
Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders

Abstract
1. Introduction
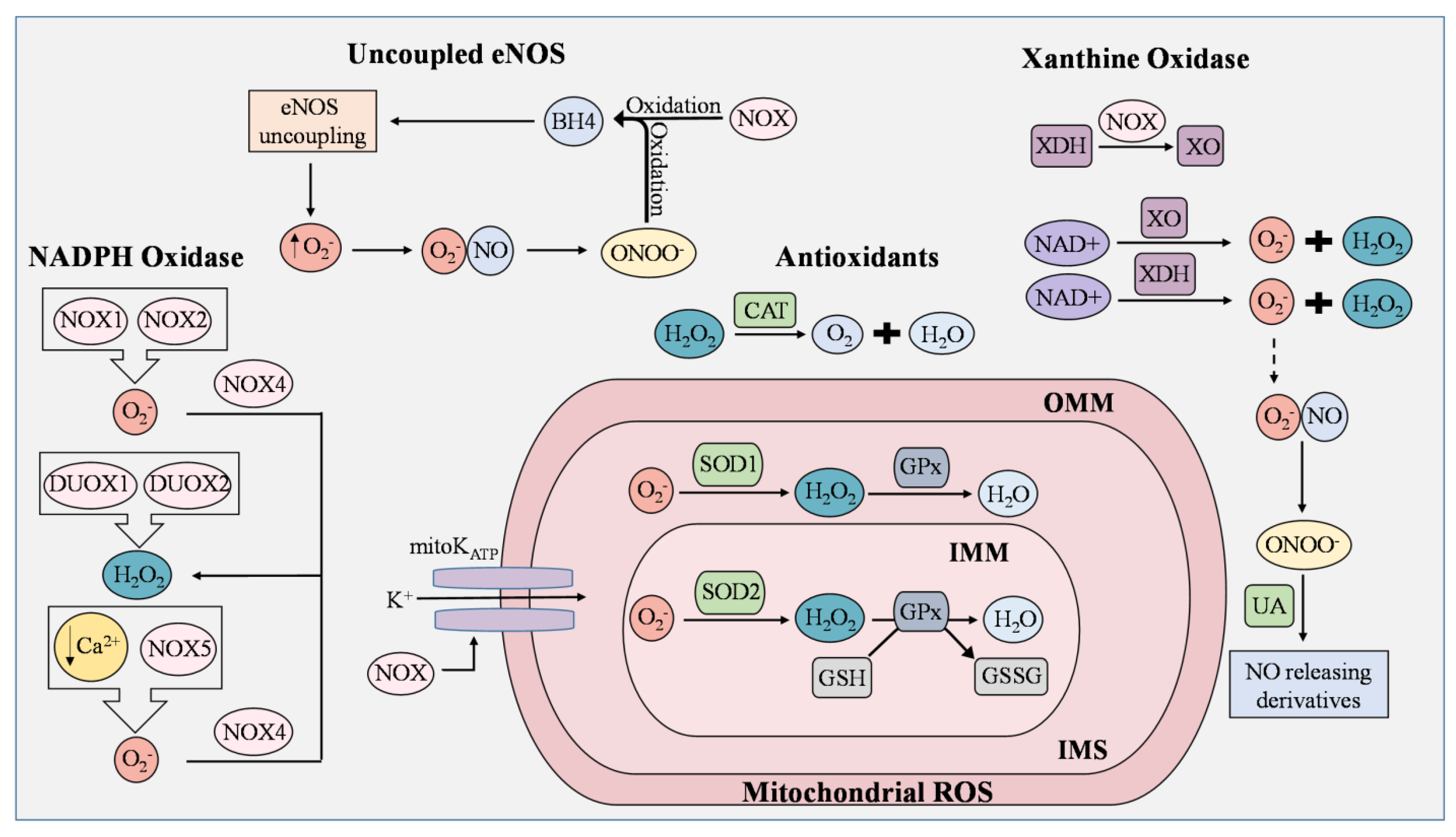
2. Generation of ROS in Endothelial Cells
2.1. Uncoupled eNOS
2.2. NADPH
2.3. Mitochondrial Electron Transport Chain
2.4. Xanthine Oxidases
2.5. Antioxidant/Defense Systems in ECs for ROS
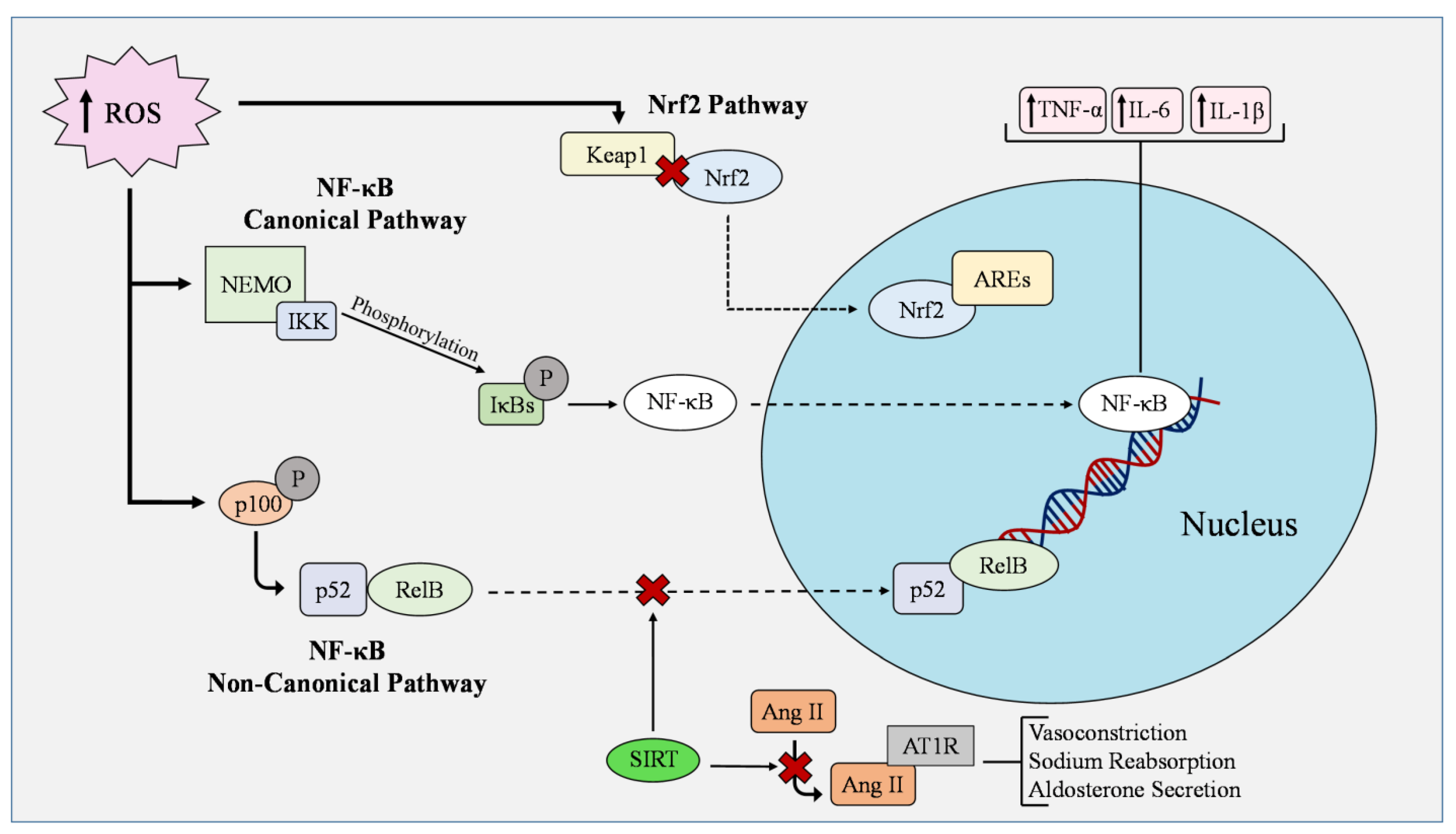
3. Pathways Regulated by ROS
3.1. Regulation of NF-κB
3.2. Regulation of Nrf2
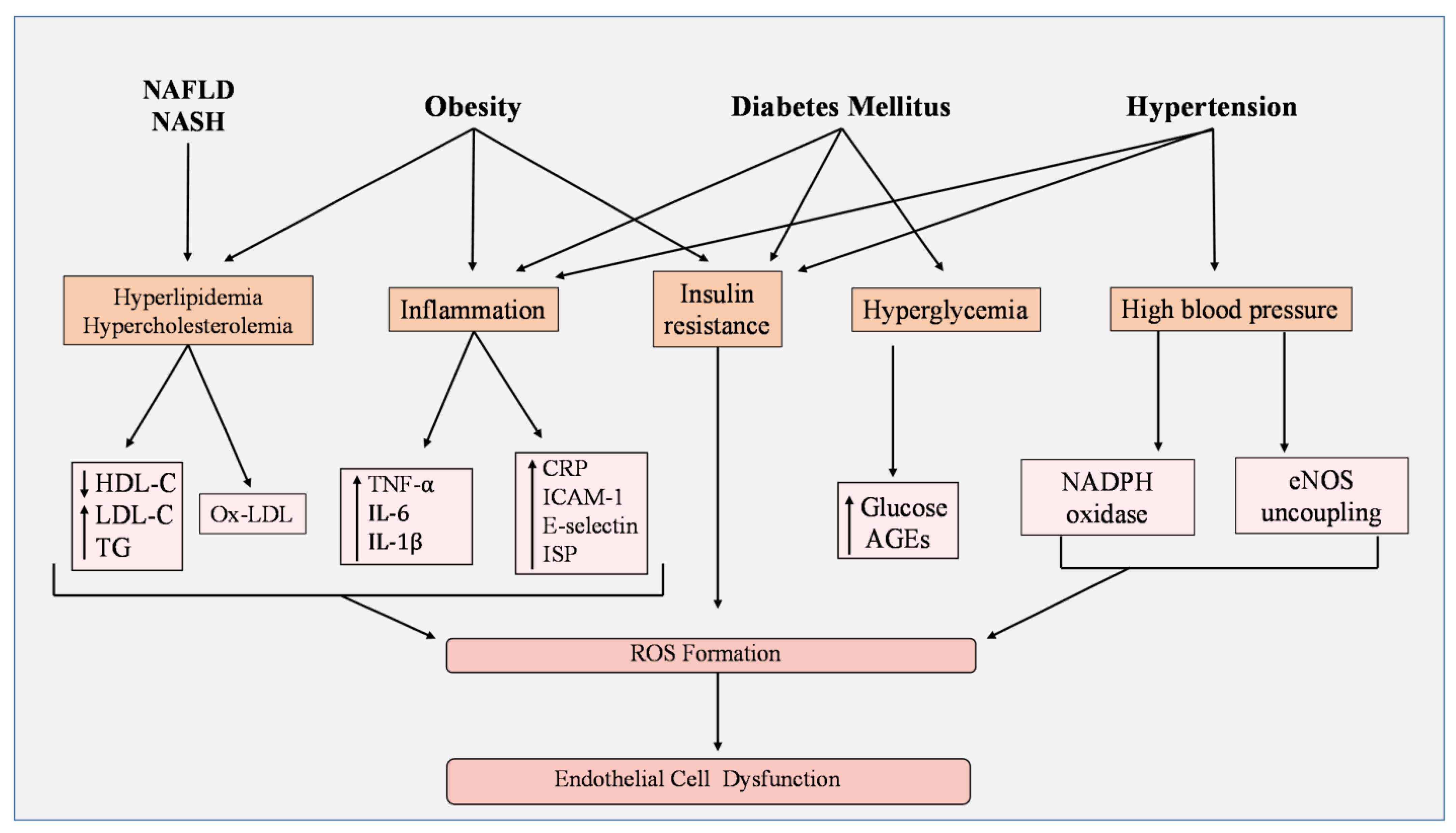
4. Endothelial Cell Damage by ROS in Metabolic Disorders
5. ROS in Endothelial Cells under Metabolic Stress
5.1. Hypertension
5.2. Obesity
5.3. Diabetes Mellitus
5.4. Hyperlipidemia
5.5. Non-Alcoholic Fatty Liver Disease
6. Redox-Modulation of Current Market Drugs
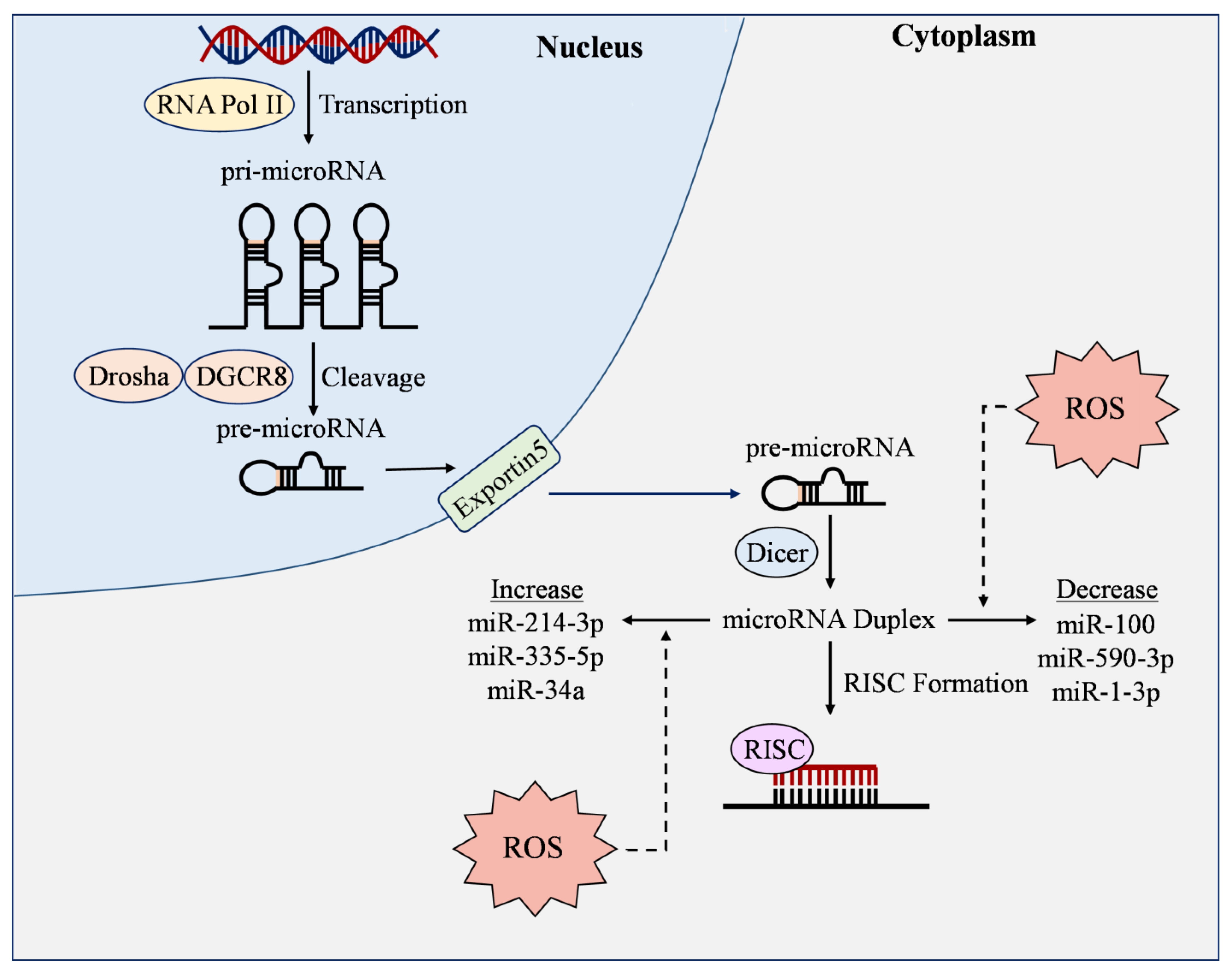
7. MicroRNAs and ROS
7.1. ROS-Responsive MicroRNAs
7.2. Regulation of ROS by MicroRNAs
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Celotto, A.M.; Liu, Z.; Vandemark, A.P.; Palladino, M.J. A novel Drosophila SOD2 mutant demonstrates a role for mitochondrial ROS in neurodevelopment and disease. Brain Behav. 2012, 2, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lin, Z.; Guan, L.; Gaughan, G.; Lin, G. Antioxidant enzymes regulate reactive oxygen species during pod elongation in Pisum sativum and Brassica chinensis. PLoS ONE 2014, 9, e87588. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Taverne, Y.J.; Bogers, A.J.; Duncker, D.J.; Merkus, D. Reactive oxygen species and the cardiovascular system. Oxid. Med. Cell. Longev. 2013, 2013, 862423. [Google Scholar] [CrossRef]
- Rotariu, D.; Babes, E.E.; Tit, D.M.; Moisi, M.; Bustea, C.; Stoicescu, M.; Radu, A.F.; Vesa, C.M.; Behl, T.; Bungau, A.F.; et al. Oxidative stress-Complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomed. Pharmacother. 2022, 152, 113238. [Google Scholar] [CrossRef]
- Bautch, V.L.; Caron, K.M. Blood and lymphatic vessel formation. Cold Spring Harb. Perspect. Biol. 2015, 7, a008268. [Google Scholar] [CrossRef]
- Aman, J.; Weijers, E.M.; van Nieuw Amerongen, G.P.; Malik, A.B.; van Hinsbergh, V.W. Using cultured endothelial cells to study endothelial barrier dysfunction: Challenges and opportunities. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L453–L466. [Google Scholar] [CrossRef]
- Hanson, M.; Gluckman, P. Endothelial dysfunction and cardiovascular disease: The role of predictive adaptive responses. Heart 2005, 91, 864–866. [Google Scholar] [CrossRef]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Alhayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941. [Google Scholar] [CrossRef] [PubMed]
- Gallo, G.; Volpe, M.; Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 2021, 8, 798958. [Google Scholar] [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef]
- K, R.B.; Tay, Y. The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer. Int. J. Mol. Sci. 2019, 20, 5335. [Google Scholar] [CrossRef]
- Ilieva, M.; Panella, R.; Uchida, S. MicroRNAs in Cancer and Cardiovascular Disease. Cells 2022, 11, 3551. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Warwick, T.; Noack, K.; Malacarne, P.F.; Cooper, A.J.L.; Weissmann, N.; Schroder, K.; Brandes, R.P.; Rezende, F. Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells. Antioxidants 2022, 11, 434. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium 2016, 60, 108–114. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, J.; Piao, H.; Zhu, Z.; Wei, R.; Liu, K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 2022, 13, 1039241. [Google Scholar] [CrossRef]
- Loperena, R.; Harrison, D.G. Oxidative Stress and Hypertensive Diseases. Med. Clin. N. Am. 2017, 101, 169–193. [Google Scholar] [CrossRef]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2019, 10, 1568. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef]
- Luczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathway in the Pathogenesis of Autoimmune Rheumatic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1417981. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, Y.; Ramprasath, T.; Zou, M.H. Oxidative Stress, GTPCH1, and Endothelial Nitric Oxide Synthase Uncoupling in Hypertension. Antioxid. Redox Signal. 2021, 34, 750–764. [Google Scholar] [CrossRef]
- Li, H.; Nguyen, H.; Meda Venkata, S.P.; Koh, J.Y.; Kowluru, A.; Li, L.; Rossi, N.F.; Chen, W.; Wang, J.M. Novel Role of GPR35 (G-Protein-Coupled Receptor 35) in the Regulation of Endothelial Cell Function and Blood Pressure. Hypertension 2021, 78, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef]
- Schroder, K. Isoform specific functions of Nox protein-derived reactive oxygen species in the vasculature. Curr. Opin. Pharmacol. 2010, 10, 122–126. [Google Scholar] [CrossRef]
