
Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease


Abstract
1. Introduction
2. The Role of Epigenetic Mechanisms in Regulating PPARγ Regulatory Function in NAFLD
2.1. DNA Methylation
2.2. Histone Modifications
2.2.1. Histone Methylation/Demethylation
2.2.2. Histone Acetylation/Deacetylation
2.3. Noncoding RNAs
2.3.1. miRNAs-PPARγ Axis
2.3.2. lncRNAs-PPARγ Axis
2.3.3. circRNAs-PPARγ Axis
3. Limitations and Future Perspectives
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Galoosian, A.; Kaswala, D.; Li, A.A.; Gadiparthi, C.; Cholankeril, G.; Kim, D.; Ahmed, A. Nonalcoholic Fatty Liver Disease: Epidemiology, Liver Transplantation Trends and Outcomes, and Risk of Recurrent Disease in the Graft. J. Clin. Transl. Hepatol. 2018, 6, 420–424. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Mingolla, L.; Rigolon, R.; Pichiri, I.; Cavalieri, V.; Zoppini, G.; Lippi, G.; Bonora, E.; Targher, G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular disease in adult patients with type 1 diabetes. Int. J. Cardiol. 2016, 225, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Ratziu, V.; Rinella, M.; Beuers, U.; Loomba, R.; Anstee, Q.M.; Harrison, S.; Francque, S.; Sanyal, A.; Newsome, P.N.; Younossi, Z. The times they are a-changin’ (for NAFLD as well). J. Hepatol. 2020, 73, 1307–1309. [Google Scholar] [CrossRef]
- Kim, D.; Ahmed, A. Reply to: “NAFLD vs. MAFLD-It is not the name but the disease that decides the outcome in fatty liver”. J. Hepatol. 2022, 76, 477–478. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Ratziu, V. Starting the battle to control non-alcoholic steatohepatitis. Lancet 2015, 385, 922–924. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 10, 1224–1234. [Google Scholar] [CrossRef]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef]
- Vidal-Puig, A.; Jimenez-Liñan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef]
- Chen, Y.; Jimenez, A.R.; Medh, J.D. Identification and regulation of novel PPAR-gamma splice variants in human THP-1 macrophages. Biochim. Biophys. Acta 2006, 1759, 32–43. [Google Scholar] [CrossRef]
- Wu, C.W.; Chu, E.S.; Lam, C.N.; Cheng, A.S.; Lee, C.W.; Wong, V.W.; Sung, J.J.; Yu, J. PPARgamma is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010, 17, 790–798. [Google Scholar] [CrossRef]
- Pettinelli, P.; Videla, L.A. Up-Regulation of PPAR-γ mRNA Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to SREBP-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Lima-Cabello, E.; García-Mediavilla, M.V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lozano-Rodríguez, T.; Fernández-Bermejo, M.; Olcoz, J.L.; González-Gallego, J.; García-Monzón, C.; Sánchez-Campos, S. Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clin. Sci. 2011, 120, 239–250. [Google Scholar] [CrossRef]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef]
- Lutchman, G.; Promrat, K.; Kleiner, D.E.; Heller, T.; Ghany, M.G.; Yanovski, J.A.; Liang, T.J.; Hoofnagle, J.H. Changes in serum adipokine levels during pioglitazone treatment for nonalcoholic steatohepatitis: Relationship to histological improvement. Clin. Gastroenterol. Hepatol. 2006, 4, 1048–1052. [Google Scholar] [CrossRef]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Foley, D.L.; Craig, J.M.; Morley, R.; Olsson, C.A.; Dwyer, T.; Smith, K.; Saffery, R. Prospects for epigenetic epidemiology. Am. J. Epidemiol. 2009, 169, 389–400. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef]
