochondrion 2015, 20, 22–33. [Google Scholar] [CrossRef]
- Chicco, A.J.; Schneider, C.M.; Hayward, R. Voluntary Exercise Protects against Acute Doxorubicin Cardiotoxicity in the Isolated Perfused Rat Heart. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2005, 289, R424–R431. [Google Scholar] [CrossRef] [PubMed]
- Shirinbayan, V.; Roshan, V.D. Pretreatment Effect of Running Exercise on HSP70 and DOX-Induced Cardiotoxicity. Asian Pac. J. Cancer Prev. 2012, 13, 5849–5855. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Roshan, V.D. Modulatory Effect of Aerobic Exercise Training on Doxorubicin-Induced Cardiotoxicity in Rats with Different Ages. Cardiovasc. Toxicol. 2018, 18, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kappus, H.; Muliawan, H.; Scheulen, M.E. In Vivo Studies on Adriamycin-Induced Lipid Peroxidation and Effects of Ferrous Ions. Dev. Toxicol. Environ. Sci. 1980, 8, 635–638. [Google Scholar]
- Younus, H. Therapeutic Potentials of Superoxide Dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Rogan, K.J.; Sung, M.M.; Zordoky, B.N.; Haykowsky, M.J.; Young, M.E.; Jones, L.W.; Dyck, J.R.B. Both Aerobic Exercise and Resveratrol Supplementation Attenuate Doxorubicin-Induced Cardiac Injury in Mice. Am. J. Physiol.-Endocrinol. Metabol. 2013, 305, E243–E253. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, C.M.; Martins, M.A.; Alves, R.; Nascimento, A.L.R.; Botti, G.C.R.M.; Rocha, V.N.; Matsuura, C. Aerobic Exercise Training Attenuates Doxorubicin-Induced Ultrastructural Changes in Rat Ventricular Myocytes. Life Sci. 2021, 264, 118698. [Google Scholar] [CrossRef]
- Farzanegi, P.; Asadi, M.; Abdi, A.; Etemadian, M.; Amani, M.; Amrollah, V.; Shahri, F.; Gholami, V.; Abdi, Z.; Moradi, L.; et al. Swimming Exercise in Combination with Garlic Extract Administration as a Therapy against Doxorubicin-Induced Hepatic, Heart and Renal Toxicity to Rats. Toxin Rev. 2020, 39, 434–443. [Google Scholar] [CrossRef]
- Ascensão, A.; Magalhães, J.; Soares, J.M.C.; Ferreira, R.; Neuparth, M.J.; Marques, F.; Oliveira, P.J.; Duarte, J.A. Moderate Endurance Training Prevents Doxorubicin-Induced in Vivo Mitochondriopathy and Reduces the Development of Cardiac Apoptosis. Am. J. Physiol.-Heart Circ. Phisyol. 2005, 289, H722–H731. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; Smuder, A.J.; Min, K.; Tümer, N.; Powers, S.K. Short-Term Exercise Training Protects against Doxorubicin-Induced Cardiac Mitochondrial Damage Independent of HSP72. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H1515–H1524. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, J. Cardioprotective Effects of Aerobic Regular Exercise against Doxorubicin—Induced Oxidative Stress in Rat. Afr. J. Pharm. Pharmacol. 2012, 6, 2380–2388. [Google Scholar] [CrossRef]
- Ascensão, A.; Magalhães, J.; Soares, J.; Ferreira, R.; Neuparth, M.; Marques, F.; Oliveira, J.; Duarte, J. Endurance Training Attenuates Doxorubicin-Induced Cardiac Oxidative Damage in Mice. Int. J. Cardiol. 2005, 100, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, T.; Yoo, S.-K.; Kim, S.-W.; Hwang, S.-Y.; Sohn, S.-H.; Kim, I.-W.; Kim, S.-K. Cordycepin (3′-Deoxyadenosine) Attenuates Age-Related Oxidative Stress and Ameliorates Antioxidant Capacity in Rats. Exp. Gerontol. 2012, 47, 979–987. [Google Scholar] [CrossRef]