- Fulton, D.J.; Barman, S.A. Clarity on the Isoform-Specific Roles of NADPH Oxidases and NADPH Oxidase-4 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 579–581. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Zhao, T.; Wang, Y.; Li, Z.; Xu, X.; Lei, S.; Huang, L.; Xu, L.; Zhang, M.; Yang, L. Associations of noise kurtosis, genetic variations in NOX3 and lifestyle factors with noise-induced hearing loss. Environ. Health 2020, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Fernandez-Irigoyen, J.; Ainzua, E.; Martinez-Azcona, M.; Cortes, A.; Roncal, C.; Orbe, J.; Santamaria, E.; Zalba, G. NADPH Oxidase 5 (NOX5) Overexpression Promotes Endothelial Dysfunction via Cell Apoptosis, Migration, and Metabolic Alterations in Human Brain Microvascular Endothelial Cells (hCMEC/D3). Antioxidants 2022, 11, 2147. [Google Scholar] [CrossRef]
- Van der Vliet, A.; Danyal, K.; Heppner, D.E. Dual oxidase: A novel therapeutic target in allergic disease. Br. J. Pharmacol. 2018, 175, 1401–1418. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Chen, Q.M. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radic. Biol. Med. 2022, 179, 133–143. [Google Scholar] [CrossRef]
- Tang, S.P.; Mao, X.L.; Chen, Y.H.; Yan, L.L.; Ye, L.P.; Li, S.W. Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 2022, 13, 870239. [Google Scholar] [CrossRef]
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharmacol. Rev. 2020, 72, 801–828. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Cheng, J.D. Uric Acid and Cardiovascular Disease: An Update from Molecular Mechanism to Clinical Perspective. Front. Pharmacol. 2020, 11, 582680. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, J.; Jiao, H.; Oluwabiyi, C.; Li, H.; Zhao, J.; Zhou, Y.; Wang, X.; Lin, H. Enterocyte synthesizes and secrets uric acid as antioxidant to protect against oxidative stress via the involvement of Nrf pathway. Free Radic. Biol. Med. 2022, 179, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, H.; Sun, Q.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. The Role of Oxidative Stress in Hyperuricemia and Xanthine Oxidoreductase (XOR) Inhibitors. Oxid. Med. Cell. Longev. 2021, 2021, 1470380. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Liu, M.; Sun, X.; Chen, B.; Dai, R.; Xi, Z.; Xu, H. Insights into Manganese Superoxide Dismutase and Human Diseases. Int. J. Mol. Sci. 2022, 23, 5893. [Google Scholar] [CrossRef]
- Banks, C.J.; Andersen, J.L. Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 2019, 26, 101270. [Google Scholar] [CrossRef]
- Coates, L.; Sullivan, B. The macromolecular neutron diffractometer at the spallation neutron source. Methods Enzymol. 2020, 634, 87–99. [Google Scholar] [CrossRef]
- Tak, L.J.; Kim, H.Y.; Ham, W.K.; Agrahari, G.; Seo, Y.; Yang, J.W.; An, E.J.; Bang, C.H.; Lee, M.J.; Kim, H.S.; et al. Superoxide Dismutase 3-Transduced Mesenchymal Stem Cells Preserve Epithelial Tight Junction Barrier in Murine Colitis and Attenuate Inflammatory Damage in Epithelial Organoids. Int. J. Mol. Sci. 2021, 22, 6431. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Zandi, P.; Schnug, E. Reactive Oxygen Species, Antioxidant Responses and Implications from a Microbial Modulation Perspective. Biology 2022, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1alpha and reactive oxygen species. Cell Death Dis. 2021, 12, 77. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, H.; Wu, X.; Wang, Q.; Chen, M.; Feng, Z.; Chen, H. The functions of glutathione peroxidase in ROS homeostasis and fruiting body development in Hypsizygus marmoreus. Appl. Microbiol. Biotechnol. 2020, 104, 10555–10570. [Google Scholar] [CrossRef] [PubMed]
- Prasai, P.K.; Shrestha, B.; Orr, A.W.; Pattillo, C.B. Decreases in GSH:GSSG activate vascular endothelial growth factor receptor 2 (VEGFR2) in human aortic endothelial cells. Redox Biol. 2018, 19, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. J. Diabetes Res. 2020, 2020, 7489795. [Google Scholar] [CrossRef]
- Hemmati-Dinarvand, M.; Saedi, S.; Valilo, M.; Kalantary-Charvadeh, A.; Alizadeh Sani, M.; Kargar, R.; Safari, H.; Samadi, N. Oxidative stress and Parkinson’s disease: Conflict of oxidant-antioxidant systems. Neurosci. Lett. 2019, 709, 134296. [Google Scholar] [CrossRef]
- Xu, F.; Xu, J.; Xiong, X.; Deng, Y. Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep. 2019, 24, 70–74. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef]
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 2009, 119, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Kairisalo, M.; Korhonen, L.; Blomgren, K.; Lindholm, D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-κB activation. Biochem. Biophys. Res. Commun. 2007, 364, 138–144. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Cantoni, O.; Zito, E.; Guidarelli, A.; Fiorani, M.; Ghezzi, P. Mitochondrial ROS, ER Stress, and Nrf2 Crosstalk in the Regulation of Mitochondrial Apoptosis Induced by Arsenite. Antioxidants 2022, 11, 1034. [Google Scholar] [CrossRef]
