
Overexpression of GREM1 Improves the Survival Capacity of Aged Cardiac Mesenchymal Progenitor Cells via Upregulation of the ERK/NRF2-Associated Antioxidant Signal Pathway

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Human Heart Tissue Collection and Δψmlow-hMPCs’ Isolation
2.3. Cell Culture
2.4. Lentiviral Production and Infection of hMPCs
2.5. ERK Inhibition
2.6. Cell Viability Assay
2.7. Senescence-Associated Beta-Galactosidase Assay
2.8. FACS Analysis for Apoptotic Assay
2.9. Cell Proliferation Assay with BrdU Incorporation
2.10. Measurement of ROS and Mitochondrial Membrane Potential
2.11. RNA Isolation and Quantitative Analysis
2.12. Immunoblotting
2.13. Statistical Analysis
3. Results
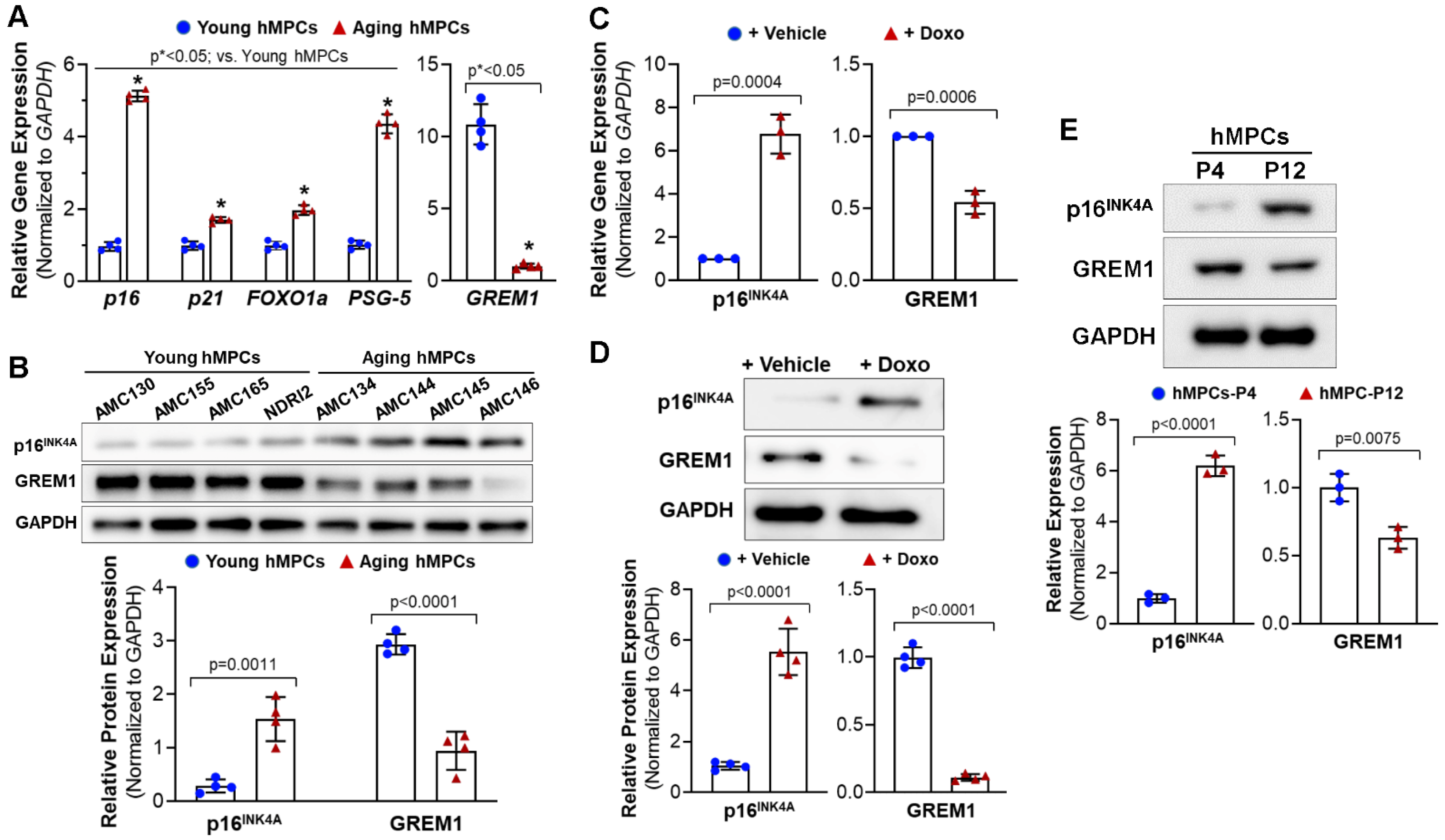
3.1. GREM1 Expression Is Associated with Cell Aging and Cell Senescence
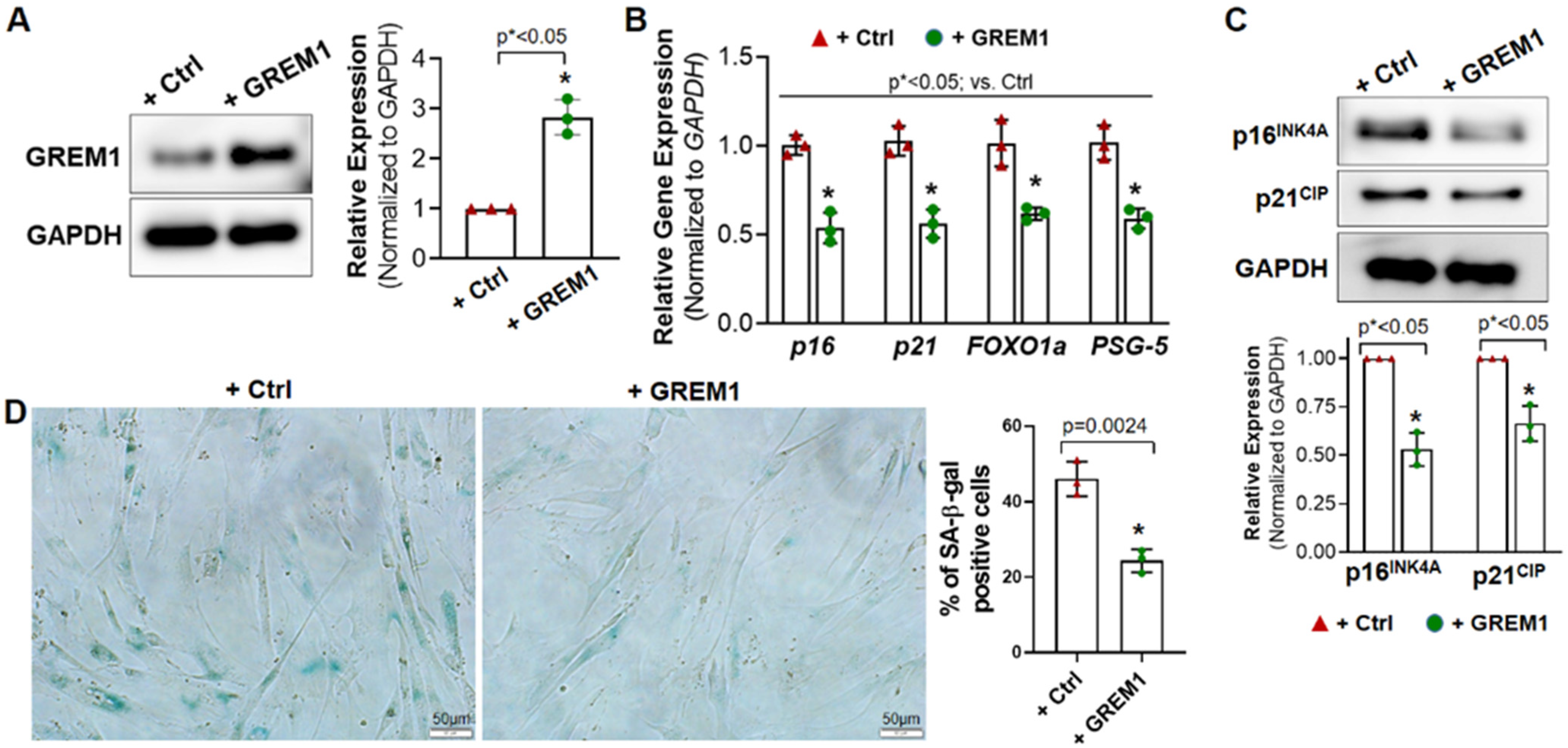
3.2. Overexpression of GREM1 Reverses the Senescent Phenotype of hMPCs
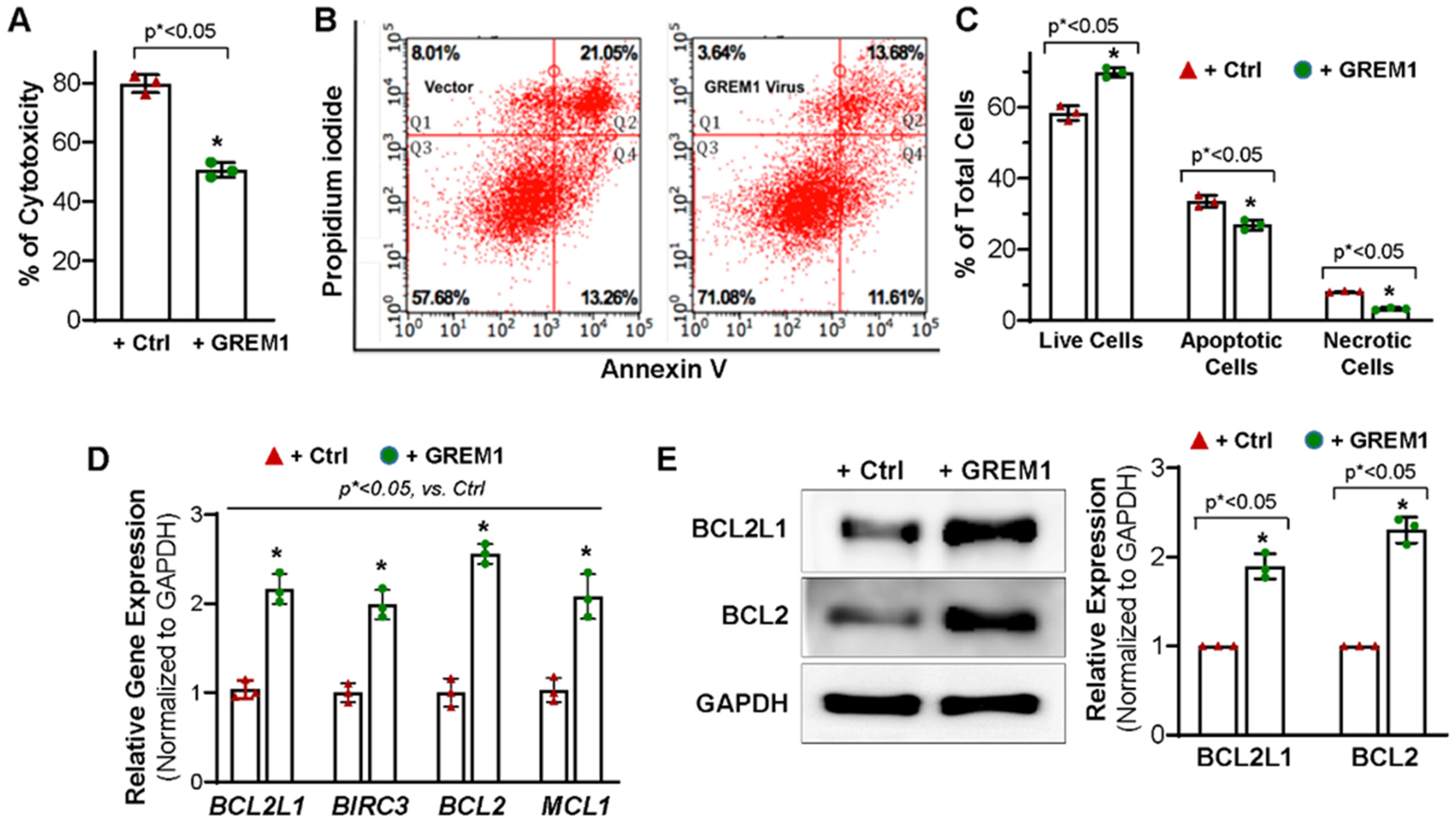
3.3. Overexpression of GREM1 Exhibits an Anti-Apoptotic Effect in Aging hMPCs
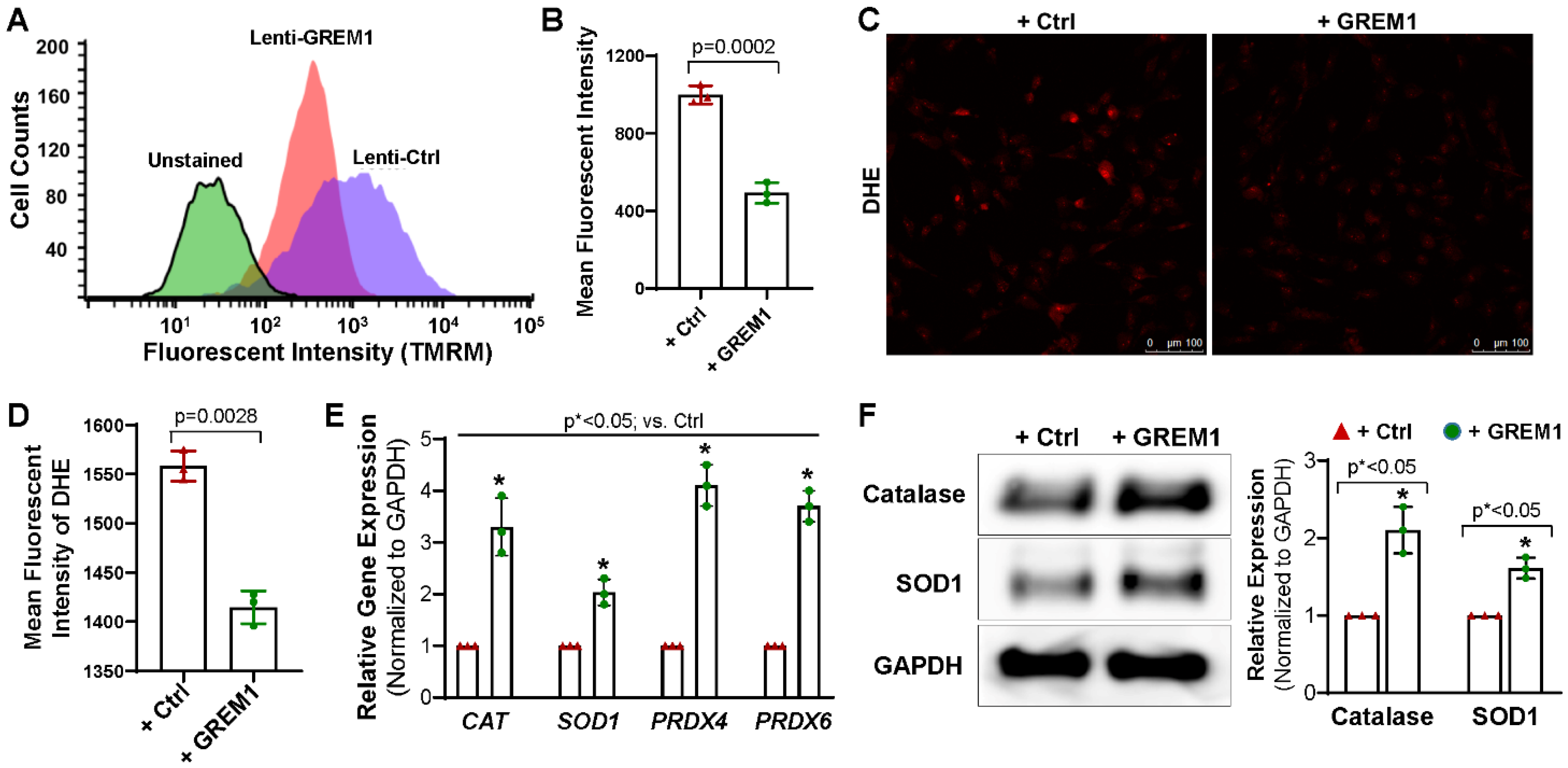
3.4. Overexpression of GREM1 Reduces ROS Generation and Increase the Expression of Antioxidants
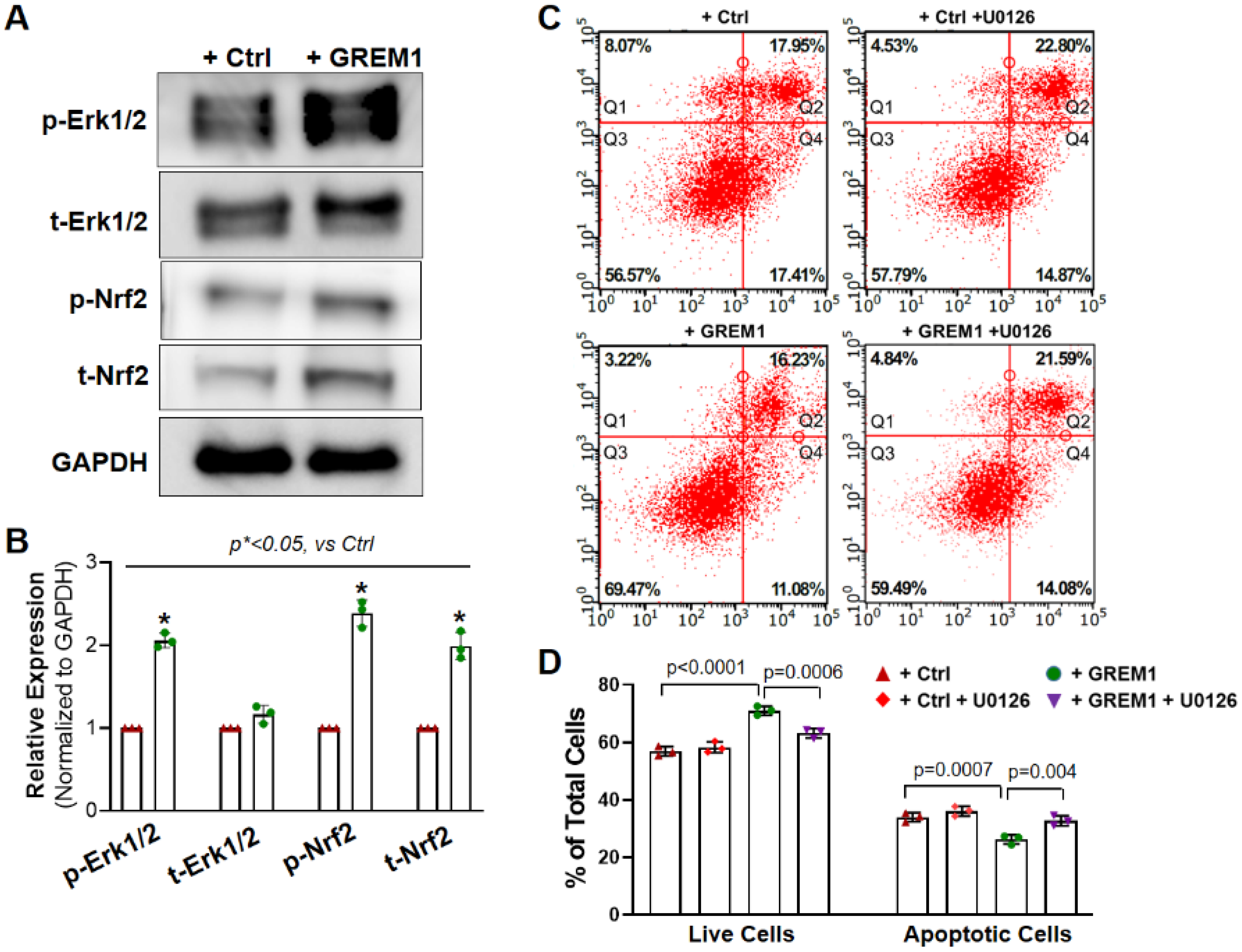
3.5. Activation of NRF2/ERK Signal Pathway Is Associated with the Cytoprotective Effect of Overexpression of GREM1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report from the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, L.C.; Carr, C.; Chang, K.C.; Lin, S.Z.; Clarke, K. Stem cell-based therapy for ischemic heart disease. Cell Transplant. 