
A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice

 ,
,  , and
, and
Abstract
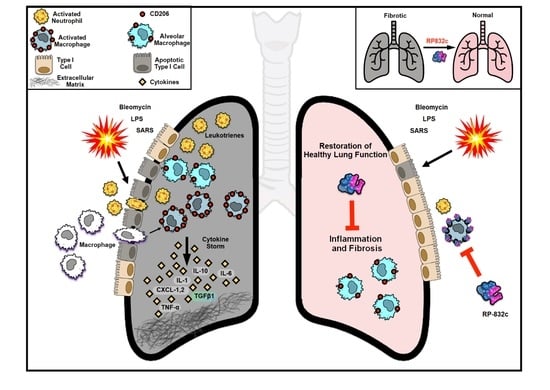
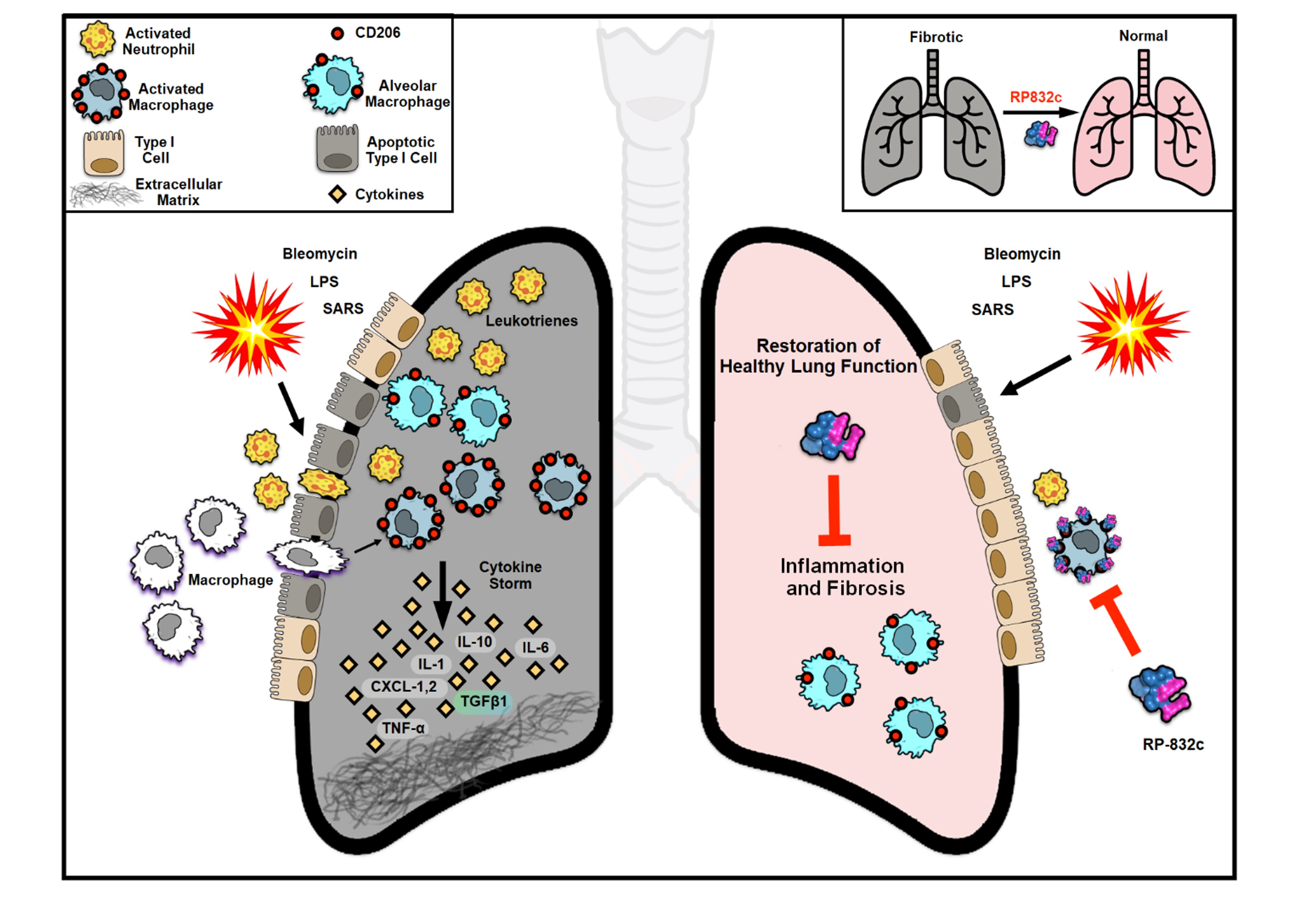
1. Introduction
2. Materials and Methods
2.1. Cell Culture of Primary Cells
2.2. Cell Viability Assay
2.3. Animal Experiments
2.4. Histological and Immunostaining Evaluation
2.5. Immunofluorescence
2.6. Quantitative Real-Time PCR
2.7. Statistical Analysis
3. Results
3.1. RP-832c Targets CD206 Positive Macrophages
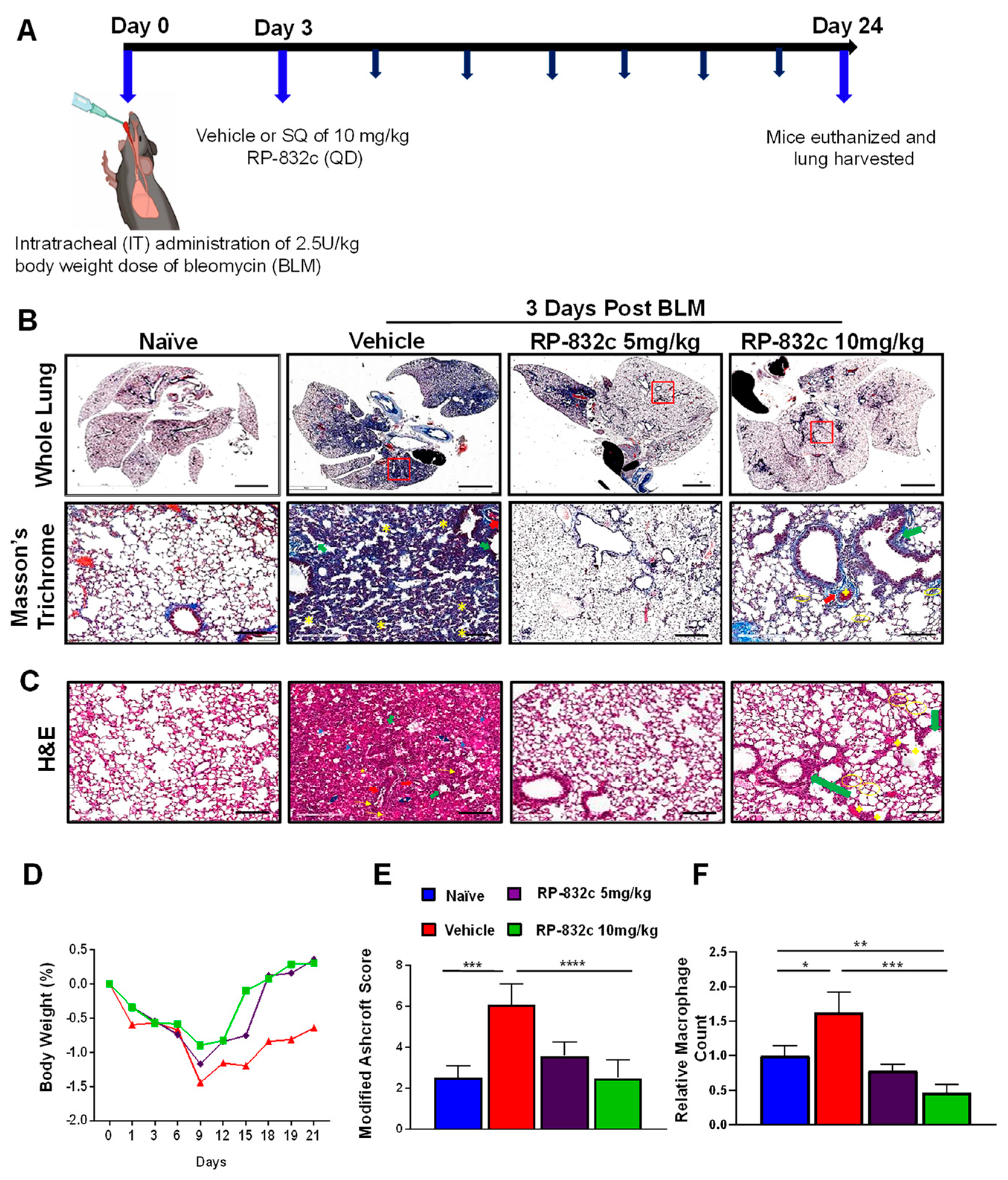
3.2. RP-832c Prevented Fibrosis in an Early Model of BLM-Induced Lung Fibrosis
3.3. RP-832c Peptide Treated Firosis and Reduced Expression of CD206 and Fibrosis Markers in a Late Model of BLM-Induced Lung Fibrosis Model
3.4. RP-832c Lacks Significant Toxicity
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirani, N.; Antonicelli, F.; Strieter, R.M.; Wiesener, M.S.; Ratcliffe, P.J.; Haslett, C.; Donnelly, S.C. The regulation of interleukin-8 by hypoxia in human macrophages--a potential role in the pathogenesis of acute respiratory distress syndrome (ARDS). Mol. Med. 2001, 7, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Carrillo, G.; Estrada, A.; Mejia, M.; Becerril, C.; Cisneros, J.; Gaxiola, M.; Pérez-Padilla, R.; Navarro, C.; Richards, T.; et al. Accelerated variant of idiopathic pulmonary fibrosis: Clinical behavior and gene expression pattern. PLoS ONE 2007, 2, e482. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, L.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology, and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Geng, Y.; Li, L.; Li, X.; Yan, X.; Fang, Y.; Li, X.; Dong, S.; Liu, X.; Yang, X.; et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J. Exp. Med. 2015, 212, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- McCubbrey, A.L.; Barthel, L.; Mohning, M.P.; Redente, E.F.; Mould, K.J.; Thomas, S.M.; Leach, S.M.; Danhorn, T.; Gibbings, S.L.; Jakubzick, C.V.; et al. Deletion of c-FLIP from CD11b(hi) Macrophages Prevents the Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2018, 58, 66–78. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at the single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.-T.; Hughes, J.; Sethi, T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef]
- Zhou, X.; Franklin, R.A.; Adler, M.; Jacox, J.B.; Bailis, W.; Shyer, J.A.; Flavell, R.A.; Mayo, A.; Alon, U.; Medzhitov, R. Circuit design features of a stable two-cell system. Cell 2018, 172, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGFβ. Sci. Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, M.; Herzog, E.L.; et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Leicester, K.L.; Olynyk, J.K.; Brunt, E.M.; Britton, R.S.; Bacon, B.R. CD14-positive hepatic monocytes/macrophages increase in hereditary hemochromatosis. Liver Int. 2004, 24, 446–451. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1 (LPS+) vs. Classically and M2 (LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A macrophage-pericyte axis directs tissue restoration via amphiregulin-induced transforming growth factor beta activation. Immunity 2019, 50, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.H.; Groves, P.; Olver, S.; Baz, A.; Doolan, D.L.; Kelso, A.; Kienzle, N. IFN-gamma inhibits IL-4-induced type 2 cytokine expression by CD8 T cells in vivo and modulates the anti-tumor response. J. Immunol. 2010, 185, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Benveniste, E.N. IL-4-activated STAT-6 inhibits IFN-gamma-induced CD40 gene expression in macrophages/microglia. J. Immunol. 2000, 165, 6235–6243. [Google Scholar] [CrossRef] [PubMed]
- Gurujeyalakshmi, G.; Giri, S.N. Molecular mechanisms of antifibrotic effect of interferon-gamma in bleomycin-mouse model of lung fibrosis: Downregulation of TGF-beta and procollagen I and III gene expression. Exp. Lung Res. 1995, 21, 791–808. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defense peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef]