- Zhuang, K.; Tang, H.; Guo, H.; Yuan, S. Geraniol prevents Helicobacterium pylori-induced human gastric cancer signalling by enhancing peroxiredoxin-1 expression in GES-1 cells. Microb. Pathog. 2023, 174, 105937. [Google Scholar] [CrossRef] [PubMed]
- Ben Ammar, R.; Mohamed, M.E.; Alfwuaires, M.; Abdulaziz Alamer, S.; Bani Ismail, M.; Veeraraghavan, V.P.; Sekar, A.K.; Ksouri, R.; Rajendran, P. Anti-Inflammatory Activity of Geraniol Isolated from Lemon Grass on Ox-LDL-Stimulated Endothelial Cells by Upregulation of Heme Oxygenase-1 via PI3K/Akt and Nrf-2 Signaling Pathways. Nutrients 2022, 14, 4817. [Google Scholar] [CrossRef]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; Yazbi, A.E.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. 2022, 27, 105. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.G.; Nadruz, W., Jr.; Monica, F.Z. Endothelial and vascular smooth muscle dysfunction in hypertension. Biochem. Pharmacol. 2022, 205, 115263. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, Inflammation, Immune System, and Hypertension. Am. J. Hypertens. 2021, 34, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Maruhashi, T.; Noma, K.; Kihara, Y. Oxidative stress and endothelial dysfunction: Clinical evidence and therapeutic implications. Trends. Cardiovasc. Med. 2014, 24, 165–169. [Google Scholar] [CrossRef]
- Huang, Y.; Song, C.; He, J.; Li, M. Research progress in endothelial cell injury and repair. Front. Pharmacol. 2022, 13, 997272. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, 992. [Google Scholar] [CrossRef]
- Akhigbe, R.; Ajayi, A. The impact of reactive oxygen species in the development of cardiometabolic disorders: A review. Lipids Health Dis. 2021, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.; Verma, H.K.; Lakkakula, S.; Merchant, N.; Kadir, F.; Rahman, S.; Jeffree, M.S.; Lakkakula, B.; Rao, P.V. Biomarkers of Oxidative Stress Tethered to Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2022, 2022, 9154295. [Google Scholar] [CrossRef]
- Nassir, F. NAFLD: Mechanisms, Treatments, and Biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef]
- Balta, S. Endothelial Dysfunction and Inflammatory Markers of Vascular Disease. Curr. Vasc. Pharmacol. 2021, 19, 243–249. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARgamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef]
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J. Nutr. 2017, 147, 506–513. [Google Scholar] [CrossRef]
- Hu, R.; Wang, M.Q.; Ni, S.H.; Wang, M.; Liu, L.Y.; You, H.Y.; Wu, X.H.; Wang, Y.J.; Lu, L.; Wei, L.B. Salidroside ameliorates endothelial inflammation and oxidative stress by regulating the AMPK/NF-κB/NLRP3 signaling pathway in AGEs-induced HUVECs. Eur. J. Pharmacol. 2020, 867, 172797. [Google Scholar] [CrossRef]
- Shediwah, F.M.H.; Naji, K.M.; Gumaih, H.S.; Alhadi, F.A.; Al-Hammami, A.L.; D’Souza, M.R. Antioxidant and antihyperlipidemic activity of Costus speciosus against atherogenic diet-induced hyperlipidemia in rabbits. J. Integr. Med. 2019, 17, 181–191. [Google Scholar] [CrossRef]
- Khalil, M.; Khalifeh, H.; Baldini, F.; Salis, A.; Damonte, G.; Daher, A.; Voci, A.; Vergani, L. Antisteatotic and antioxidant activities of Thymbra spicata L. extracts in hepatic and endothelial cells as in vitro models of non-alcoholic fatty liver disease. J. Ethnopharmacol. 2019, 239, 111919. [Google Scholar] [CrossRef]
- Krzeminska, J.; Wronka, M.; Mlynarska, E.; Franczyk, B.; Rysz, J. Arterial Hypertension-Oxidative Stress and Inflammation. Antioxidants 2022, 11, 172. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef]
- Man, A.W.C.; Li, H.; Xia, N. The Role of Sirtuin1 in Regulating Endothelial Function, Arterial Remodeling and Vascular Aging. Front. Physiol. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Liu, X.; Feng, H.; Zhao, S.; Gao, H. Grape seed proanthocyanidin extracts enhance endothelial nitric oxide synthase expression through 5′-AMP activated protein kinase/Surtuin 1-Krupple like factor 2 pathway and modulate blood pressure in ouabain induced hypertensive rats. Biol. Pharm. Bull. 2012, 35, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Strand, S.; Schlufter, F.; Siuda, D.; Reifenberg, G.; Kleinert, H.; Forstermann, U.; Li, H. Role of SIRT1 and FOXO factors in eNOS transcriptional activation by resveratrol. Nitric Oxide 2013, 32, 29–35. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Dell’Omo, G.; Crescenti, D.; Vantaggiato, C.; Parravicini, C.; Borroni, A.P.; Rizzi, N.; Garofalo, M.; Pinto, A.; Recordati, C.; Scanziani, E.; et al. Inhibition of SIRT1 deacetylase and p53 activation uncouples the anti-inflammatory and chemopreventive actions of NSAIDs. Br. J. Cancer 2019, 120, 537–546. [Google Scholar] [CrossRef]
- Wang, F.; Shang, Y.; Zhang, R.; Gao, X.; Zeng, Q. A SIRT1 agonist reduces cognitive decline in type 2 diabetic rats through antioxidative and anti-inflammatory mechanisms. Mol. Med. Rep. 2019, 19, 1040–1048. [Google Scholar] [CrossRef]
