
The Muscarinic Acetylcholine M2 Receptor-Induced Nitration of p190A by eNOS Increases RhoA Activity in Cardiac Myocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies, Reagents, and Inhibitors
2.2. Isolation and Culture of Neonatal Rat Cardiac Myocytes
2.3. Immunoblot Analysis
2.4. RhoGTPase Activation Assay
2.5. Immunoprecipitation
2.6. Fluorescence Determination of Nitric Oxide Production in NRCM
2.7. Measurement of the Substrate Specificity of p190A and p190B towards Rac1 and RhoA
2.8. Statistical Analysis
3. Results and Discussion
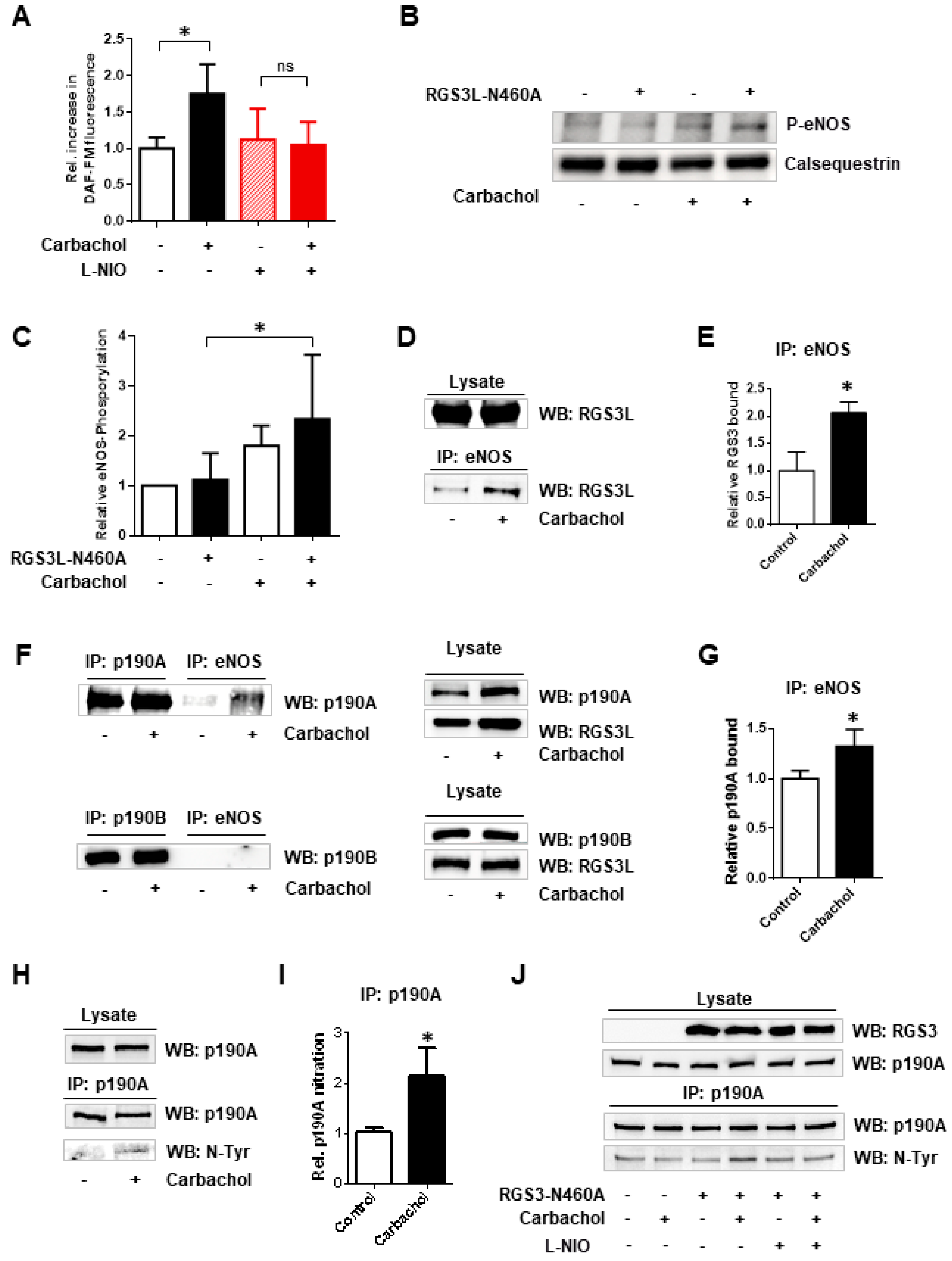
3.1. The Substrate Preference of p190A towards RhoGTPases, as Well as the Complex Formation with RGS3L, Depends on Nitration via eNOS-Derived Peroxynitrite Formation
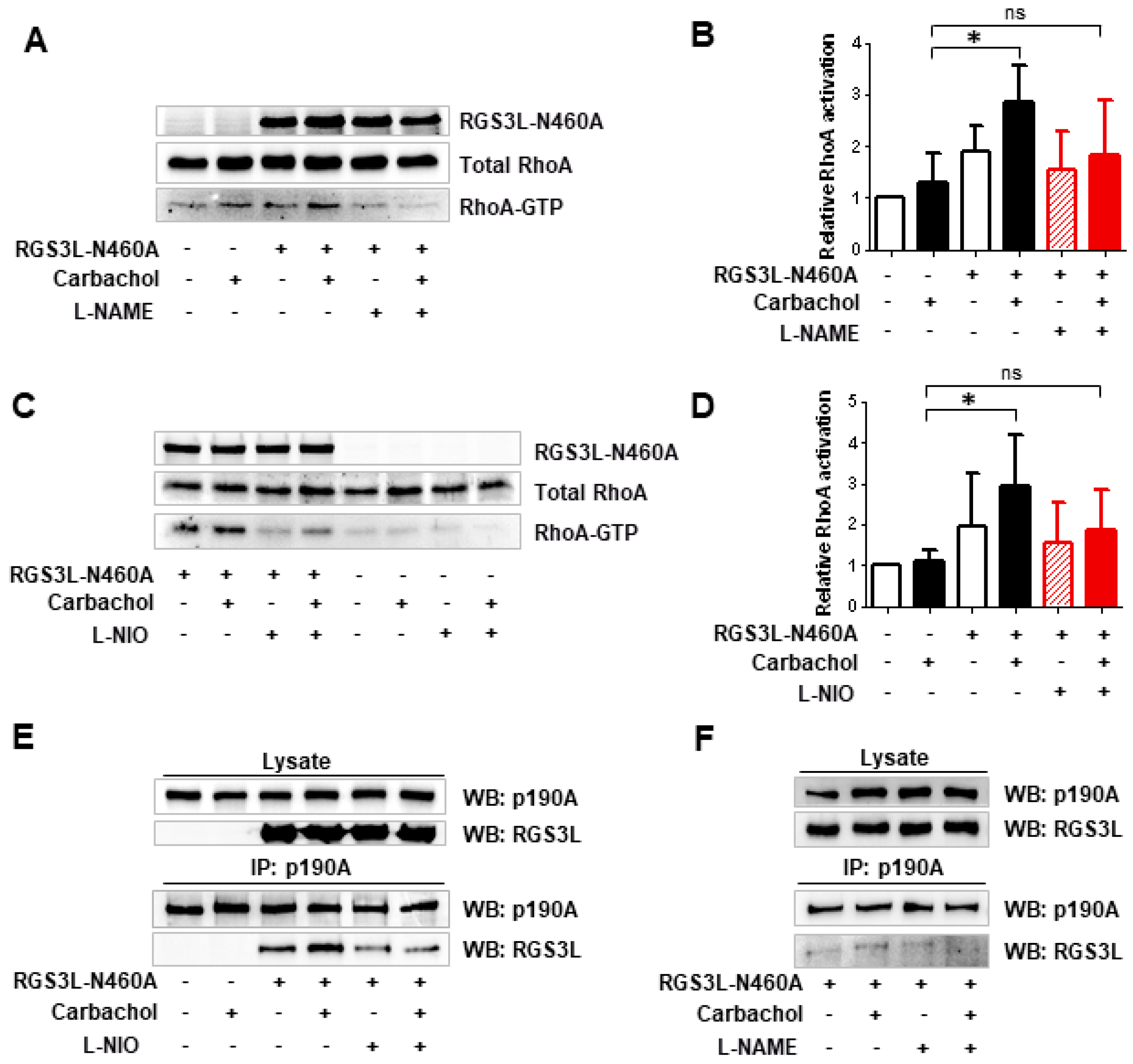
3.2. The RGS3L-Dependent RhoA Activation after Carbachol Stimulation in NRCM Likely Occurs at Caveolae
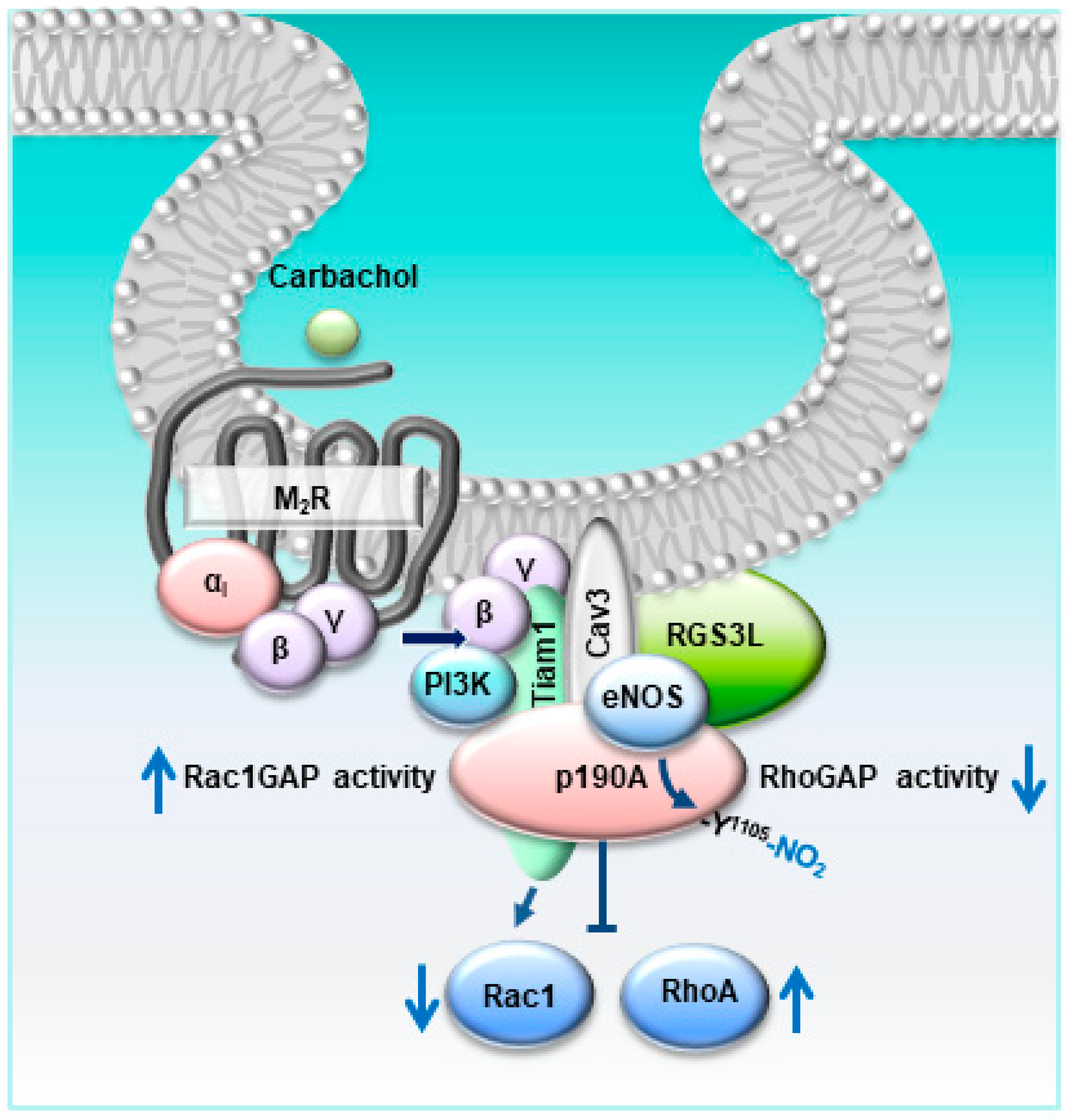
4. Conclusions, Study Limitations, and Further Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Umar, S.; van der Laarse, A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol. Cell. Biochem. 2010, 333, 191–201. [Google Scholar] [CrossRef]
- Casadei, B.; Sears, C.E. Nitric-oxide-mediated regulation of cardiac contractility and stretch responses. Prog. Biophys. Mol. Biol. 2003, 82, 67–80. [Google Scholar] [CrossRef]
- Yang, R.; Beqiri, D.; Shen, J.B.; Redden, J.M.; Dodge-Kafka, K.; Jacobson, K.A.; Liang, B.T. P2X4 receptor-eNOS signaling pathway in cardiac myocytes as a novel protective mechanism in heart failure. Comput. Struct. Biotechnol. J. 2015, 13, 1–7. [Google Scholar] [CrossRef]