- Zaiou, M.; Amrani, R.; Rihn, B.; Hajri, T. Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Biomedicines 2021, 9, 1256. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.-S.; Chan, H.L.-Y.; Cheng, A.S.-L. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 2013, 23, 471–482. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- De Mello, V.D.; Matte, A.; Perfilyev, A.; Männistö, V.; Rönn, T.; Nilsson, E.; Käkelä, P.; Ling, C.; Pihlajamäki, J. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017, 12, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator–activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Zeybel, M.; Hardy, T.; Wong, Y.K.; Mathers, J.C.; Fox, C.R.; Gackowska, A.; Oakley, F.; Burt, A.D.; Wilson, C.L.; Anstee, Q.M.; et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat. Med. 2012, 18, 1369–1377. [Google Scholar] [CrossRef]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenet. 2015, 7, 25. [Google Scholar] [CrossRef]
- Yigit, B.; Boyle, M.; Ozler, O.; Erden, N.; Tutucu, F.; Hardy, T.; Bergmann, C.; Distler, J.H.W.; Adali, G.; Dayangac, M.; et al. Plasma cell-free DNA methylation: A liquid biomarker of hepatic fibrosis. Gut 2018, 67, 1907–1908. [Google Scholar] [CrossRef]
- Hajri, T.; Zaiou, M.; Fungwe, T.V.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Laird, A.; Thomson, J.P.; Harrison, D.J.; Meehan, R.R. 5-hydroxymethylcytosine profiling as an indicator of cellular state. Epigenomics 2013, 5, 655–669. [Google Scholar] [CrossRef]
- Pirola, C.J.; Scian, R.; Gianotti, T.F.; Dopazo, H.; Rohr, C.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic Modifications in the Biology of Nonalcoholic Fatty Liver Disease: The Role of DNA Hydroxymethylation and TET Proteins. Medicine 2015, 94, e1480. [Google Scholar] [CrossRef]
- Tong, J.; Han, C.J.; Zhang, J.Z.; He, W.Z.; Zhao, G.J.; Cheng, X.; Zhang, L.; Deng, K.Q.; Liu, Y.; Fan, H.F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 2019, 69, 2471–2488. [Google Scholar] [CrossRef]
- Lee, J.; Song, J.H.; Park, J.H.; Chung, M.Y.; Lee, S.H.; Jeon, S.B.; Park, S.H.; Hwang, J.T.; Choi, H.K. Dnmt1/Tet2-mediated changes in Cmip methylation regulate the development of nonalcoholic fatty liver disease by controlling the Gbp2-Pparγ-CD36 axis. Exp. Mol. Med. 2023, 55, 143–157. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.W. Epigenetics in non-alcoholic fatty liver disease. Mol. Asp. Med. 2017, 54, 78–88. [Google Scholar] [CrossRef]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARγ2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef]
- Jun, H.J.; Kim, J.; Hoang, M.H.; Lee, S.J. Hepatic lipid accumulation alters global histone h3 lysine 9 and 4 trimethylation in the peroxisome proliferator-activated receptor alpha network. PLoS ONE 2012, 7, e44345. [Google Scholar] [CrossRef]
- Fan, Z.; Li, L.; Li, M.; Zhang, X.; Hao, C.; Yu, L.; Zeng, S.; Xu, H.; Fang, M.; Shen, A.; et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology 2017, 65, 1904–1919. [Google Scholar] [CrossRef]
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Colak, Y.; Yesil, A.; Mutlu, H.H.; Caklili, O.T.; Ulasoglu, C.; Senates, E.; Takir, M.; Kostek, O.; Yilmaz, Y.; Yilmaz Enc, F.; et al. A potential treatment of non-alcoholic fatty liver disease with SIRT1 activators. J. Gastrointestin. Liver Dis. 2014, 23, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J.; Liu, X.S.; et al. PPARγ and C/EBP Factors Orchestrate Adipocyte Biology via Adjacent Binding on a Genome-Wide Scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef]
- Kraakman, M.J.; Liu, Q.; Postigo-Fernandez, J.; Ji, R.; Kon, N.; Larrea, D.; Namwanje, M.; Fan, L.; Chan, M.; Area-Gomez, E.; et al. PPARγ deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects. J. Clin. Investig. 2018, 128, 2600–2612. [Google Scholar] [CrossRef]
- Zahr, T.; Liu, L.; Chan, M.; Zhou, Q.; Cai, B.; He, Y.; Aaron, N.; Accili, D.; Sun, L.; Qiang, L. PPARγ (Peroxisome Proliferator-Activated Receptor γ) Deacetylation Suppresses Aging-Associated Atherosclerosis and Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 30–44. [Google Scholar] [CrossRef]
- Liu, L.; Fan, L.; Chan, M.; Kraakman, M.J.; Yang, J.; Fan, Y.; Aaron, N.; Wan, Q.; Carrillo-Sepulveda, M.A.; Tall, A.R.; et al. PPARγ Deacetylation Confers the Antiatherogenic Effect and Improves Endothelial Function in Diabetes Treatment. Diabetes 2020, 69, 1793–1803. [Google Scholar] [CrossRef]