- He, X.; Zeng, H.; Chen, J.X. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J. Cell. Physiol. 2019, 234, 2252–2265. [Google Scholar] [CrossRef]
- Ghatage, T.; Goyal, S.G.; Dhar, A.; Bhat, A. Novel therapeutics for the treatment of hypertension and its associated complications: Peptide-and nonpeptide-based strategies. Hypertens. Res. 2021, 44, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Banday, A.A.; Lokhandwala, M.F. Loss of biphasic effect on Na/K-ATPase activity by angiotensin II involves defective angiotensin type 1 receptor-nitric oxide signaling. Hypertension 2008, 52, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Colak, E.; Pap, D. The role of oxidative stress in the development of obesity and obesity-related metabolic disorders. J. Med. Biochem. 2021, 40, 1–9. [Google Scholar] [CrossRef]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, L.; Zheng, H.; Zeng, Y. Cytokines secreted from adipose tissues mediate tumor proliferation and metastasis in triple negative breast cancer. BMC Cancer 2022, 22, 886. [Google Scholar] [CrossRef]
- Zhang, H.; Potter, B.J.; Cao, J.M.; Zhang, C. Interferon-gamma induced adipose tissue inflammation is linked to endothelial dysfunction in type 2 diabetic mice. Basic Res. Cardiol. 2011, 106, 1135–1145. [Google Scholar] [CrossRef]
- Jakubiak, G.K.; Osadnik, K.; Lejawa, M.; Osadnik, T.; Golawski, M.; Lewandowski, P.; Pawlas, N. “Obesity and Insulin Resistance” Is the Component of the Metabolic Syndrome Most Strongly Associated with Oxidative Stress. Antioxidants 2021, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef] [PubMed]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 70. [Google Scholar] [CrossRef]
- Rudnicka, E.; Suchta, K.; Grymowicz, M.; Calik-Ksepka, A.; Smolarczyk, K.; Duszewska, A.M.; Smolarczyk, R.; Meczekalski, B. Chronic Low Grade Inflammation in Pathogenesis of PCOS. Int. J. Mol. Sci. 2021, 22, 3789. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Estato, V.; Castro-Faria-Neto, H.C.; Tibirica, E. Obesity-Related Inflammation and Endothelial Dysfunction in COVID-19: Impact on Disease Severity. J. Inflamm. Res. 2021, 14, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Flores-Cortez, Y.A.; Barragan-Bonilla, M.I.; Mendoza-Bello, J.M.; Gonzalez-Calixto, C.; Flores-Alfaro, E.; Espinoza-Rojo, M. Interplay of retinol binding protein 4 with obesity and associated chronic alterations. Mol. Med. Rep. 2022, 26, 244. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; El-Atat, F.A.; McFarlane, S.I.; Aneja, A.; Sowers, J.R. Cardiometabolic syndrome: Pathophysiology and treatment. Curr. Hypertens. Rep. 2003, 5, 393–401. [Google Scholar] [CrossRef]
- Gonzalez-Chavez, A.; Chavez-Fernandez, J.A.; Elizondo-Argueta, S.; Gonzalez-Tapia, A.; Leon-Pedroza, J.I.; Ochoa, C. Metabolic Syndrome and Cardiovascular Disease: A Health Challenge. Arch. Med. Res. 2018, 49, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Fluitt, M.B.; Mohit, N.; Gambhir, K.K.; Nunlee-Bland, G. To the Future: The Role of Exosome-Derived microRNAs as Markers, Mediators, and Therapies for Endothelial Dysfunction in Type 2 Diabetes Mellitus. J. Diabetes Res. 2022, 2022, 5126968. [Google Scholar] [CrossRef]
- Calles-Escandon, J.; Cipolla, M. Diabetes and endothelial dysfunction: A clinical perspective. Endocr. Rev. 2001, 22, 36–52. [Google Scholar] [CrossRef]
- Takeda, Y.; Matoba, K.; Sekiguchi, K.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Endothelial Dysfunction in Diabetes. Biomedicines 2020, 8, 182. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Huang, L.; Chen, Z.; Chen, R.; Lin, L.; Ren, L.; Zhang, M.; Liu, L. Increased fatty acid metabolism attenuates cardiac resistance to β-adrenoceptor activation via mitochondrial reactive oxygen species: A potential mechanism of hypoglycemia-induced myocardial injury in diabetes. Redox Biol. 2022, 52, 102320. [Google Scholar] [CrossRef]
- Odegaard, A.O.; Jacobs, D.R., Jr.; Sanchez, O.A.; Goff, D.C., Jr.; Reiner, A.P.; Gross, M.D. Oxidative stress, inflammation, endothelial dysfunction and incidence of type 2 diabetes. Cardiovasc. Diabetol. 2016, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Balbaa, M.; Abdulmalek, S.A.; Khalil, S. Oxidative stress and expression of insulin signaling proteins in the brain of diabetic rats: Role of Nigella sativa oil and antidiabetic drugs. PLoS ONE 2017, 12, e0172429. [Google Scholar] [CrossRef] [PubMed]
- Han, W.M.; Chen, X.C.; Li, G.R.; Wang, Y. Acacetin Protects Against High Glucose-Induced Endothelial Cells Injury by Preserving Mitochondrial Function via Activating Sirt1/Sirt3/AMPK Signals. Front. Pharmacol. 2020, 11, 607796. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Li, C.; Reif, M.M.; Craige, S.M.; Kant, S.; Keaney, J.F., Jr. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric Oxide 2016, 55–56, 45–53. [Google Scholar] [CrossRef]