- Bejma, J.; Ramires, P.; Ji, L.L. Free Radical Generation and Oxidative Stress with Ageing and Exercise: Differential Effects in the Myocardium and Liver. Acta Physiol. Scand. 2000, 169, 343–351. [Google Scholar] [CrossRef]
- Kregel, K.C. Invited Review: Heat Shock Proteins: Modifying Factors in Physiological Stress Responses and Acquired Thermotolerance. J. Appl. Physiol. 2002, 92, 2177–2186. [Google Scholar] [CrossRef]
- Peng, W.; Zhang, Y.; Zheng, M.; Cheng, H.; Zhu, W.; Cao, C.-M.; Xiao, R.-P. Cardioprotection by CaMKII-ΔB Is Mediated by Phosphorylation of Heat Shock Factor 1 and Subsequent Expression of Inducible Heat Shock Protein 70. Circ. Res. 2010, 106, 102–110. [Google Scholar] [CrossRef]
- Tanimoto, T.; Parseghian, M.H.; Nakahara, T.; Kawai, H.; Narula, N.; Kim, D.; Nishimura, R.; Weisbart, R.H.; Chan, G.; Richieri, R.A.; et al. Cardioprotective Effects of HSP72 Administration on Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2017, 70, 1479–1492. [Google Scholar] [CrossRef]
- Marber, M.S.; Mestril, R.; Chi, S.H.; Sayen, M.R.; Yellon, D.M.; Dillmann, W.H. Overexpression of the Rat Inducible 70-KD Heat Stress Protein in a Transgenic Mouse Increases the Resistance of the Heart to Ischemic Injury. J. Clin. Investig. 1995, 95, 1446–1456. [Google Scholar] [CrossRef]
- Chicco, A.J.; Schneider, C.M.; Hayward, R. Exercise Training Attenuates Acute Doxorubicin-Induced Cardiac Dysfunction. J. Cardiovasc. Pharmacol. 2006, 47, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.L.; Gao, X.; Du, X.-J.; Boey, E.J.H.; Matsumoto, A.; Bernardo, B.C.; Kiriazis, H.; Cemerlang, N.; Tan, J.W.; Tham, Y.K.; et al. Phosphoinositide 3-Kinase P110α Is a Master Regulator of Exercise-Induced Cardioprotection and PI3K Gene Therapy Rescues Cardiac Dysfunction. Circ. Heart Fail. 2012, 5, 523–534. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-Kinase(P110α) Plays a Critical Role for the Induction of Physiological, but Not Pathological, Cardiac Hypertrophy. Proc. Nat. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Prasad, S.V.N.; Mao, L.; Noma, T.; Yan, Z.; Kim, H.-S.; Smithies, O.; Rockman, H.A. Intermittent Pressure Overload Triggers Hypertrophy-Independent Cardiac Dysfunction and Vascular Rarefaction. J. Clin. Investig. 2006, 116, 1547–1560. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Sapra, G.; Patterson, N.L.; Cemerlang, N.; Kiriazis, H.; Ueyama, T.; Febbraio, M.A.; McMullen, J.R. Long-Term Overexpression of Hsp70 Does Not Protect against Cardiac Dysfunction and Adverse Remodeling in a MURC Transgenic Mouse Model with Chronic Heart Failure and Atrial Fibrillation. PLoS ONE 2015, 10, e0145173. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the Failing Heart. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1360–1372. [Google Scholar] [CrossRef]
- Neely, J.R.; Rovetto, M.J.; Oram, J.F. Myocardial Utilization of Carbohydrate and Lipids. Prog. Cardiovasc. Dis. 1972, 15, 289–329. [Google Scholar] [CrossRef]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-Induced Mitophagy and Mitochondrial Damage Is Associated with Dysregulation of the PINK1/Parkin Pathway. Toxicol. Vitr. 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Wang, F.; Chandra, J.; Kleinerman, E.S. Exercise Intervention Decreases Acute and Late Doxorubicin-induced Cardiotoxicity. Cancer Med. 2021, 10, 7572–7584. [Google Scholar] [CrossRef]