- Rodrigues, B.; McNeill, J.H. Cardiac dysfunction in isolated perfused hearts from spontaneously diabetic BB rats. Can. J. Physiol. Pharmacol. 1990, 68, 514–518. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell. Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Knutson, S.K.; Chyla, B.J.; Amann, J.M.; Bhaskara, S.; Huppert, S.S.; Hiebert, S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008, 27, 1017–1028. [Google Scholar] [CrossRef]
- Ferrari, A.; Longo, R.; Fiorino, E.; Silva, R.; Mitro, N.; Cermenati, G.; Gilardi, F.; Desvergne, B.; Andolfo, A.; Magagnotti, C.; et al. HDAC3 is a molecular brake of the metabolic switch supporting white adipose tissue browning. Nat. Commun. 2017, 8, 93. [Google Scholar] [CrossRef]
- Whitt, J.; Woo, V.; Lee, P.; Moncivaiz, J.; Haberman, Y.; Denson, L.; Tso, P.; Alenghat, T. Disruption of Epithelial HDAC3 in Intestine Prevents Diet-Induced Obesity in Mice. Gastroenterology 2018, 155, 501–513. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Ren, W.; Ren, J.; Lan, X.; Wang, F.; Li, H.; Zhang, F.; Han, Y.; Song, T.; et al. High expression of liver histone deacetylase 3 contributes to high-fat-diet-induced metabolic syndrome by suppressing the PPAR-γ and LXR-α-pathways in E3 rats. Mol. Cell. Endocrinol. 2011, 344, 69–80. [Google Scholar] [CrossRef]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef]
- Cao, Y.; Xue, Y.; Xue, L.; Jiang, X.; Wang, X.; Zhang, Z.; Yang, J.; Lu, J.; Zhang, C.; Wang, W.; et al. Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J. Hepatol. 2013, 59, 1299–1306. [Google Scholar] [CrossRef]
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858. [Google Scholar] [CrossRef]
- Vilà, L.; Elias, I.; Roca, C.; Ribera, A.; Ferré, T.; Casellas, A.; Lage, R.; Franckhauser, S.; Bosch, F. AAV8-mediated Sirt1 gene transfer to the liver prevents high carbohydrate diet-induced nonalcoholic fatty liver disease. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2014, 1, 14039. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Deng, X.Q.; Chen, L.L.; Li, N.X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 2007, 27, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.; Fiore, D.; Basciani, S.; Persichetti, A.; Contini, S.; Lubrano, C.; Salvatori, L.; Lenzi, A.; Gnessi, L. Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 2015, 49, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M., Jr.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Li, P.; Ruan, X.; Yang, L.; Kiesewetter, K.; Zhao, Y.; Luo, H.; Chen, Y.; Gucek, M.; Zhu, J.; Cao, H. A Liver-Enriched Long Non-Coding RNA, lncLSTR, Regulates Systemic Lipid Metabolism in Mice. Cell Metab. 2015, 21, 455–467. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Barbier, O.; Smalling, R.; Tsuchiya, H.; Lee, S.; Delker, D.; Zou, A.; Hagedorn, C.H.; Wang, L. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci. Rep. 2016, 6, 20559. [Google Scholar] [CrossRef]
- Zaiou, M. Noncoding RNAs as additional mediators of epigenetic regulation in nonalcoholic fatty liver disease. World J. Gastroenterol. 2022, 28, 5111–5128. [Google Scholar] [CrossRef]
- Liu, C.H.; Ampuero, J.; Gil-Gómez, A.; Montero-Vallejo, R.; Rojas, Á.; Muñoz-Hernández, R.; Gallego-Durán, R.; Romero-Gómez, M. miRNAs in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2018, 69, 1335–1348. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Q.; Zhou, W.; Liu, T.; Yang, L.; Zheng, P.; Zhang, L.; Ji, G. Integrated analysis of hepatic mRNA and miRNA profiles identified molecular networks and potential biomarkers of NAFLD. Sci. Rep. 2018, 8, 7628. [Google Scholar] [CrossRef]
- Lv, X.; Yan, J.; Jiang, J.; Zhou, X.; Lu, Y.; Jiang, H. MicroRNA-27a-3p suppression of peroxisome proliferator-activated receptor-γ contributes to cognitive impairments resulting from sevoflurane treatment. J. Neurochem. 2017, 143, 306–319. [Google Scholar] [CrossRef]