- Long, Y.; Liu, X.; Tan, X.Z.; Jiang, C.X.; Chen, S.W.; Liang, G.N.; He, X.M.; Wu, J.; Chen, T.; Xu, Y. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118- induced pyroptosis in endothelial cells. Ecotoxicol. Environ. Saf. 2020, 189, 109937. [Google Scholar] [CrossRef]
- Karr, S. Epidemiology and management of hyperlipidemia. Am. J. Manag. Care 2017, 23, S139–S148. [Google Scholar]
- Yao, Y.S.; Li, T.D.; Zeng, Z.H. Mechanisms underlying direct actions of hyperlipidemia on myocardium: An updated review. Lipids Health Dis. 2020, 19, 23. [Google Scholar] [CrossRef]
- Hill, M.F.; Bordoni, B. Hyperlipidemia; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Amiya, E. Interaction of hyperlipidemia and reactive oxygen species: Insights from the lipid-raft platform. World J. Cardiol. 2016, 8, 689–694. [Google Scholar] [CrossRef]
- Csonka, C.; Sarkozy, M.; Pipicz, M.; Dux, L.; Csont, T. Modulation of Hypercholesterolemia-Induced Oxidative/Nitrative Stress in the Heart. Oxid. Med. Cell. Longev. 2016, 2016, 3863726. [Google Scholar] [CrossRef] [PubMed]
- Sozen, E.; Ozer, N.K. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini-review. Redox Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Lei, Y.; Liu, Y.; Tan, F.; Li, S.; Wang, X.; Xu, M.; Cai, W.; Du, B.; Xu, F.; et al. Vaccarin prevents ox-LDL-induced HUVEC EndMT, inflammation and apoptosis by suppressing ROS/p38 MAPK signaling. Am. J. Transl. Res. 2019, 11, 2140–2154. [Google Scholar]
- Rahman, T.; Hamzan, N.S.; Mokhsin, A.; Rahmat, R.; Ibrahim, Z.O.; Razali, R.; Thevarajah, M.; Nawawi, H. Enhanced status of inflammation and endothelial activation in subjects with familial hypercholesterolaemia and their related unaffected family members: A case control study. Lipids Health Dis. 2017, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Ganjali, S.; Keshavarz, R.; Hosseini, S.; Mansouri, A.; Mannarino, M.R.; Pirro, M.; Jamialahmadi, T.; Sahebkar, A. Evaluation of Oxidative Stress Status in Familial Hypercholesterolemia. J. Clin. Med. 2021, 10, 5867. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.Y.; Roh, Y.S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 11, 91. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Masarone, M.; Gravina, A.G.; Di Sarno, R.; Tuccillo, C.; Cossiga, V.; Lama, S.; Stiuso, P.; Morisco, F.; et al. Evaluation of the Effect Derived from Silybin with Vitamin D and Vitamin E Administration on Clinical, Metabolic, Endothelial Dysfunction, Oxidative Stress Parameters, and Serological Worsening Markers in Nonalcoholic Fatty Liver Disease Patients. Oxid. Med. Cell. Longev. 2019, 2019, 8742075. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Liu, P.; Yang, F.; Wang, X.; Zheng, W.; Sun, W. Hesperetin ameliorates hepatic oxidative stress and inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic acid-induced HepG2 cells and a rat model of high-fat diet-induced NAFLD. Food Funct. 2021, 12, 3898–3918. [Google Scholar] [CrossRef]
- Ali Sangouni, A.; Abdollahi, S.; Mozaffari-Khosravi, H. Effect of resveratrol supplementation on hepatic steatosis and cardiovascular indices in overweight subjects with type 2 diabetes: A double-blind, randomized controlled trial. BMC Cardiovasc. Disord. 2022, 22, 212. [Google Scholar] [CrossRef]
- Bagul, P.K.; Deepthi, N.; Sultana, R.; Banerjee, S.K. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J. Nutr. Biochem. 2015, 26, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef] [PubMed]
- Yew, W.W.; Chang, K.C.; Chan, D.P. Oxidative Stress and First-Line Antituberculosis Drug-Induced Hepatotoxicity. Antimicrob. Agents. Chemother. 2018, 62, e02637-17. [Google Scholar] [CrossRef]
- Vujic, T.; Schvartz, D.; Furlani, I.L.; Meister, I.; Gonzalez-Ruiz, V.; Rudaz, S.; Sanchez, J.C. Oxidative Stress and Extracellular Matrix Remodeling Are Signature Pathways of Extracellular Vesicles Released upon Morphine Exposure on Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3926. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, E.J.; Lim, K.M. Ibuprofen Increases the Hepatotoxicity of Ethanol through Potentiating Oxidative Stress. Biomol. Ther. 2021, 29, 205–210. [Google Scholar] [CrossRef]
- Osmanlioglu, H.O.; Yildirim, M.K.; Akyuva, Y.; Yildizhan, K.; Naziroglu, M. Morphine Induces Apoptosis, Inflammation, and Mitochondrial Oxidative Stress via Activation of TRPM2 Channel and Nitric Oxide Signaling Pathways in the Hippocampus. Mol. Neurobiol. 2020, 57, 3376–3389. [Google Scholar] [CrossRef]
- Zhang, Q.; Qu, H.; Chen, Y.; Luo, X.; Chen, C.; Xiao, B.; Ding, X.; Zhao, P.; Lu, Y.; Chen, A.F.; et al. Atorvastatin Induces Mitochondria-Dependent Ferroptosis via the Modulation of Nrf2-xCT/GPx4 Axis. Front. Cell Dev. Biol. 2022, 10, 806081. [Google Scholar] [CrossRef]