- Yamamoto, E.; Hirata, Y.; Tokitsu, T.; Kusaka, H.; Sakamoto, K.; Yamamuro, M.; Kaikita, K.; Watanabe, H.; Hokimoto, S.; Sugiyama, S.; et al. The pivotal role of eNOS uncoupling in vascular endothelial dysfunction in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2015, 190, 335–337. [Google Scholar] [CrossRef]
- Flaherty, M.P.; Brown, M.; Grupp, I.L.; Schultz, J.E.; Murphree, S.S.; Jones, W.K. eNOS deficient mice develop progressive cardiac hypertrophy with altered cytokine and calcium handling protein expression. Cardiovasc. Toxicol. 2007, 7, 165–177. [Google Scholar] [CrossRef]
- Feron, O.; Zhao, Y.Y.; Kelly, R.A. The ins and outs of caveolar signaling. m2 muscarinic cholinergic receptors and eNOS activation versus neuregulin and ErbB4 signaling in cardiac myocytes. Ann. N. Y. Acad. Sci. 1999, 874, 11–19. [Google Scholar] [CrossRef]
- Feron, O.; Balligand, J.L. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc. Res. 2006, 69, 788–797. [Google Scholar] [CrossRef]
- Schwencke, C.; Braun-Dullaeus, R.C.; Wunderlich, C.; Strasser, R.H. Caveolae and caveolin in transmembrane signaling: Implications for human disease. Cardiovasc. Res. 2006, 70, 42–49. [Google Scholar] [CrossRef]
- Roth, D.M.; Patel, H.H. Role of caveolae in cardiac protection. Pediatr. Cardiol. 2011, 32, 329–333. [Google Scholar] [CrossRef]