2013, 22, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Behfar, A.; Terzic, A. Stem cells versus senescence: The yin and yang of cardiac health. J. Am. Coll. Cardiol. 2015, 65, 148–150. [Google Scholar] [CrossRef]
- RajendranNair, D.S.; Karunakaran, J.; Nair, R.R. Differential response of human cardiac stem cells and bone marrow mesenchymal stem cells to hypoxia-reoxygenation injury. Mol. Cell. Biochem. 2017, 425, 139–153. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Bolli, R. Cell therapy for heart failure: A comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ. Res. 2013, 113, 810–834. [Google Scholar] [CrossRef] [PubMed]
- Zwetsloot, P.P.; Vegh, A.M.; Jansen of Lorkeers, S.J.; van Hout, G.P.; Currie, G.L.; Sena, E.S.; Gremmels, H.; Buikema, J.W.; Goumans, M.J.; Macleod, M.R.; et al. Cardiac Stem Cell Treatment in Myocardial Infarction: A Systematic Review and Meta-Analysis of Preclinical Studies. Circ. Res. 2016, 118, 1223–1232. [Google Scholar] [CrossRef]
- Oskouei, B.N.; Lamirault, G.; Joseph, C.; Treuer, A.V.; Landa, S.; Da Silva, J.; Hatzistergos, K.; Dauer, M.; Balkan, W.; McNiece, I.; et al. Increased potency of cardiac stem cells compared with bone marrow mesenchymal stem cells in cardiac repair. Stem Cells Transl. Med. 2012, 1, 116–124. [Google Scholar] [CrossRef]