- Baffy, G. MicroRNAs in Nonalcoholic Fatty Liver Disease. J. Clin. Med. 2015, 4, 1977–1988. [Google Scholar] [CrossRef]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef]
- Sun, C.; Huang, F.; Liu, X.; Xiao, X.; Yang, M.; Hu, G.; Liu, H.; Liao, L. miR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCR. Int. J. Mol. Med. 2015, 35, 847–853. [Google Scholar] [CrossRef]
- Loyer, X.; Paradis, V.; Hénique, C.; Vion, A.-C.; Colnot, N.; Guerin, C.L.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARα expression. Gut 2015, 65, 1882–1894. [Google Scholar] [CrossRef]
- Benhamouche-Trouillet, S.; Postic, C. Emerging role of miR-21 in non-alcoholic fatty liver disease. Gut 2016, 65, 1781–1783. [Google Scholar] [CrossRef]
- Pillai, S.S.; Lakhani, H.V.; Zehra, M.; Wang, J.; Dilip, A.; Puri, N.; O’Hanlon, K.; Sodhi, K. Predicting Nonalcoholic Fatty Liver Disease through a Panel of Plasma Biomarkers and MicroRNAs in Female West Virginia Population. Int. J. Mol. Sci. 2020, 21, 6698. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Y.; Wu, X.; Jiang, L.; Yang, S.; Ding, Z.; Fang, Z.; Hua, H.; Kirby, M.S.; Shou, J. A circulating microRNA signature as noninvasive diagnostic and prognostic biomarkers for nonalcoholic steatohepatitis. BMC Genom. 2018, 19, 188. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Afonso, M.B.; Simão, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.; et al. miR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 2017, 8, e2825. [Google Scholar] [CrossRef]
- Almohawes, Z.N.; El-Kott, A.; Morsy, K.; Shati, A.A.; El-Kenawy, A.E.; Khalifa, H.S.; Elsaid, F.G.; Abd-Lateif, A.M.; Abu-Zaiton, A.; Ebealy, E.R.; et al. Salidroside inhibits insulin resistance and hepatic steatosis by downregulating miR-21 and subsequent activation of AMPK and upregulation of PPARα in the liver and muscles of high fat diet-fed rats. Arch. Physiol. Biochem. 2022, 1–18. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, F.; Zhang, Y.; Zhang, X.; Chen, J.; Jiang, Y. PPARγ attenuates hepatic inflammation and oxidative stress of non-alcoholic steatohepatitis via modulating the miR-21-5p/SFRP5 pathway. Mol. Med. Rep. 2021, 24, 823. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S.; et al. MicroRNA-27a Regulates Lipid Metabolism and Inhibits Hepatitis C Virus Replication in Human Hepatoma Cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, A.Y.; Lee, H.W.; Son, Y.H.; Lee, G.Y.; Lee, J.W.; Lee, Y.K.; Kim, J.B. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARgamma expression. Biochem. Biophys. Res. Commun. 2010, 392, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, W.; Zhou, M.; Tang, Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci. Rep. 2017, 7, 14493. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ge, G.; Pan, T.; Wen, D.; Gan, J. A pilot study of serum microRNAs panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e105192. [Google Scholar] [CrossRef]
- Gerhard, G.S. Micro RNAs in the development of non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 226–234. [Google Scholar] [CrossRef]
- Zhang, J.; Powell, C.; Kay, M.; Sonkar, R.; Meruvu, S.; Choudhury, M. Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. Int. J. Mol. Sci. 2021, 22, 1822. [Google Scholar] [CrossRef]
- Zhu, D.; Lyu, L.; Shen, P.; Wang, J.; Chen, J.; Sun, X.; Chen, L.; Zhang, L.; Zhou, Q.; Duan, Y. rSjP40 protein promotes PPARγ expression in LX-2 cells through microRNA-27b. FASEB J. 2018, 32, 4798–4803. [Google Scholar] [CrossRef]
- Jennewein, C.; von Knethen, A.; Schmid, T.; Brüne, B. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA destabilization. J. Biol. Chem. 2010, 285, 11846–11853. [Google Scholar] [CrossRef]