- Steinstraesser, L.; Kraneburg, U.M.; Hirsch, T.; Kesting, M.; Steinau, H.U.; Jacobsen, F.; Al-Benna, S. Host defense peptides as effector molecules of the innate immune response: A sledgehammer for drug resistance? Int. J. Mol. Sci. 2009, 10, 3951–3970. [Google Scholar] [CrossRef]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12, eaax6337. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, Y.H.; Kim, K.H.; Lee, S.H.; Cha, J.Y.; Shin, E.K.; Jung, S.; Jang, A.S.; Park, S.W.; Uh, S.T.; et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Investig. 2007, 117, 3786–3799. [Google Scholar]
- Kim, T.H.; Lee, Y.H.; Kim, K.H.; Lee, S.H.; Cha, J.Y.; Shin, E.K.; Jung, S.; Jang, A.S.; Park, S.W.; Uh, S.T.; et al. Role of Lung Apolipoprotein A-I in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon cancer stem cells: Promise of targeted therapy. Gastro 2010, 138, 2151–2162. [Google Scholar] [CrossRef] [PubMed]
- Lierova, A.; Jelicova, M.; Nemcova, M.; Proksova, M.; Pejchal, J.; Zarybnicka, L.; Sinkorova, Z. Cytokines and radiation-induced pulmonary injuries. J. Radiat. Res. 2018, 59, 709–753. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Frederick, E.; Hausburg, M.; Goldberg, L.; Hoke, M.; Roshon, M.; Mains, C.; Bar-Or, D. The novel immunomodulatory biologic LMWF5A for pharmacological attenuation of the “cytokine storm” in COVID-19 patients: A hypothesis. Patient Saf. Surg. 2020, 14, 21. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 2011, 89, 557–563. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef]
- Kaku, Y.; Imaoka, H.; Morimatsu, Y.; Komohara, Y.; Ohnishi, K.; Oda, H.; Takenaka, S.; Matsuoka, M.; Kawayama, T.; Takeya, M.; et al. Overexpression of CD163, CD204, and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PLoS ONE 2014, 9, e87400. [Google Scholar] [CrossRef]