- Khatiwala, R.; Cai, C. Strategies to Enhance the Effectiveness of Adult Stem Cell Therapy for Ischemic Heart Diseases Affecting the Elderly Patients. Stem Cell Rev. 2016, 12, 214–223. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; He, P.; Bennett, E.; Haggard, E.; Ma, J.; Cai, C. Mitochondrial Membrane Potential Identifies a Subpopulation of Mesenchymal Progenitor Cells to Promote Angiogenesis and Myocardial Repair. Cells 2022, 11, 1713. [Google Scholar] [CrossRef]
- Sanz-Ruiz, R.; Fernandez-Aviles, F. Autologous and allogeneic cardiac stem cell therapy for cardiovascular diseases. Pharmacol. Res. 2017, 27, 92–100. [Google Scholar] [CrossRef]
- Ni, N.C.; Li, R.K.; Weisel, R.D. The promise and challenges of cardiac stem cell therapy. Semin. Thorac. Cardiovasc. Surg. 2014, 26, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yu, S.P.; Fraser, J.L.; Lu, Z.; Ogle, M.E.; Wang, J.A.; Wei, L. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 2008, 135, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Diaz Canestro, C.; Libby, P.; Luscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Sussman, M.A. Rejuvenating the senescent heart. Curr. Opin. Cardiol. 2015, 30, 235–239. [Google Scholar] [CrossRef]
- Cesselli, D.; Beltrami, A.P.; D’Aurizio, F.; Marcon, P.; Bergamin, N.; Toffoletto, B.; Pandolfi, M.; Puppato, E.; Marino, L.; Signore, S.; et al. Effects of age and heart failure on human cardiac stem cell function. Am. J. Pathol. 2011, 179, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.; Sussman, M.A. Cardiac Hegemony of Senescence. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2013, 2, 247–254. [Google Scholar] [CrossRef]