- Hsu, C.C.; Lai, C.Y.; Lin, C.Y.; Yeh, K.Y.; Her, G.M. MicroRNA-27b Depletion Enhances Endotrophic and Intravascular Lipid Accumulation and Induces Adipocyte Hyperplasia in Zebrafish. Int. J. Mol. Sci. 2017, 19, 93. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Fei, B.Y.; Shao, D.; Pan, Y.; Mo, Z.H.; Sun, B.Z.; Zhang, D.; Zheng, X.; Zhang, M.; et al. MiR-27a promotes hepatocellular carcinoma cell proliferation through suppression of its target gene peroxisome proliferator-activated receptor γ Chin. Med. J. 2015, 128, 941–947. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, F.; Zhao, Y.; Zhang, J.; Dawulieti, J.; Pan, Y.; Cui, L.; Sun, M.; Shao, D.; Li, M.; et al. Self-assembled dual fluorescence nanoparticles for CD44-targeted delivery of anti-miR-27a in liver cancer theranostics. Theranostics 2018, 8, 3808–3823. [Google Scholar] [CrossRef]
- Zarfeshani, A.; Ngo, S.; Sheppard, A.M. MicroRNA Expression Relating to Dietary-Induced Liver Steatosis and NASH. J. Clin. Med. 2015, 4, 1938–1950. [Google Scholar] [CrossRef]
- Castro, R.E.; Ferreira, D.M.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef]
- Meng, F.; Glaser, S.S.; Francis, H.; Yang, F.; Han, Y.; Stokes, A.; Staloch, D.; McCarra, J.; Liu, J.; Venter, J.; et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am. J. Pathol. 2012, 181, 804–817. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating MicroRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Salvoza, N.C.; Klinzing, D.C.; Gopez-Cervantes, J.; Baclig, M.O. Association of Circulating Serum miR-34a and miR-122 with Dyslipidemia among Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0153497. [Google Scholar] [CrossRef]
- Braza-Boïls, A.; Marí-Alexandre, J.; Molina, P.; Arnau, M.A.; Barceló-Molina, M.; Domingo, D.; Girbes, J.; Giner, J.; Martínez-Dolz, L.; Zorio, E. Deregulated hepatic microRNAs underlie the association between non-alcoholic fatty liver disease and coronary artery disease. Liver Int. 2016, 36, 1221–1229. [Google Scholar] [CrossRef]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef]
- Guo, X.-Y.; Sun, F.; Chen, J.-N.; Wang, Y.-Q.; Pan, Q.; Fan, J.-G. circRNA_0046366 inhibits hepatocellular steatosis by normalization of PPAR signaling. World J. Gastroenterol. 2018, 24, 323–337. [Google Scholar] [CrossRef]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor γ. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Estep, M.; Armistead, D.; Hossain, N.; Elarainy, H.; Goodman, Z.; Baranova, A.; Chandhoke, V.; Younossi, Z.M. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 32, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Hanin, G.; Yayon, N.; Tzur, Y.; Haviv, R.; Bennett, E.R.; Udi, S.; Krishnamoorthy, Y.R.; Kotsiliti, E.; Zangen, R.; Efron, B.; et al. miRNA-132 induces hepatic steatosis and hyperlipidaemia by synergistic multitarget suppression. Gut 2018, 67, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714. [Google Scholar] [CrossRef]
- Csak, T.; Bala, S.; Lippai, D.; Kodys, K.; Catalano, D.; Iracheta-Vellve, A.; Szabo, G. MicroRNA-155 Deficiency Attenuates Liver Steatosis and Fibrosis without Reducing Inflammation in a Mouse Model of Steatohepatitis. PLoS ONE 2015, 10, e0129251. [Google Scholar] [CrossRef]
- Miller, A.M.; Gilchrist, D.S.; Nijjar, J.; Araldi, E.; Ramírez, C.M.; Lavery, C.A.; Fernandez-Hernando, C.; McInnes, I.B.; Kurowska-Stolarska, M. MiR-155 Has a Protective Role in the Development of Non-Alcoholic Hepatosteatosis in Mice. PLoS ONE 2013, 8, e72324. [Google Scholar] [CrossRef]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, Z.J.; Jia, Y.J.; Yang, Y.L.; Xue, Y.M. Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of diabetic kidney disease. J. Transl. Med. 2018, 16, 146. [Google Scholar] [CrossRef]
- Skårn, M.; Namløs, H.M.; Noordhuis, P.; Wang, M.-Y.; Meza-Zepeda, L.A.; Myklebost, O. Adipocyte Differentiation of Human Bone Marrow-Derived Stromal Cells Is Modulated by MicroRNA-155, MicroRNA-221, and MicroRNA-222. Stem Cells Dev. 2012, 21, 873–883. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef]