- Feron, O.; Smith, T.W.; Michel, T.; Kelly, R.A. Dynamic targeting of the agonist-stimulated m2 muscarinic acetylcholine receptor to caveolae in cardiac myocytes. J. Biol. Chem. 1997, 272, 17744–17748. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Han, X.; Kelly, R.A. Muscarinic cholinergic signaling in cardiac myocytes: Dynamic targeting of M2AChR to sarcolemmal caveolae and eNOS activation. Life Sci. 1999, 64, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Levay, M.K.; Krobert, K.A.; Vogt, A.; Ahmad, A.; Jungmann, A.; Neuber, C.; Pasch, S.; Hansen, A.; Muller, O.J.; Lutz, S.; et al. RGS3L allows for an M2 muscarinic receptor-mediated RhoA-dependent inotropy in cardiomyocytes. Basic Res. Cardiol. 2022, 117, 8. [Google Scholar] [CrossRef]
- Héraud, C.; Pinault, M.; Lagrée, V.; Moreau, V. p190RhoGAPs, the ARHGAP35- and ARHGAP5-Encoded Proteins, in Health and Disease. Cells 2019, 8, 351. [Google Scholar] [CrossRef]
- Guegan, F.; Tatin, F.; Leste-Lasserre, T.; Drutel, G.; Genot, E.; Moreau, V. p190B RhoGAP regulates endothelial-cell-associated proteolysis through MT1-MMP and MMP2. J. Cell Sci. 2008, 121, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.R.; Komarova, Y.A.; Vogel, S.M.; Gao, X.; Bonini, M.G.; Rajasingh, J.; Zhao, Y.Y.; Brovkovych, V.; Malik, A.B. Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. J. Cell Biol. 2011, 193, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Lutz, S.; Rumenapp, U.; Han, L.; Jakobs, K.H.; Schmidt, M.; Wieland, T. Regulator of G-protein signalling 3 redirects prototypical Gi-coupled receptors from Rac1 to RhoA activation. Cell. Signal. 2007, 19, 1229–1237. [Google Scholar] [CrossRef]
- Noren, N.K.; Arthur, W.T.; Burridge, K. Cadherin engagement inhibits RhoA via p190RhoGAP. J. Biol. Chem. 2003, 278, 13615–13618. [Google Scholar] [CrossRef]