- Moore, B.B.; Paine, R., 3rd; Christensen, P.J.; Moore, T.A.; Sitterding, S.; Ngan, R.; Wilke, C.A.; Kuziel, W.A.; Toews, G.B. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J. Immunol. 2001, 167, 4368–4377. [Google Scholar] [CrossRef]
- Xu, J.; Flaczyk, A.; Neal, L.M.; Fa, Z.; Cheng, D.; Ivey, M.; Moore, B.B.; Curtis, J.L.; Osterholzer, J.J.; Olszewski, M.A. Exploitation of Scavenger Receptor, Macrophage Receptor with Collagenous Structure, by Cryptococcus neoformans Promotes Alternative Activation of Pulmonary Lymph Node CD11b (+) Conventional Dendritic Cells and Non-Protective Th2 Bias. Front. Immunol. 2017, 8, 1231. [Google Scholar] [CrossRef] [PubMed]
- Salazar, F.; Hall, L.; Negm, O.H.; Awuah, D.; Tighe, P.J.; Shakib, F.; Ghaemmaghami, A.M. The mannose receptor negatively modulates the Toll-like receptor 4-aryl hydrocarbon receptor-indoleamine 2,3-dioxygenase axis in dendritic cells affecting T helper cell polarization. J. Allergy Clin. Immunol. 2016, 137, 1841–1851.e1842. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.N.; Hyde, D.M.; A Hollinger, M. Effect of antibody to transforming growth factor beta on bleomycin-induced accumulation of lung collagen in mice. Thorax 1993, 48, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell. Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef]
- Paracuellos, P.; Briggs, D.C.; Carafoli, F.; Lončar, T.; Hohenester, E. Insights into Collagen Uptake by C-type Mannose Receptors from the Crystal Structure of Endo180 Domains 1-4. Structure 2015, 23, 2133–2142. [Google Scholar] [CrossRef]
- Chang, C.H.; Juan, Y.H.; Hu, H.C.; Kao, K.C.; Lee, C.S. Reversal of lung fibrosis: An unexpected finding in survivor of acute respiratory distress syndrome. QJM Int. J. Med. 2018, 111, 47–48. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saline | RP-832c 50 mg/kg | |||
|---|---|---|---|---|
| Parameter | n = 5 | n = 10 | Mean Difference | p Value |