- Tryggestad, J.B.; Teague, A.M.; Sparling, D.P.; Jiang, S.; Chernausek, S.D. Macrophage-Derived MicroRNA-155 Increases in Obesity and Influences Adipocyte Metabolism by Targeting Peroxisome Proliferator-Activated Receptor Gamma. Obesity 2019, 27, 1856–1864. [Google Scholar] [CrossRef]
- Wang, D.R.; Wang, B.; Yang, M.; Liu, Z.L.; Sun, J.; Wang, Y.; Sun, H.; Xie, L.J. Suppression of miR-30a-3p Attenuates Hepatic Steatosis in Non-alcoholic Fatty Liver Disease. Biochem. Genet. 2020, 58, 691–704. [Google Scholar] [CrossRef]
- Mittal, S.; Inamdar, S.; Acharya, J.; Pekhale, K.; Kalamkar, S.; Boppana, R.; Ghaskadbi, S. miR-3666 inhibits development of hepatic steatosis by negatively regulating PPARγ. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158777. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 1–17. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Sookoian, S.; Rohr, C.; Salatino, A.; Dopazo, H.; Fernandez Gianotti, T.; Castaño, G.O.; Pirola, C.J. Genetic variation in long noncoding RNAs and the risk of nonalcoholic fatty liver disease. Oncotarget 2017, 8, 22917–22926. [Google Scholar] [CrossRef]
- Lan, X.; Wu, L.; Wu, N.; Chen, Q.; Li, Y.; Du, X.; Wei, C.; Feng, L.; Li, Y.; Osoro, E.K.; et al. Long Noncoding RNA lnc-HC Regulates PPARγ-Mediated Hepatic Lipid Metabolism through miR-130b-3p. Mol. Ther. Nucleic Acids 2019, 18, 954–965. [Google Scholar] [CrossRef]
- Xu, B.M.; Xiao, L.; Kang, C.M.; Ding, L.; Guo, F.X.; Li, P.; Lu, Z.F.; Wu, Q.; Xu, Y.J.; Bai, H.L.; et al. LncRNA AC096664.3/PPAR-γ/ABCG1-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis. J. Cell. Biochem. 2019, 120, 13775–13782. [Google Scholar] [CrossRef]
- Liu, J.; Tang, T.; Wang, G.D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, BSR20181722. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Y.; Shu, L.; Zhu, Y.; Peng, Q.; Ran, L.; Wu, J.; Luo, Y.; Zuo, G.; Luo, J.; et al. Long non-coding RNA (lncRNA) H19 induces hepatic steatosis through activating MLXIPL and mTORC1 networks in hepatocytes. J. Cell. Mol. Med. 2020, 24, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yu, D.; Nian, X.; Liu, J.; Koenig, R.J.; Xu, B.; Sheng, L. LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL). Sci. Rep. 2016, 6, 35531. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.X.; Li, B.Z.; Mitchell, L.A.; Wu, Y.; Qi, X.; Jin, Z.; Jia, B.; Wang, X.; Zeng, B.X.; Liu, H.M.; et al. “Perfect” designer chromosome V and behavior of a ring derivative. Science 2017, 355, eaaf4704. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Gerin, I.; Miao, H.; Vu-Phan, D.; Johnson, C.N.; Xu, R.; Chen, X.W.; Cawthorn, W.P.; MacDougald, O.A.; Koenig, R.J. Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS ONE 2010, 5, e14199. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Ding, J.; Chen, A.; Song, Y.; Xu, C.; Tian, F.; Zhao, J. Aerobic exercise improves adipogenesis in diet-induced obese mice via the lncSRA/p38/JNK/PPARγ pathway. Nutr. Res. 2022, 105, 20–32. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; NASH CRN. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2016, 63, 1155–1169. [Google Scholar] [CrossRef]
- Jiang, Y.; Peng, J.; Song, J.; He, J.; Jiang, M.; Wang, J.; Ma, L.; Wang, Y.; Lin, M.; Wu, H.; et al. Loss of Hilnc prevents diet-induced hepatic steatosis through binding of IGF2BP2. Nat. Metab. 2021, 3, 1569–1584. [Google Scholar] [CrossRef]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Zaiou, M. The Emerging Role and Promise of Circular RNAs in Obesity and Related Metabolic Disorders. Cells 2020, 9, 1473. [Google Scholar] [CrossRef]
- Jin, X.; Feng, C.Y.; Xiang, Z.; Chen, Y.P.; Li, Y.M. CircRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of nonalcoholic steatohepatitis. Oncotarget 2016, 7, 66455–66467. [Google Scholar] [CrossRef]
- Guo, X.Y.; Chen, J.N.; Sun, F.; Wang, Y.Q.; Pan, Q.; Fan, J.G. circRNA_0046367 Prevents Hepatoxicity of Lipid Peroxidation: An Inhibitory Role against Hepatic Steatosis. Oxid. Med. Cell. Longev. 2017, 2017, 3960197. [Google Scholar] [CrossRef]