- Liang, R.; Ghaffari, S. Stem cells, redox signaling, and stem cell aging. Antioxid. Redox Signal. 2014, 20, 1902–1916. [Google Scholar] [CrossRef]
- Kaur, G.; Cai, C. Current Progress in the Rejuvenation of Aging Stem/Progenitor Cells for Improving the Therapeutic Effectiveness of Myocardial Repair. Stem Cells Int. 2018, 2018, 9308301. [Google Scholar] [CrossRef]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef]
- Mitola, S.; Ravelli, C.; Moroni, E.; Salvi, V.; Leali, D.; Ballmer-Hofer, K.; Zammataro, L.; Presta, M. Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 2010, 116, 3677–3680. [Google Scholar] [CrossRef]
- Chen, B.; Athanasiou, M.; Gu, Q.; Blair, D.G. Drm/Gremlin transcriptionally activates p21(Cip1) via a novel mechanism and inhibits neoplastic transformation. Biochem. Biophys. Res. Commun. 2002, 295, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Van Vlodrop, I.J.H.; Baldewijns, M.M.L.; Smits, K.M.; Schouten, L.J.; van Neste, L.; van Criekinge, W.; van Poppel, H.; Lerut, E.; Schuebel, K.E.; Ahuja, N.; et al. Prognostic Significance of Gremlin1 (GREM1) Promoter CpG Island Hypermethylation in Clear Cell Renal Cell Carcinoma. Am. J. Pathol. 2010, 176, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Mitola, S.; Moroni, E.; Belleri, M.; Nicoli, S.; Coltrini, D.; Peri, F.; Pessi, A.; Orsatti, L.; Talamo, F.; et al. Bone morphogenic protein antagonist Drm/gremlin is a novel proangiogenic factor. Blood 2007, 109, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Kho, A.T.; Schatton, T.; Murphy, G.F.; Molloy, M.J.; Zhan, Q.; Ramoni, M.F.; Frank, M.H.; Kohane, I.S.; Gussoni, E. Regulation of myogenic progenitor proliferation in human fetal skeletal muscle by BMP4 and its antagonist Gremlin. J. Cell Biol. 2006, 175, 99–110. [Google Scholar] [CrossRef]