- Ascensão, A.; Lumini-Oliveira, J.; Machado, N.G.; Ferreira, R.M.; Gonçalves, I.O.; Moreira, A.C.; Marques, F.; Sardão, V.A.; Oliveira, P.J.; Magalhães, J. Acute Exercise Protects against Calcium-Induced Cardiac Mitochondrial Permeability Transition Pore Opening in Doxorubicin-Treated Rats. Clin. Sci. 2010, 120, 37–49. [Google Scholar] [CrossRef]
- Halestrap, A.P. What Is the Mitochondrial Permeability Transition Pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin Induces Mitochondrial Permeability Transition and Contractile Dysfunction in the Human Myocardium. Mitochondrion 2011, 11, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Marques-Aleixo, I.; Santos-Alves, E.; Torrella, J.R.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Exercise and Doxorubicin Treatment Modulate Cardiac Mitochondrial Quality Control Signaling. Cardiovasc. Toxicol. 2018, 18, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.B.; Huertas, A.M.; Hinkley, J.M.; Ichinoseki-Sekine, N.; Christou, D.D.; Smuder, A.J. Mitochondrial Accumulation of Doxorubicin in Cardiac and Diaphragm Muscle Following Exercise Preconditioning. Mitochondrion 2019, 45, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome Proliferator–Activated Receptor γ Coactivator-1 Promotes Cardiac Mitochondrial Biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Dhar, S.S.; Ongwijitwat, S.; Wong-Riley, M.T.T. Nuclear Respiratory Factor 1 Regulates All Ten Nuclear-Encoded Subunits of Cytochrome c Oxidase in Neurons*. J. Biol. Chem. 2008, 283, 3120–3129. [Google Scholar] [CrossRef]
- Ongwijitwat, S.; Liang, H.L.; Graboyes, E.M.; Wong-Riley, M.T.T. Nuclear Respiratory Factor 2 Senses Changing Cellular Energy Demands and Its Silencing Down-Regulates Cytochrome Oxidase and Other Target Gene MRNAs. Gene 2006, 374, 39–49. [Google Scholar] [CrossRef]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.-H.; et al. Transcriptional Coactivator PGC-1α Controls the Energy State and Contractile Function of Cardiac Muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1α Activation by Melatonin Attenuates Acute Doxorubicin Cardiotoxicity via Alleviating Mitochondrial Oxidative Damage and Apoptosis. Free. Radic. Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; Smuder, A.J.; Powers, S.K. Effects of Short-Term Endurance Exercise Training on Acute Doxorubicin-Induced FoxO Transcription in Cardiac and Skeletal Muscle. J. Appl. Physiol. 2014, 117, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, M.; Zhang, Y.; Li, X. Trimetazidine Attenuates Exhaustive Exercise-Induced Myocardial Injury in Rats via Regulation of the Nrf2/NF-ΚB Signaling Pathway. Front. Pharmacol. 2019, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute Exercise Stress Activates Nrf2/ARE Signaling and Promotes Antioxidant Mechanisms in the Myocardium. Free. Radic. Biol Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, W.; Niu, T.