- Cai, H.; Jiang, Z.; Yang, X.; Lin, J.; Cai, Q.; Li, X. Circular RNA HIPK3 contributes to hyperglycemia and insulin homeostasis by sponging miR-192-5p and upregulating transcription factor forkhead box O1. Endocr. J. 2020, 67, 397–408. [Google Scholar] [CrossRef]
- Lu, J.; Pang, L.; Zhang, B.; Gong, Z.; Song, C. Silencing circANKRD36 inhibits streptozotocin-induced insulin resistance and inflammation in diabetic rats by targeting miR-145 via XBP1. Inflamm. Res. 2021, 70, 695–704. [Google Scholar] [CrossRef]
- Guo, X.Y.; He, C.X.; Wang, Y.Q.; Sun, C.; Li, G.M.; Su, Q.; Pan, Q.; Fan, J.G. Circular RNA Profiling and Bioinformatic Modeling Identify Its Regulatory Role in Hepatic Steatosis. BioMed Res. Int. 2017, 2017, 5936171. [Google Scholar] [CrossRef]
- Yuan, X.; Li, Y.; Wen, S.; Xu, C.; Wang, C.; He, Y.; Zhou, L. CircLDLR acts as a sponge for miR-667-5p to regulate SIRT1 expression in non-alcoholic fatty liver disease. Lipids Health Dis. 2022, 21, 127. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 Promotes Fat Mobilization in White Adipocytes by Repressing PPAR-Gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Lin, X.; Du, Y.; Lu, W.; Gui, W.; Sun, S.; Zhu, Y.; Wang, G.; Eserberg, D.T.; Zheng, F.; Zhou, J.; et al. CircRNF111 Protects Against Insulin Resistance and Lipid Deposition via Regulating miR-143-3p/IGF2R Axis in Metabolic Syndrome. Front. Cell Dev. Biol. 2021, 9, 663148. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C.; Song, G.G. Meta-analysis of associations between the peroxisome proliferator-activated receptor-γ Pro12Ala polymorphism and susceptibility to nonalcoholic fatty liver disease, rheumatoid arthritis, and psoriatic arthritis. Genet. Test. Mol. Biomark. 2014, 18, 341–348. [Google Scholar] [CrossRef]
- Xie, R.; Tang, S.; Yang, Y. Associations of peroxisome proliferator-activated receptor-γ Pro12Ala polymorphism with non-alcoholic fatty liver disease: A meta-analysis. J. Diabetes Complicat. 2022, 36, 108261. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, Y.-Y.; Nie, Y.-Q.; Sha, W.-H.; Du, Y.-L.; Lai, X.-B.; Zhou, Y.-J. Effect of peroxisome proliferator-activated receptors-gamma and co-activator-1alpha genetic polymorphisms on plasma adiponectin levels and susceptibility of non-alcoholic fatty liver disease in Chinese people. Liver Int. 2008, 28, 385–392. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Li, Y.Y.; Nie, Y.Q.; Yang, H.; Zhan, Q.; Huang, J.; Shi, S.L.; Lai, X.B.; Huang, H.L. Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. J. Gastroenterol. Hepatol. 2010, 25, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Flessa, C.M.; Nasiri-Ansari, N.; Kyrou, I.; Leca, B.M.; Lianou, M.; Chatzigeorgiou, A.; Kaltsas, G.; Kassi, E.; Randeva, H.S. Genetic and Diet-Induced Animal Models for Non-Alcoholic Fatty Liver Disease (NAFLD) Research. Int. J. Mol. Sci. 2022, 23, 15791. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Horii, T.; Morita, S.; Hatada, I. Generation of Epigenetic Disease Model Mice by Targeted Demethylation of the Epigenome. Methods Mol. Biol. 2023, 2577, 255–268. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; LIDO Study Group. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Skat-Rørdam, J.; Højland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Yang, X.; Frank, J.; Gonzalez, F.J.; Huang, M.; Bi, H. Nuclear receptors and non-alcoholic fatty liver disease: An update. Liver Res. 2020, 4, 88–93. [Google Scholar] [CrossRef]
- Ahmadian, M.; Myoung Suh, J.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Romualdo, G.R.; Valente, L.C.; Sprocatti, A.C.; Bacil, G.P.; de Souza, I.P.; Rodrigues, J.; Rodrigues, M.A.M.; Vinken, M.; Cogliati, B.; Barbisan, L.F. Western diet-induced mouse model of non-alcoholic fatty liver disease associated with metabolic outcomes: Features of gut microbiome-liver-adipose tissue axis. Nutrition 2022, 103–104, 111836. [Google Scholar] [CrossRef]