| RBC/mm3 | 8.3 ± 0.6 | 8.5 ± 0.7 | 0.2 | 0.7295 |
| WBC/μL | 5.7 ± 1.8 | 7.7 ± 3.2 | 2.0 | 0.2163 |
| Total Protein (g/dL) | 5.6 ± 0.8 | 5.9 ± 0.5 | 0.3 | 0.5797 |
| AST (mg/dL) | 140.0 ± 75.5 | 91.2 ± 21.7 | −48.8 | 0.1237 |
| Phosphorus (mg/dL) | 8.8 ± 2.4 | 7.7 ± 0.5 | −1.1 | 0.2623 |
| Creatinine (mg/dL) | 0.2 ± 0 | 0.2 ± 0 | 0.0 | - |
| ALT (mg/dL) | 35.8 ± 18.9 | 27.2 ± 10 | −8.6 | 0.3808 |
| Albumin (g/dL) | 2.9 ± 0.3 | 3.1± 0.3 | 0.2 | 0.3982 |
| BUN (mg/dL) | 22.4 ± 3.4 | 24.0 ± 4.3 | 1.6 | 0.3087 |
| Globulin (g/dL) | 2.6 ± 0.5 | 2.7 ± 0.2 | 0.1 | 0.4413 |
| Alb/Globulin Ratio | 1.1 ± 0.2 | 1.2 ± 0.1 | 0.1 | 0.3295 |
| BUN/Creatinine Ratio | 111.9 ± 17 | 120.1 ± 21.6 | 8.2 | 0.3087 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghebremedhin, A.; Salam, A.B.; Adu-Addai, B.; Noonan, S.; Stratton, R.; Ahmed, M.S.U.; Khantwal, C.; Martin, G.R.; Lin, H.; Andrews, C.; et al. A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice. Cells 2023, 12, 1254. https://doi.org/10.3390/cells12091254
Ghebremedhin A, Salam AB, Adu-Addai B, Noonan S, Stratton R, Ahmed MSU, Khantwal C, Martin GR, Lin H, Andrews C, et al. A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice. Cells. 2023; 12(9):1254. https://doi.org/10.3390/cells12091254
Chicago/Turabian StyleGhebremedhin, Anghesom, Ahmad Bin Salam, Benjamin Adu-Addai, Steve Noonan, Richard Stratton, Md Shakir Uddin Ahmed, Chandra Khantwal, George R. Martin, Huixian Lin, Chris Andrews, and et al. 2023. "A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice" Cells 12, no. 9: 1254. https://doi.org/10.3390/cells12091254
APA StyleGhebremedhin, A., Salam, A. B., Adu-Addai, B., Noonan, S., Stratton, R., Ahmed, M. S. U., Khantwal, C., Martin, G. R., Lin, H., Andrews, C., Karanam, B., Rudloff, U., Lopez, H., Jaynes, J., & Yates, C. (2023). A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice. Cells, 12(9), 1254. https://doi.org/10.3390/cells12091254