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Li, H.; Janicki, J.S.; Wang, X.L.; et al. Nrf2 Deficiency Exaggerates Doxorubicin-Induced Cardiotoxicity and Cardiac Dysfunction. Oxid. Med. Cell. Longev. 2014, 2014, 748524. [Google Scholar] [CrossRef]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Azami, N.; Hamzehlou, S.; Farahani, M.V.; Hushmandi, K.; Ashrafizadeh, M.; et al. Nrf2 Signaling Pathway in Chemoprotection and Doxorubicin Resistance: Potential Application in Drug Discovery. Antioxidants 2021, 10, 349. [Google Scholar] [CrossRef]
- Nicolay, K.; Timmers, R.J.M.; Spoelstra, E.; Neut, R.V.D.; Fok, J.J.; Huigen, Y.M.; Verkleij, A.J.; Kruijff, B.D. The Interaction of Adriamycin with Cardiolipin in Model and Rat Liver Mitochondrial Membranes. Biochim. Biophys. Acta (BBA)—Biomembr. 1984, 778, 359–371. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Morton, A.B.; Hall, S.E.; Smuder, A.J. Effects of Doxorubicin on Cardiac Muscle Subsarcolemmal and Intermyofibrillar Mitochondria. Mitochondrion 2017, 34, 9–19. [Google Scholar] [CrossRef]
- Cogswell, A.M.; Stevens, R.J.; Hood, D.A. Properties of Skeletal Muscle Mitochondria Isolated from Subsarcolemmal and Intermyofibrillar Regions. Am. J. Physiol.-Cell Physiol. 1993, 264, C383–C389. [Google Scholar] [CrossRef]
- Wang, F.; Iskra, B.; Kleinerman, E.; Alvarez-Florez, C.; Andrews, T.; Shaw, A.; Chandra, J.; Schadler, K.; Aune, G.J. Aerobic Exercise During Early Murine Doxorubicin Exposure Mitigates Cardiac Toxicity. J. Pediatr. Hematol. Sol. Oncol. 2018, 40, 208–215. [Google Scholar] [CrossRef]
- Jensen, B.T.; Lien, C.-Y.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Exercise Mitigates Cardiac Doxorubicin Accumulation and Preserves Function in the Rat. J. Cardiovasc. Pharmacol. 2013, 62, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Zutz, A.; Gompf, S.; Schägger, H.; Tampé, R. Mitochondrial ABC Proteins in Health and Disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 681–690. [Google Scholar] [CrossRef]
- Dell’Acqua, G.; Polishchuck, R.; Fallon, J.T.; Gordon, J.W. Cardiac Resistance to Adriamycin in Transgenic Mice Expressing a Rat Alpha-Cardiac Myosin Heavy Chain/Human Multiple Drug Resistance 1 Fusion Gene. Hum. Gene Ther. 1999, 10, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- van Asperen, J.; van Tellingen, O.; Tijssen, F.; Schinkel, A.H.; Beijnen, J.H. Increased Accumulation of Doxorubicin and Doxorubicinol in Cardiac Tissue of Mice Lacking Mdr1a P-Glycoprotein. Br. J. Cancer 1999, 79, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell. Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Alihemmati, A.; Ebadi, F.; Moghadaszadeh, M.; Asadi, M.; Zare, P.; Badalzadeh, R. Effects of High-Intensity Interval Training on the Expression of MicroRNA-499 and pro- and Anti-Apoptotic Genes in Doxorubicin-Cardiotoxicity in Rats. J. Electrocardiol. 2019, 55, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Chicco, A.J.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Low-Intensity Exercise Training during Doxorubicin Treatment Protects against Cardiotoxicity. J. Appl. Physiol. 2006, 100, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kwon, I.; Jang, Y.; Cosio-Lima, L.; Barrington, P. Endurance Exercise Attenuates Doxorubicin-Induced Cardiotoxicity. Med. Sci. Sports Exerc. 2019, 52, 25–36. [Google Scholar] [CrossRef]