- Ning, D.; Yang, X.; Wang, T.; Jiang, Q.; Yu, J.; Wang, D. Atorvastatin treatment ameliorates cardiac function and remodeling induced by isoproterenol attack through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 2021, 574, 39–47. [Google Scholar] [CrossRef]
- Dang, H.; Song, B.; Dong, R.; Zhang, H. Atorvastatin reverses the dysfunction of human umbilical vein endothelial cells induced by angiotensin II. Exp. Ther. Med. 2018, 16, 5286–5297. [Google Scholar] [CrossRef]
- Gowd, V.; Kanika; Jori, C.; Chaudhary, A.A.; Rudayni, H.A.; Rashid, S.; Khan, R. Resveratrol and resveratrol nano-delivery systems in the treatment of inflammatory bowel disease. J. Nutr. Biochem. 2022, 109, 109101. [Google Scholar] [CrossRef]
- Liu, J.; Yao, L.; Wang, Y. Resveratrol alleviates preeclampsia-like symptoms in rats through a mechanism involving the miR-363-3p/PEDF/VEGF axis. Microvasc. Res. 2023, 146, 104451. [Google Scholar] [CrossRef] [PubMed]
- Petrella, C.; Carito, V.; Carere, C.; Ferraguti, G.; Ciafre, S.; Natella, F.; Bello, C.; Greco, A.; Ralli, M.; Mancinelli, R.; et al. Oxidative stress inhibition by resveratrol in alcohol-dependent mice. Nutrition 2020, 79–80, 110783. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; O’Meara, M.; Zhang, X.; Zhang, K.; Seyoum, B.; Yi, Z.; Kaufman, R.J.; Monks, T.J.; Wang, J.M. Ameliorating Methylglyoxal-Induced Progenitor Cell Dysfunction for Tissue Repair in Diabetes. Diabetes 2019, 68, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.; Viggiani, G.; Chen, Y.W.; Coulis, G.; Castaldi, A. MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 4370. [Google Scholar] [CrossRef] [PubMed]
- Kura, B.; Szeiffova Bacova, B.; Kalocayova, B.; Sykora, M.; Slezak, J. Oxidative Stress-Responsive MicroRNAs in Heart Injury. Int. J. Mol. Sci. 2020, 21, 358. [Google Scholar] [CrossRef]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid. Med. Cell. Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Gu, C.; Draga, D.; Zhou, C.; Su, T.; Zou, C.; Gu, Q.; Lahm, T.; Zheng, Z.; Qiu, Q. miR-590-3p Inhibits Pyroptosis in Diabetic Retinopathy by Targeting NLRP1 and Inactivating the NOX4 Signaling Pathway. Invest. Ophthalmol. Vis. Sci. 2019, 60, 4215–4223. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Jiang, Z.; Bai, H.; Du, Z. miR-100 alleviates the inflammatory damage and apoptosis of H2O2-induced human umbilical vein endothelial cells via inactivation of Notch signaling by targeting MMP9. Vascular 2022, 30, 151–161. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, J.; Yang, X.; Lai, P.; Mou, Y.; Deng, J.; Li, X.; Wang, H.; Liu, X.; Zhou, L.; et al. H2O2 down-regulates SIRT7’s protective role of endothelial premature dysfunction via microRNA-335-5p. Biosci. Rep. 2022, 42, BSR20211775. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Cheng, M.; Yang, Z.; Qiao, L.; Yang, Y.; Deng, Y.; Zhang, C.; Mi, T. AGEs induce endothelial cells senescence and endothelial barrier dysfunction via miR-1-3p/MLCK signaling pathways. Gene 2023, 851, 147030. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Liu, W.; Xu, Q.; Liu, H.L. MicroRNA-486-5p functions as a diagnostic marker for carotid artery stenosis and prevents endothelial dysfunction through inhibiting inflammation and oxidative stress. Bioengineered 2022, 13, 8667–8675. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, W.; Zhang, Y.; Yang, J. MicroRNA-20a protects human aortic endothelial cells from Ox-LDL-induced inflammation through targeting TLR4 and TXNIP signaling. Biomed. Pharmacother. 2018, 103, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Li, P.; Li, J.; He, R.; Cheng, G.; Li, Y. Downregulation of microRNA-34a inhibits oxidized low-density lipoprotein-induced apoptosis and oxidative stress in human umbilical vein endothelial cells. Int. J. Mol. Med. 2018, 42, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Huang, P.; Wu, T.; Chen, L.; Guo, R. Inhibition of miR-214-3p Protects Endothelial Cells from ox-LDL-Induced Damage by Targeting GPX4. Biomed. Res. Int. 2021, 2021, 9919729. [Google Scholar] [CrossRef]
- Wu, J.; Liang, W.; Tian, Y.; Ma, F.; Huang, W.; Jia, Y.; Jiang, Z.; Wu, H. Inhibition of P53/miR-34a improves diabetic endothelial dysfunction via activation of SIRT1. J. Cell. Mol. Med. 2019, 23, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Gong, Z.; Bi, Z. Inhibition of miR-383 suppresses oxidative stress and improves endothelial function by increasing sirtuin 1. Braz. J. Med. Biol. Res. 2020, 53, e8616. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, C.; Wang, Y. miR-126 overexpression attenuates oxygen-glucose deprivation/reperfusion injury by inhibiting oxidative stress and inflammatory response via the activation of SIRT1/Nrf2 signaling pathway in human umbilical vein endothelial cells. Mol. Med. Rep. 2021, 23, 165. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Ren, K.; Liu, S.H.; Li, W.M.; Huang, C.J.; Yang, X.H. MicroRNA-140-5p aggravates hypertension and oxidative stress of atherosclerosis via targeting Nrf2 and Sirt2. Int. J. Mol. Med. 2019, 43, 839–849. [Google Scholar] [CrossRef]