- Shekels, L.L.; Colvin Wanshura, L.E.; Xie, Y.; Nelson, M.S.; Stephenson, E.J.; Khan, S.A.; Gupta, P. The effects of Gremlin1 on human umbilical cord blood hematopoietic progenitors. Blood Cells Mol. Dis. 2015, 54, 103–109. [Google Scholar] [CrossRef]
- Kami, D.; Shiojima, I.; Makino, H.; Matsumoto, K.; Takahashi, Y.; Ishii, R.; Naito, A.T.; Toyoda, M.; Saito, H.; Watanabe, M.; et al. Gremlin enhances the determined path to cardiomyogenesis. PLoS ONE 2008, 3, e2407. [Google Scholar] [CrossRef]
- Xiang, Q.; Hong, D.; Liao, Y.; Cao, Y.; Liu, M.; Pang, J.; Zhou, J.; Wang, G.; Yang, R.; Wang, M.; et al. Overexpression of Gremlin1 in Mesenchymal Stem Cells Improves Hindlimb Ischemia in Mice by Enhancing Cell Survival. J. Cell. Physiol. 2016, 232, 996–1007. [Google Scholar] [CrossRef]
- Cai, C.X.; Teng, L.; Vu, D.; He, J.Q.; Guo, Y.R.; Li, Q.H.; Tang, X.L.; Rokosh, G.; Bhatnagar, A.; Bolli, R. The Heme Oxygenase 1 Inducer (CoPP) Protects Human Cardiac Stem Cells against Apoptosis through Activation of the Extracellular Signal-regulated Kinase (ERK)/NRF2 Signaling Pathway and Cytokine Release. J. Biol. Chem. 2012, 287, 33720–33732. [Google Scholar] [CrossRef]