- Walklate, J.; Ferrantini, C.; Johnson, C.A.; Tesi, C.; Poggesi, C.; Geeves, M.A. Alpha and Beta Myosin Isoforms and Human Atrial and Ventricular Contraction. Cell. Mol. Life Sci. 2021, 78, 7309–7337. [Google Scholar] [CrossRef] [PubMed]
- Herron, T.J.; McDonald, K.S. Small Amounts of α-Myosin Heavy Chain Isoform Expression Significantly Increase Power Output of Rat Cardiac Myocyte Fragments. Circ. Res. 2002, 90, 1150–1152. [Google Scholar] [CrossRef] [PubMed]
- Hydock, D.S.; Lien, C.-Y.; Schneider, C.M.; Hayward, R. Exercise Preconditioning Protects against Doxorubicin-Induced Cardiac Dysfunction. Med. Sci. Sports Exerc. 2008, 40, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Hydock, D.S.; Lien, C.-Y.; Jensen, B.T.; Schneider, C.M.; Hayward, R. Exercise Preconditioning Provides Long-Term Protection Against Early Chronic Doxorubicin Cardiotoxicity. Integr. Cancer Ther. 2011, 10, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Pfannenstiel, K.; Hayward, R. Effects of Resistance Exercise Training on Doxorubicin-Induced Cardiotoxicity. J. Cardiovasc. Pharmacol. 2018, 71, 332–339. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef]
- Meléndez, G.C.; Jordan, J.H.; D’Agostino, R.B.; Vasu, S.; Hamilton, C.A.; Hundley, W.G. Progressive 3-Month Increase in LV Myocardial ECV after Anthracycline-Based Chemotherapy. JACC Cardiovasc. Imaging 2017, 10, 708–709. [Google Scholar] [CrossRef]
- Jordan, J.H.; Vasu, S.; Morgan, T.M.; D’Agostino, R.B.D., Jr.; Meléndez, G.C.; Hamilton, C.A.; Arai, A.E.; Liu, S.; Liu, C.-Y.; Lima, J.A.C.; et al. Anthracycline-Associated T1 Mapping Characteristics Are Elevated Independent of the Presence of Cardiovascular Comorbidities in Cancer Survivors. Circ. Cardiovasc. Imaging 2018, 9, e004325. [Google Scholar] [CrossRef]
- Gomes-Santos, I.L.; Jordão, C.P.; Passos, C.S.; Brum, P.C.; Oliveira, E.M.; Chammas, R.; Camargo, A.A.; Negrão, C.E. Exercise Training Preserves Myocardial Strain and Improves Exercise Tolerance in Doxorubicin-Induced Cardiotoxicity. Front. Cardiovasc. Med. 2021, 8, 605993. [Google Scholar] [CrossRef]
- Yang, H.-L.; Hsieh, P.-L.; Hung, C.-H.; Cheng, H.-C.; Chou, W.-C.; Chu, P.-M.; Chang, Y.-C.; Tsai, K.-L. Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-Caused Cardiac Dysfunction through Inhibition of Cardiac Fibrosis and Inflammation. Cancers 2020, 12, 1102. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Scully, R.E.; Stevenson, K.E.; Franco, V.I.; Neuberg, D.S.; Colan, S.D.; Silverman, L.B.; Moslehi, J.J.; Cheng, S.; Sallan, S.E. Hearts Too Small for Body Size after Doxorubicin for Childhood ALL: Grinch Syndrome. J. Clin. Oncol. 2014, 32 (Suppl. S15), 10021. [Google Scholar] [CrossRef]
- Neilan, T.G.; Coelho-Filho, O.R.; Pena-Herrera, D.; Shah, R.V.; Jerosch-Herold, M.; Francis, S.A.; Moslehi, J.; Kwong, R.Y. Left Ventricular Mass in Patients with a Cardiomyopathy after Treatment with Anthracyclines. Am. J. Cardiol. 2012, 110, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, H.; Tang, M.; Ryu, Y.-C.; Wang, X. A Therapeutic Dose of Doxorubicin Activates Ubiquitin-Proteasome System-Mediated Proteolysis by Acting on Both the Ubiquitination Apparatus and Proteasome. Am. J. Physiol.-Heart Circ. Physiol. 2008, 295, H2541–H2550. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and Molecular Mechanisms of Muscle Atrophy. Dis. Model. Mech. 2012, 6, 25–39. [Google Scholar] [CrossRef]