- Avvisato, R.; Mone, P.; Jankauskas, S.S.; Varzideh, F.; Kansakar, U.; Gambardella, J.; De Luca, A.; Matarese, A.; Santulli, G. miR-4432 Targets FGFBP1 in Human Endothelial Cells. Biology 2023, 12, 459. [Google Scholar] [CrossRef]
- Ma, C.; Yang, L.; Gao, Q.; Wang, L. miR-602 Activates NRF2 Antioxidant Pathways to Protect HBMECs from OGD/R-Induced Oxidative Stress via Targeting KEAP1 and HRD1. Dis. Markers 2022, 2022, 6967573. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Lin, A.; Wang, C. Adipocyte-Derived Exosomal LINC00968 Promotes Mouse Retina Microvascular Endothelial Cell Dysfunction in a High-Glucose Environment by Modulating the miR-361-5p/TRAF3 Axis. Horm. Metab. Res. 2023, 55, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, W.; Fan, Z.; Yang, C.; Wang, W.; Xiong, M.; Ma, C.; Yang, J. MicroRNA-24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho-1 signaling pathway. Atherosclerosis 2019, 290, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef]
- Zhan, D.; Zhao, J.; Shi, Q.; Lou, J.; Wang, W. 25-hydroxyvitamin D3 inhibits oxidative stress and ferroptosis in retinal microvascular endothelial cells induced by high glucose through down-regulation of miR-93. BMC Ophthalmol. 2023, 23, 22. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, C.; Liu, Y.; Bai, F.; Tu, T.; Liu, Q. Exosomal miR-27b-3p Derived from Hypoxic Cardiac Microvascular Endothelial Cells Alleviates Rat Myocardial Ischemia/Reperfusion Injury through Inhibiting Oxidative Stress-Induced Pyroptosis via Foxo1/GSDMD Signaling. Oxid. Med. Cell. Longev. 2022, 2022, 8215842. [Google Scholar] [CrossRef]
- Wang, J.M.; Tao, J.; Chen, D.D.; Cai, J.J.; Irani, K.; Wang, Q.; Yuan, H.; Chen, A.F. MicroRNA miR-27b rescues bone marrow-derived angiogenic cell function and accelerates wound healing in type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 99–109. [Google Scholar] [CrossRef]
- Veitch, S.; Njock, M.S.; Chandy, M.; Siraj, M.A.; Chi, L.; Mak, H.; Yu, K.; Rathnakumar, K.; Perez-Romero, C.A.; Chen, Z.; et al. MiR-30 promotes fatty acid beta-oxidation and endothelial cell dysfunction and is a circulating biomarker of coronary microvascular dysfunction in pre-clinical models of diabetes. Cardiovasc. Diabetol. 2022, 21, 31. [Google Scholar] [CrossRef]
- Zhou, Z.; Collado, A.; Sun, C.; Tratsiakovich, Y.; Mahdi, A.; Winter, H.; Chernogubova, E.; Seime, T.; Narayanan, S.; Jiao, T.; et al. Downregulation of Erythrocyte miR-210 Induces Endothelial Dysfunction in Type 2 Diabetes. Diabetes 2022, 71, 285–297. [Google Scholar] [CrossRef]
- Gou, L.; Zhao, L.; Song, W.; Wang, L.; Liu, J.; Zhang, H.; Huang, Y.; Lau, C.W.; Yao, X.; Tian, X.Y.; et al. Inhibition of miR-92a Suppresses Oxidative Stress and Improves Endothelial Function by Upregulating Heme Oxygenase-1 in db/db Mice. Antioxid. Redox Signal. 2018, 28, 358–370. [Google Scholar] [CrossRef]
- Li, H.; Song, D.; Liu, Q.; Li, L.; Sun, X.; Guo, J.; Li, D.; Li, P. miR-351 promotes atherosclerosis in diabetes by inhibiting the ITGB3/PIK3R1/Akt pathway and induces endothelial cell injury and lipid accumulation. Mol. Med. 2022, 28, 120. [Google Scholar] [CrossRef]
- Tang, Z.; Song, J.; Yu, Z.; Cui, K.; Ruan, Y.; Liu, Y.; Wang, T.; Wang, S.; Liu, J.; Yang, J. Inhibition of MicroRNA-92a Improved Erectile Dysfunction in Streptozotocin-Induced Diabetic Rats via Suppressing Oxidative Stress and Endothelial Dysfunction. World J. Men’s Health 2023, 41, 142–154. [Google Scholar] [CrossRef]
- Schulte, C.; Karakas, M.; Zeller, T. microRNAs in cardiovascular disease-clinical application. Clin. Chem. Lab. Med. 2017, 55, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zou, Y.; Zhang, H. MicroRNA-126-5p Facilitates Hypoxia-Induced Vascular Endothelial Cell Injury via HIPK2. Ann. Clin. Lab. Sci. 2022, 52, 918–926. [Google Scholar] [PubMed]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C., IV; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Ghose, S.; Biswas, S. Therapeutic strategies for miRNA delivery to reduce hepatocellular carcinoma. Semin. Cell Dev. Biol. 2022, 124, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Holjencin, C.; Jakymiw, A. MicroRNAs and Their Big Therapeutic Impacts: Delivery Strategies for Cancer Intervention. Cells 2022, 11, 2332. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Yashiro, R. Extracellular Vesicles as Novel Drug-Delivery Systems through Intracellular Communications. Membranes 2022, 12, 550. [Google Scholar] [CrossRef]
- Laggerbauer, B.; Engelhardt, S. MicroRNAs as therapeutic targets in cardiovascular disease. J. Clin. Investig. 2022, 132, e159179. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
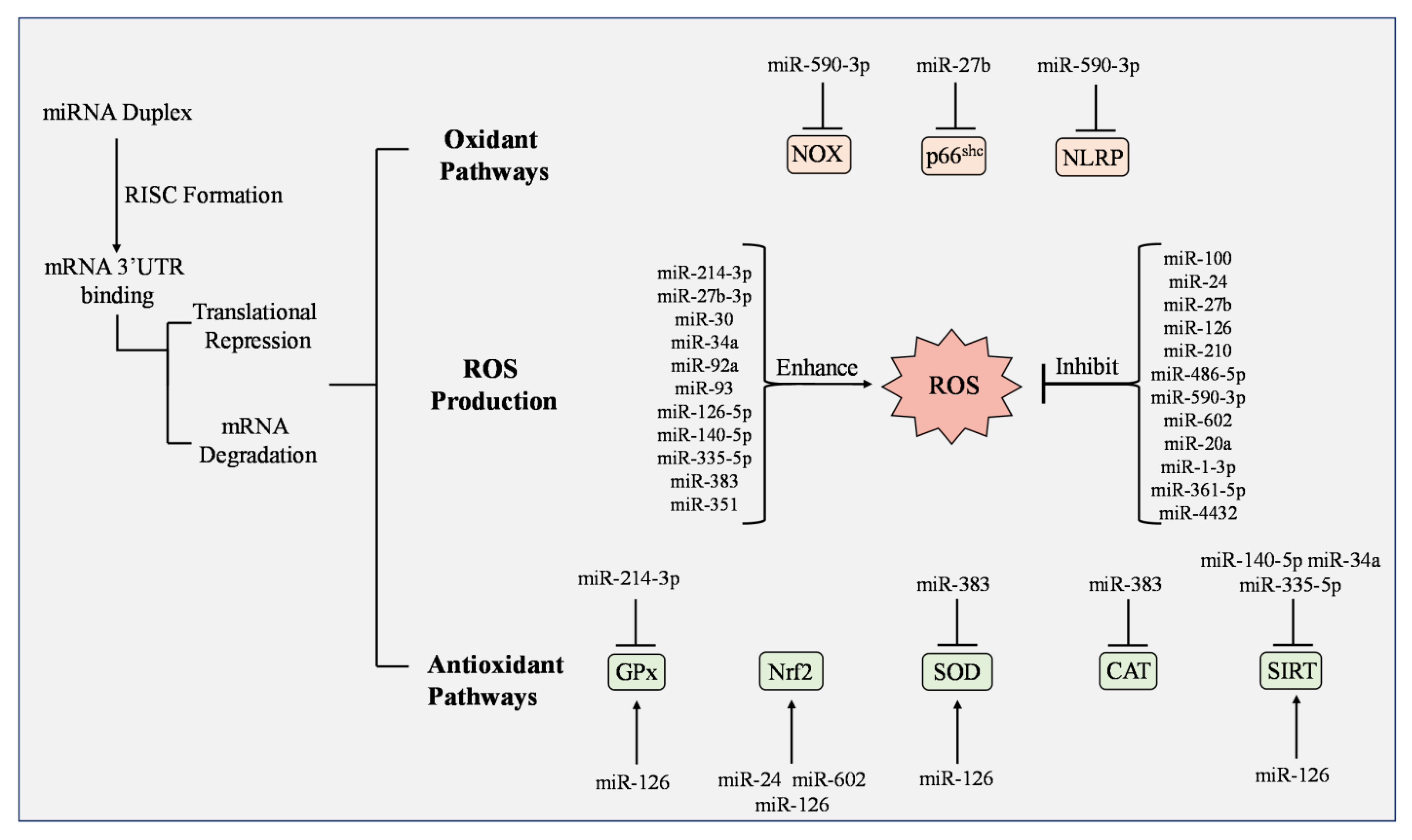{kind=link}