- Teng, L.; Bennett, E.; Cai, C. Preconditioning c-Kit-positive Human Cardiac Stem Cells with a Nitric Oxide Donor Enhances Cell Survival through Activation of Survival Signaling Pathways. J. Biol. Chem. 2016, 291, 9733–9747. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Jourd’heuil, F.L.; Qu, S.; Devejian, N.; Bennett, E.; Jourd’heuil, D.; Cai, C. Cytoglobin Promotes Cardiac Progenitor Cell Survival against Oxidative Stress via the Upregulation of the NFkappaB/iNOS Signal Pathway and Nitric Oxide Production. Sci. Rep. 2017, 7, 10754. [Google Scholar] [CrossRef]
- Cai, C.; Guo, Y.; Teng, L.; Nong, Y.; Tan, M.; Book, M.J.; Zhu, X.; Wang, X.L.; Du, J.; Wu, W.J.; et al. Preconditioning Human Cardiac Stem Cells with an HO-1 Inducer Exerts Beneficial Effects After Cell Transplantation in the Infarcted Murine Heart. Stem Cells 2015, 33, 3596–3607. [Google Scholar] [CrossRef] [PubMed]
- Kozhukharova, I.; Zemelko, V.; Kovaleva, Z.; Alekseenko, L.; Lyublinskaya, O.; Nikolsky, N. Therapeutic doses of doxorubicin induce premature senescence of human mesenchymal stem cells derived from menstrual blood, bone marrow and adipose tissue. Int. J. Hematol. 2018, 107, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxidative Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, M.S.; Kwon, Y.W.; Lee, S.; Fang, L.T.; Choi, H.; Ray, R.; Kang, H.C.; Mao, J.H.; Jablons, D.; Kim, I.J. Gremlin is overexpressed in lung adenocarcinoma and increases cell growth and proliferation in normal lung cells. PLoS ONE 2012, 7, e42264. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, P.; Wang, X.L.; Zhang, S.; Devejian, N.; Bennett, E.; Cai, C. Sulfiredoxin-1 enhances cardiac progenitor cell survival against oxidative stress via the upregulation of the ERK/NRF2 signal pathway. Free Radic. Biol. Med. 2018, 123, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Khokha, M.K.; Hsu, D.; Brunet, L.J.; Dionne, M.S.; Harland, R.M. Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat. Genet. 2003, 34, 303–307. [Google Scholar] [CrossRef]
- Michos, O.; Panman, L.; Vintersten, K.; Beier, K.; Zeller, R.; Zuniga, A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development 2004, 131, 3401–3410. [Google Scholar] [CrossRef]
- Bylund, J.B.; Trinh, L.T.; Awgulewitsch, C.P.; Paik, D.T.; Jetter, C.; Jha, R.; Zhang, J.; Nolan, K.; Xu, C.; Thompson, T.B.; et al. Coordinated Proliferation and Differentiation of Human-Induced Pluripotent Stem Cell-Derived Cardiac Progenitor Cells Depend on Bone Morphogenetic Protein Signaling Regulation by GREMLIN 2. Stem Cells Dev. 2017, 26, 678–693. [Google Scholar] [CrossRef]
- Maciel, T.T.; Melo, R.S.; Schor, N.; Campos, A.H. Gremlin promotes vascular smooth muscle cell proliferation and migration. J. Mol. Cell. Cardiol. 2008, 44, 370–379. [Google Scholar] [CrossRef]
- Curran, S.P.; Hickey, F.B.; Watson, A.; Godson, C.; Brazil, D.P. Deletion of Gremlin1 increases cell proliferation and migration responses in mouse embryonic fibroblasts. Cell. Signal. 2012, 24, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Code No. | Gender | Age (Years) | Disease | Tissue Used for Cell Sorting |
|---|---|---|---|---|
| AMC130 | Male | 2 | ASD | Atrial septum |
| AMC155 | Male | 4 | ASD | Atrial septum |
| AMC165 | Female | 8 | ASD | Atrial septum |