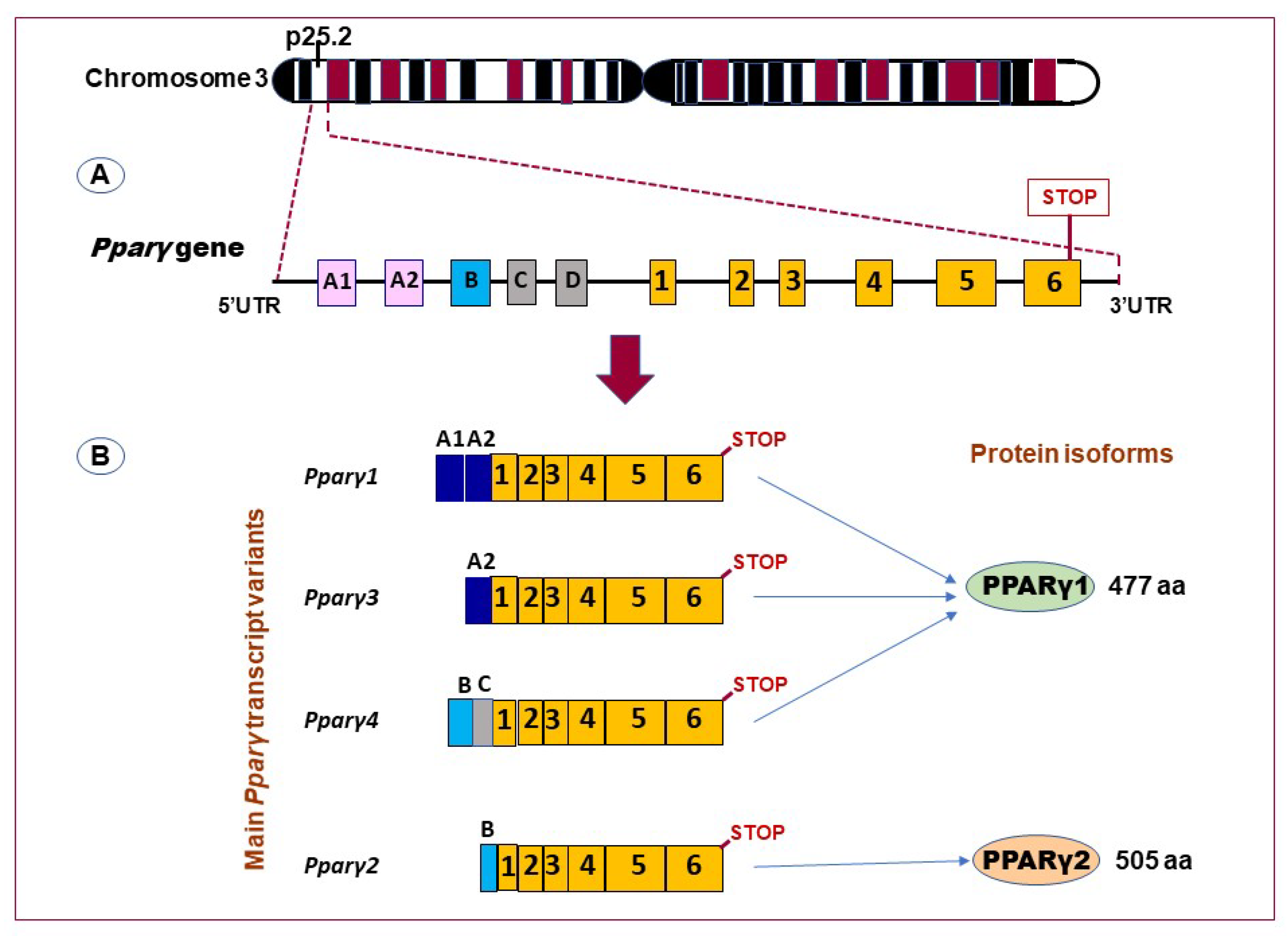{kind=link}
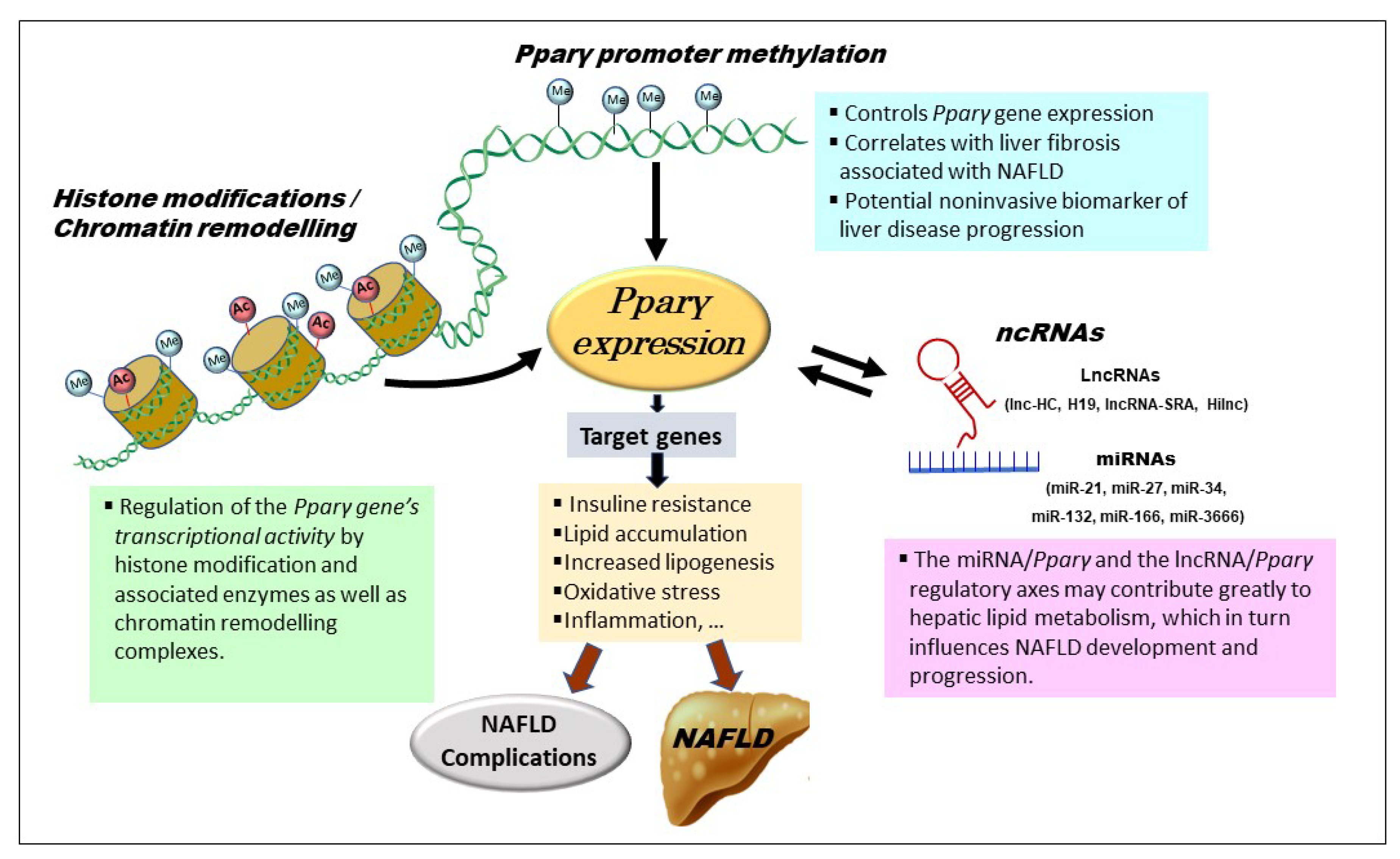{kind=link}
| Epigenetic Change | Biological Effect | Reference |
|---|---|---|
| Pparα/Pparγ/ methylation | DNA methylation of Pparα and Pparγ can distinguish between mild and severe NAFLD-associated fibrosis | [45] |
| Pparγ promoter methylation | HFD and palmitic acid alter global and Pparγ promoter DNA methylation, resulting in Pparγ expression and enhanced lipid retention in the liver, which leads to the development of NAFLD | [47] |
| Pparγ promoter methylation | Pparγ promoter hypermethylation levels in plasma-free DNA could be used as a non-invasive method to differentiate between NAFLD patients with mild and severe fibrosis | [48] |
| Pparγ promoter methylation | Methylation levels of Pparγ correlate with liver fibrosis in rat model as well as in NAFLD patients | [44,48] |
| Epigenetic Effector | Biological Effect | Reference |
|---|---|---|
| MLL4 | Murine steatosis caused by excessive feeding is regulated by the histone H3 lysine 4 methyltransferase MLL4/KMT2D via PPARγ2 | [56] |
| Suv39h2 | SUV39H2 expression in hepatocytes, mice, and human livers is induced by pro-NASH stimuli, and thus contributes to NASH pathogenesis by suppressing Pparγ and Sirt1 expression. | [58] |
| JMJD1A | JMJD1A promotes PPARγ expression by regulating the demethylation of Pparγ gene and thus inhibit HSCs activation and fibrosis | [60] |
| JMJD2B | JMJD2B promotes the development of hepatic steatosis by upregulating PPARγ2 and steatosis target genes. | [61] |
| miRNA | Target or Pathway | Pathophysiological Processes | References |
|---|---|---|---|
| miR-21 | PPARγ/SFR5 | PPARγ prevents inflammation and oxidative stress in mouse models and human tissue samples from NASH patients by modulating the miR21-5p/SFRP5 pathway | [101] |
| miR-27 | PPARγ | Disrupting endogenous miR-27b activity in Zebrafish causes lipid accumulation and increased PPARγ expression in the liver, resulting in the early onset of NAFLD and NASH | [110] |
| miR-34 | PPARγ | By targeting PPARγ, miR-34a/c activation may be associated with liver fibrosis | [122] |
| miR-132 | PPARγ/MeCP/EZH2 | miR132/MeCP2/EZH2 axis is involved in the regulation of liver fibrosis | [125] |
| miR-155 | PPARγ | miR-155 contained in obese ATM-exosomes, contributes to insulin resistance and glucose intolerance via a mechanism that is most likely related to direct suppression of its target gene Pparγ | [131] |
| miR-3666 | PPARγ | miR-3666 inhibits the development of hepatic steatosis by negatively regulating PPARγ | [134] |
| LncRNA | Targeted or Pathway | Pathophysiological Processes | References |
|---|---|---|---|
| Lnc-HC | miR-130-3p/PPARγ | LncRNA regulates hepatic lipid droplets accumulation via miR-130-3p/PPARγ pathway, which may help to prevent NAFLD | [138] |
| H19 | miR-130a/PPARγ | H19 promotes hepatic lipogenesis and the progression of NAFLD via miR-130a/PPARγ axis | [140] |
| LncRNA-SRA | PPARγ | LncRNA-SRA promotes hepatic steatosis by repressing the expression of adipose triglyceride lipase | [141] |
| Hilnc | IGF22BP2/PPARγ | The loss of Hilnc prevents diet-induced hepatic steatosis by inhibiting PPARγ | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaiou, M. Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Cells 2023, 12, 1205. https://doi.org/10.3390/cells12081205
Zaiou M. Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Cells. 2023; 12(8):1205. https://doi.org/10.3390/cells12081205
Chicago/Turabian StyleZaiou, Mohamed. 2023. "Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease" Cells 12, no. 8: 1205. https://doi.org/10.3390/cells12081205
APA StyleZaiou, M. (2023). Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Cells, 12(8), 1205. https://doi.org/10.3390/cells12081205