| MicroRNA | Potential Impact on ROS | Cardiovascular/ Metabolic Disorder | Key Experimental Findings | References |
|---|---|---|---|---|
| miR-1-3p | reduces | Advanced glycation end-products (AGEs) | Reduced expression by AGEs impaired EC layer integrity; miR targets and represses myosin light chain kinase | [172] |
| miR-20a | reduces | Atherosclerosis | Ox-LDL reduces miR-20a expression in ECs; overexpressed miR-20a reduced ROS generation under Ox-LDL treatment | [174] |
| miR-24 | reduces | Diabetes Mellitus | Activated the Nrf2/HO-1 signaling pathway | [184] |
| miR-27b | reduces | Diabetes | miR-27b suppresses mitochondrial ROS and p66shc expression, accelerates wound closure, and improves angiogenesis | [188] |
| miR-27b-3p | enhances | Hypoxia | Inhibition reduced oxidative stress | [187] |
| miR-30 | enhances | Diabetes Mellitus | Overexpression increased fatty-acid β-oxidation, ROS generation, and lipid peroxidation; expression of eNOS was also downregulated | [189] |
| miR-34a | enhances | Diabetes Mellitus | Inhibition reduced oxidative stress; inhibition of P53 increased miR-34a expression and reduced SIRT1 expression | [177] |
| miR-34a | enhances | Atherosclerosis | Knockdown protected against Ox-LDL apoptosis and ROS by inhibiting the mitochondrial apoptotic pathway | [175] |
| miR-92a | enhances | Diabetes Mellitus | Inhibition reduced ROS generation | [191] |
| miR-92a | enhances | Diabetes Mellitus Erectile Dysfunction | Inhibition reduced oxidative stress and improved EC dysfunction | [193] |
| miR-93 | enhances | Diabetic Retinopathy | Overexpression inhibited 25 (OH) D3 functions by increasing ROS and upregulating Fe2+ levels | [186] |
| miR-100 | reduces | Nonatherosclerotic inflammatory disease | miR-100 was decreased in H2O2-induced ECs; overexpression attenuated inflammatory response, oxidative stress, and cell apoptosis by inactivating Notch signaling | [169] |
| miR-126 | reduces | Hypoxia | miR-126 mimic and VEGF-plasmid co-transfection improved proliferation, migration, tube-forming ability, and restored AKT phosphorylation | [194] |
| miR-126 | reduces | Ischemia | Overexpression promoted the SIRT1/Nrf2 signaling pathway, attenuated cytotoxicity and apoptosis, decreased ROS generation and malondialdehyde content, and increased superoxide dismutase and glutathione peroxidase activity | [179] |
| miR-126-5p | enhances | Hypoxia | Knockdown suppressed hypoxia-induced cell apoptosis and oxidative stress in ECs | [195] |
| miR-140-5p | enhances | Atherosclerosis | Overexpression led to increased ROS; miR-140-5p regulated Nrf2 and SIRT2 expression | [180] |
| miR-210 | reduces | Diabetes Mellitus Type 2 | Downregulated miR-210 caused EC dysfunction | [190] |
| miR-214-3p | enhances | Atherosclerosis | Ox-LDL increased miR-214-3p expression and decreased GPX4 expression; overexpression of miR decreased GPX4 expression; inhibition of miR reduced ROS levels | [176] |
| miR-335-5p | enhances | Atherosclerosis | Overexpression decreased the SIRT7 expression in ECs and induced ROS overproduction | [170] |
| miR-351 | enhances | Atherosclerosis | Lower levels of miR-351 expression reduced apoptosis, ROS generation, and lipid accumulation in ECs treated with Ox-LDL and high glucose | [192] |
| miR-361-5p | reduces | Diabetes Mellitus Obesity | Targets tumor necrosis factor receptor-associated factor 3 under high-glucose conditions | [183] |
| miR-383 | enhances | Diabetes Mellitus | Suppression reduced ROS generation and elevated CAT and SOD1 activity | [178] |
| miR-4432 | reduces | Hypertension | Targets fibroblast growth factor binding protein 2 | [181] |
| miR-486-5p | reduces | Carotid artery stenosis (CAS) | Overexpression promoted EC proliferation, suppressed cell apoptosis, and reversed the release of ROS and inflammatory factors induced by Ox-LDL | [173] |
| miR-590-3p | reduces | Diabetic Retinopathy | Inhibition upregulated NLRP1, the NOX4/ROS/TXNIP/NLRP3 pathway, and caspase-1. NLRP1 and NOX4 were confirmed as direct target genes of miR-590-3p | [168] |
| miR-602 | reduces | Diabetes Mellitus Type 2 | Reduced ROS levels, increased Nrf2 expression, and enhanced transcription activity of Nrf2/ARE | [182] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minjares, M.; Wu, W.; Wang, J.-M. Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders. Cells 2023, 12, 1341. https://doi.org/10.3390/cells12091341
Minjares M, Wu W, Wang J-M. Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders. Cells. 2023; 12(9):1341. https://doi.org/10.3390/cells12091341
Chicago/Turabian StyleMinjares, Morgan, Wendy Wu, and Jie-Mei Wang. 2023. "Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders" Cells 12, no. 9: 1341. https://doi.org/10.3390/cells12091341
APA StyleMinjares, M., Wu, W., & Wang, J.-M. (2023). Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders. Cells, 12(9), 1341. https://doi.org/10.3390/cells12091341