| NDRI2 | Female | 15 | Accident | Right atrial |
| AMC134 | Male | 65 | CAD | Right atrial appendage |
| AMC144 | Male | 52 | CAD | Right atrial appendage |
| AMC145 | Female | 81 | CAD | Right atrial appendage |
| AMC146 | Male | 84 | CAD | Right atrial appendage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, G.; Wang, X.; Li, X.; Ong, H.; He, X.; Cai, C. Overexpression of GREM1 Improves the Survival Capacity of Aged Cardiac Mesenchymal Progenitor Cells via Upregulation of the ERK/NRF2-Associated Antioxidant Signal Pathway. Cells 2023, 12, 1203. https://doi.org/10.3390/cells12081203
Kaur G, Wang X, Li X, Ong H, He X, Cai C. Overexpression of GREM1 Improves the Survival Capacity of Aged Cardiac Mesenchymal Progenitor Cells via Upregulation of the ERK/NRF2-Associated Antioxidant Signal Pathway. Cells. 2023; 12(8):1203. https://doi.org/10.3390/cells12081203
Chicago/Turabian StyleKaur, Gurleen, Xiaoliang Wang, Xiuchun Li, Hannah Ong, Xiangfei He, and Chuanxi Cai. 2023. "Overexpression of GREM1 Improves the Survival Capacity of Aged Cardiac Mesenchymal Progenitor Cells via Upregulation of the ERK/NRF2-Associated Antioxidant Signal Pathway" Cells 12, no. 8: 1203. https://doi.org/10.3390/cells12081203
APA StyleKaur, G., Wang, X., Li, X., Ong, H., He, X., & Cai, C. (2023). Overexpression of GREM1 Improves the Survival Capacity of Aged Cardiac Mesenchymal Progenitor Cells via Upregulation of the ERK/NRF2-Associated Antioxidant Signal Pathway. Cells, 12(8), 1203. https://doi.org/10.3390/cells12081203