- Pinto, A.P.; da Rocha, A.L.; Marafon, B.B.; Rovina, R.L.; Muñoz, V.R.; da Silva, L.E.C.M.; Pauli, J.R.; de Moura, L.P.; Cintra, D.E.; Ropelle, E.R.; et al. Impact of Different Physical Exercises on the Expression of Autophagy Markers in Mice. Int. J. Mol. Sci. 2021, 22, 2635. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Doxorubicin-Induced Markers of Myocardial Autophagic Signaling in Sedentary and Exercise Trained Animals. J. Appl. Physiol. 2013, 115, 176–185. [Google Scholar] [CrossRef]
- Ghignatti, P.V.d.C.; Russo, M.K.B.; Becker, T.; Guecheva, T.N.; Teixeira, L.V.; Lehnen, A.M.; Schaun, M.I.; Leguisamo, N.M. Preventive Aerobic Training Preserves Sympathovagal Function and Improves DNA Repair Capacity of Peripheral Blood Mononuclear Cells in Rats with Cardiomyopathy. Sci. Rep. 2022, 12, 6422. [Google Scholar] [CrossRef]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The Effects of Doxorubicin on Cardiac Calcium Homeostasis and Contractile Function. J. Cardiol. 2022, 80, 125–132. [Google Scholar] [CrossRef]
- Bass-Stringer, S.; Bernardo, B.C.; May, C.N.; Thomas, C.J.; Weeks, K.L.; McMullen, J.R. Adeno-Associated Virus Gene Therapy: Translational Progress and Future Prospects in the Treatment of Heart Failure. Heart Lung Circ. 2018, 27, 1285–1300. [Google Scholar] [CrossRef]
- Mushlin, P.S.; Cusack, B.J.; Boucek, R.J.; Andrejuk, T.; Li, X.; Olson, R.D. Time-related Increases in Cardiac Concentrations of Doxorubicinol Could Interact with Doxorubicin to Depress Myocardial Contractile Function. Br. J. Pharmacol. 1993, 110, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2018, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Maruhashi, T.; Noma, K.; Kihara, Y. Oxidative Stress and Endothelial Dysfunction: Clinical Evidence and Therapeutic Implications. Trends Cardiovasc Med. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Gibson, N.M.; Greufe, S.E.; Hydock, D.S.; Hayward, R. Doxorubicin-Induced Vascular Dysfunction and Its Attenuation by Exercise Preconditioning. J. Cardiovasc. Pharmacol. 2013, 62, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.-H.; Kobayashi, M.; Yang, Y.; Kleinerman, E.S. Exercise Inhibits Doxorubicin-Induced Damage to Cardiac Vessels and Activation of Hippo/YAP-Mediated Apoptosis. Cancers 2021, 13, 2740. [Google Scholar] [CrossRef]
- Chow, L.S.; Gerszten, R.E.; Taylor, J.M.; Pedersen, B.K.; van Praag, H.; Trappe, S.; Febbraio, M.A.; Galis, Z.S.; Gao, Y.; Haus, J.M.; et al. Exerkines in Health, Resilience and Disease. Nat. Rev. Endocrinol. 2022, 18, 273–289. [Google Scholar] [CrossRef]
- Priest, C.; Tontonoz, P. Inter-Organ Cross-Talk in Metabolic Syndrome. Nat. Metab. 2019, 1, 1177–1188. [Google Scholar] [CrossRef]