- Levay, M.; Bartos, B.; Ligeti, E. p190RhoGAP has cellular RacGAP activity regulated by a polybasic region. Cell. Signal. 2013, 25, 1388–1394. [Google Scholar] [CrossRef]
- Levay, M.; Krobert, K.A.; Wittig, K.; Voigt, N.; Bermudez, M.; Wolber, G.; Dobrev, D.; Levy, F.O.; Wieland, T. NSC23766, a widely used inhibitor of Rac1 activation, additionally acts as a competitive antagonist at muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2013, 347, 69–79. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Munzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, H.; Ziolo, M.T. Targeting NOS as a therapeutic approach for heart failure. Pharmacol. Ther. 2014, 142, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W. Regulation of endothelial nitric oxide synthase: Location, location, location. Annu. Rev. Physiol. 2002, 64, 749–774. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Dessy, C.; Opel, D.J.; Arstall, M.A.; Kelly, R.A.; Michel, T. Modulation of the endothelial nitric-oxide synthase-caveolin interaction in cardiac myocytes. Implications for the autonomic regulation of heart rate. J. Biol. Chem. 1998, 273, 30249–30254. [Google Scholar] [CrossRef]
- Hissa, B.; Oakes, P.W.; Pontes, B.; Ramírez-San Juan, G.; Gardel, M.L. Cholesterol depletion impairs contractile machinery in neonatal rat cardiomyocytes. Sci. Rep. 2017, 7, 43764. [Google Scholar] [CrossRef]
- Shi, C.S.; Lee, S.B.; Sinnarajah, S.; Dessauer, C.W.; Rhee, S.G.; Kehrl, J.H. Regulator of G-protein signaling 3 (RGS3) inhibits Gbeta1gamma 2-induced inositol phosphate production, mitogen-activated protein kinase activation, and Akt activation. J. Biol. Chem. 2001, 276, 24293–24300. [Google Scholar] [CrossRef]