- Steensberg, A.; Hall, G.; Osada, T.; Sacchetti, M.; Saltin, B.; Pedersen, B.K. Production of Interleukin-6 in Contracting Human Skeletal Muscles Can Account for the Exercise-induced Increase in Plasma Interleukin-6. J. Physiol. 2000, 529, 237–242. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim. Biophys. Acta (BBA)-Mol. Cell. Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- McGinnis, G.R.; Ballmann, C.; Peters, B.; Nanayakkara, G.; Roberts, M.; Amin, R.; Quindry, J.C. Interleukin-6 Mediates Exercise Preconditioning against Myocardial Ischemia Reperfusion Injury. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, H1423–H1433. [Google Scholar] [CrossRef]
- Matsushita, K.; Iwanaga, S.; Oda, T.; Kimura, K.; Shimada, M.; Sano, M.; Umezawa, A.; Hata, J.; Ogawa, S. Interleukin-6/Soluble Interleukin-6 Receptor Complex Reduces Infarct Size via Inhibiting Myocardial Apoptosis. Lab. Investig. 2005, 85, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.-I.; Hara, A. The MRNA Expression of Il6 and Pdcd1 Are Predictive and Protective Factors for Doxorubicin-Induced Cardiotoxicity. Mol. Med. Rep. 2021, 23, 113. [Google Scholar] [CrossRef]
- Sanford, J.A.; Nogiec, C.D.; Lindholm, M.E.; Adkins, J.N.; Amar, D.; Dasari, S.; Drugan, J.K.; Fernández, F.M.; Radom-Aizik, S.; Schenk, S.; et al. Molecular Transducers of Physical Activity Consortium (MoTrPAC): Mapping the Dynamic Responses to Exercise. Cell 2020, 181, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhang, H.; Lin, H.; Gao, L.; Zhang, H.; Zhang, J.; Wang, C.; Gu, J. Irisin Ameliorates Doxorubicin-Induced Cardiac Perivascular Fibrosis through Inhibiting Endothelial-to-Mesenchymal Transition by Regulating ROS Accumulation and Autophagy Disorder in Endothelial Cells. Redox Biol. 2021, 46, 102120. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Whellan, D.J.; Lee, K.L.; Keteyian, S.J.; Cooper, L.S.; Ellis, S.J.; Leifer, E.S.; Kraus, W.E.; Kitzman, D.W.; Blumenthal, J.A.; et al. Efficacy and Safety of Exercise Training in Patients with Chronic Heart Failure: HF-ACTION Randomized Controlled Trial. JAMA 2009, 301, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Meiners, B.; Shenoy, C.; Zordoky, B.N. Clinical and Preclinical Evidence of Sex-Related Differences in Anthracycline-Induced Cardiotoxicity. Biol. Sex Differ. 2018, 9, 38. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dozic, S.; Howden, E.J.; Bell, J.R.; Mellor, K.M.; Delbridge, L.M.D.; Weeks, K.L. Cellular Mechanisms Mediating Exercise-Induced Protection against Cardiotoxic Anthracycline Cancer Therapy. Cells 2023, 12, 1312. https://doi.org/10.3390/cells12091312
Dozic S, Howden EJ, Bell JR, Mellor KM, Delbridge LMD, Weeks KL. Cellular Mechanisms Mediating Exercise-Induced Protection against Cardiotoxic Anthracycline Cancer Therapy. Cells. 2023; 12(9):1312. https://doi.org/10.3390/cells12091312
Chicago/Turabian StyleDozic, Sanela, Erin J. Howden, James R. Bell, Kimberley M. Mellor, Lea M. D. Delbridge, and Kate L. Weeks. 2023. "Cellular Mechanisms Mediating Exercise-Induced Protection against Cardiotoxic Anthracycline Cancer Therapy" Cells 12, no. 9: 1312. https://doi.org/10.3390/cells12091312
APA StyleDozic, S., Howden, E. J., Bell, J. R., Mellor, K. M., Delbridge, L. M. D., & Weeks, K. L. (2023). Cellular Mechanisms Mediating Exercise-Induced Protection against Cardiotoxic Anthracycline Cancer Therapy. Cells, 12(9), 1312. https://doi.org/10.3390/cells12091312