- Hussain, R.I.; Qvigstad, E.; Birkeland, J.A.; Eikemo, H.; Glende, A.; Sjaastad, I.; Skomedal, T.; Osnes, J.B.; Levy, F.O.; Krobert, K.A. Activation of muscarinic receptors elicits inotropic responses in ventricular muscle from rats with heart failure through myosin light chain phosphorylation. Br. J. Pharmacol. 2009, 156, 575–586. [Google Scholar] [CrossRef]
- Owen, V.J.; Burton, P.B.; Mullen, A.J.; Birks, E.J.; Barton, P.; Yacoub, M.H. Expression of RGS3, RGS4 and Gi alpha 2 in acutely failing donor hearts and end-stage heart failure. Eur. Heart J. 2001, 22, 1015–1020. [Google Scholar] [CrossRef]
- Gödecke, A.; Heinicke, T.; Kamkin, A.; Kiseleva, I.; Strasser, R.H.; Decking, U.K.; Stumpe, T.; Isenberg, G.; Schrader, J. Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J. Physiol. 2001, 532, 195–204. [Google Scholar] [CrossRef]
- Gyurko, R.; Kuhlencordt, P.; Fishman, M.C.; Huang, P.L. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H971–H981. [Google Scholar] [CrossRef]
- Han, X.; Kubota, I.; Feron, O.; Opel, D.J.; Arstall, M.A.; Zhao, Y.Y.; Huang, P.; Fishman, M.C.; Michel, T.; Kelly, R.A. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 6510–6515. [Google Scholar] [CrossRef]
- Idigo, W.O.; Reilly, S.; Zhang, M.H.; Zhang, Y.H.; Jayaram, R.; Carnicer, R.; Crabtree, M.J.; Balligand, J.L.; Casadei, B. Regulation of endothelial nitric-oxide synthase (NOS) S-glutathionylation by neuronal NOS: Evidence of a functional interaction between myocardial constitutive NOS isoforms. J. Biol. Chem. 2012, 287, 43665–43673. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kai, Y.; Mano, A.; Nakamura, S.; Kakinuma, Y. A Novel Nitric Oxide Donor, S-Nitroso-NPivaloyl-D-Penicillamine, Activates a Non-Neuronal Cardiac Cholinergic System to Synthesize Acetylcholine and Augments Cardiac Function. Cell. Physiol. Biochem. 2019, 52, 922–934. [Google Scholar] [PubMed]
- Saw, E.L.; Kakinuma, Y.; Fronius, M.; Katare, R. The non-neuronal cholinergic system in the heart: A comprehensive review. J. Mol. Cell. Cardiol. 2018, 125, 129–139. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levay, M.K.; Throm, L.; Bahrami, N.; Wieland, T. The Muscarinic Acetylcholine M2 Receptor-Induced Nitration of p190A by eNOS Increases RhoA Activity in Cardiac Myocytes. Cells 2023, 12, 2432. https://doi.org/10.3390/cells12202432
Levay MK, Throm L, Bahrami N, Wieland T. The Muscarinic Acetylcholine M2 Receptor-Induced Nitration of p190A by eNOS Increases RhoA Activity in Cardiac Myocytes. Cells. 2023; 12(20):2432. https://doi.org/10.3390/cells12202432
Chicago/Turabian StyleLevay, Magdolna K., Lena Throm, Nabil Bahrami, and Thomas Wieland. 2023. "The Muscarinic Acetylcholine M2 Receptor-Induced Nitration of p190A by eNOS Increases RhoA Activity in Cardiac Myocytes" Cells 12, no. 20: 2432. https://doi.org/10.3390/cells12202432
APA StyleLevay, M. K., Throm, L., Bahrami, N., & Wieland, T. (2023). The Muscarinic Acetylcholine M2 Receptor-Induced Nitration of p190A by eNOS Increases RhoA Activity in Cardiac Myocytes. Cells, 12(20), 2432. https://doi.org/10.3390/cells12202432