
Hypercontractile Cardiac Phenotype in Mice with Migraine-Associated Mutation in the Na+,K+-ATPase α2-Isoform
 , , and
, , and 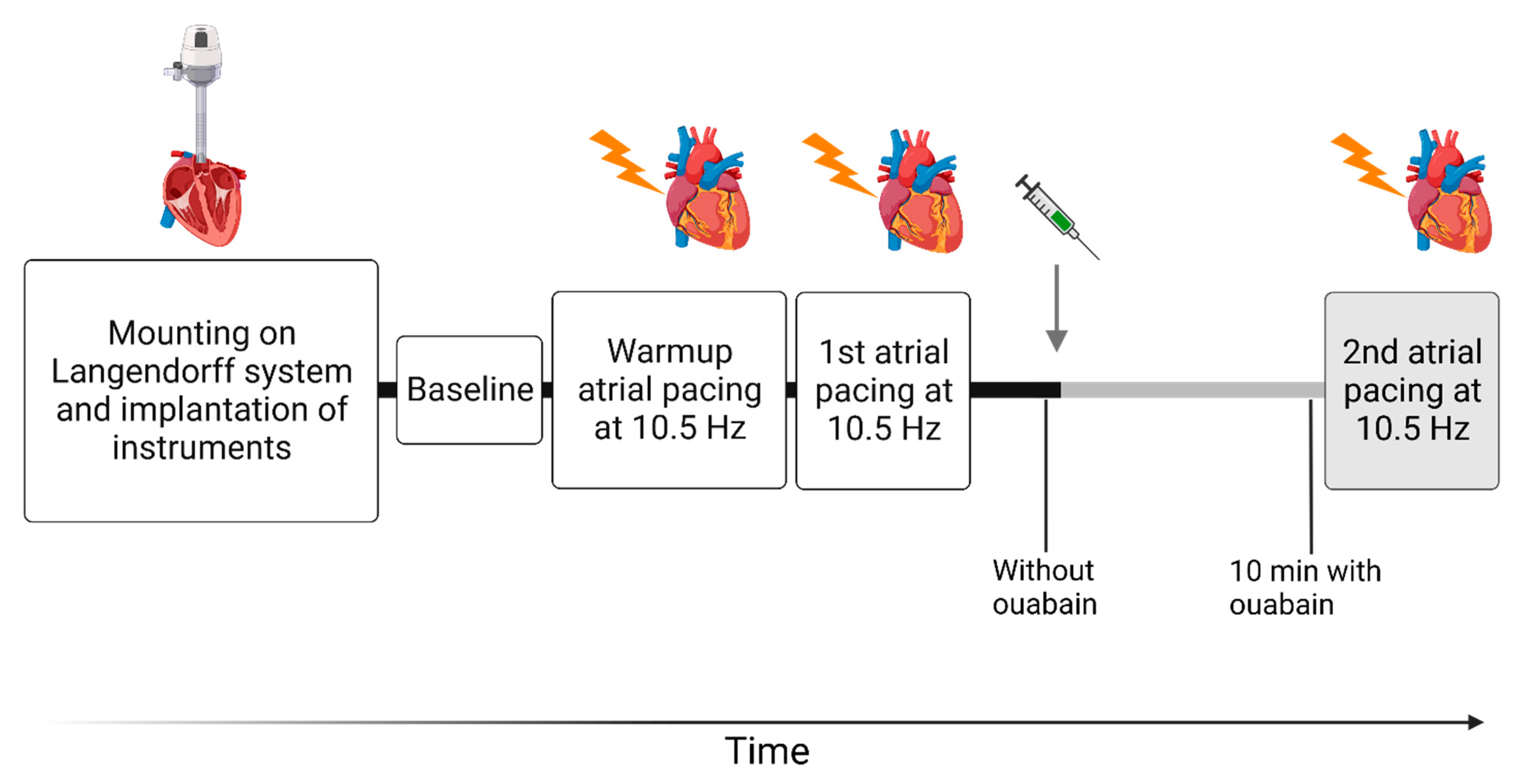{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Isolated Cardiac Perfusions
2.3. Data Acquisition and Calculations
2.4. Stereology
2.5. Statistical Analysis
3. Results
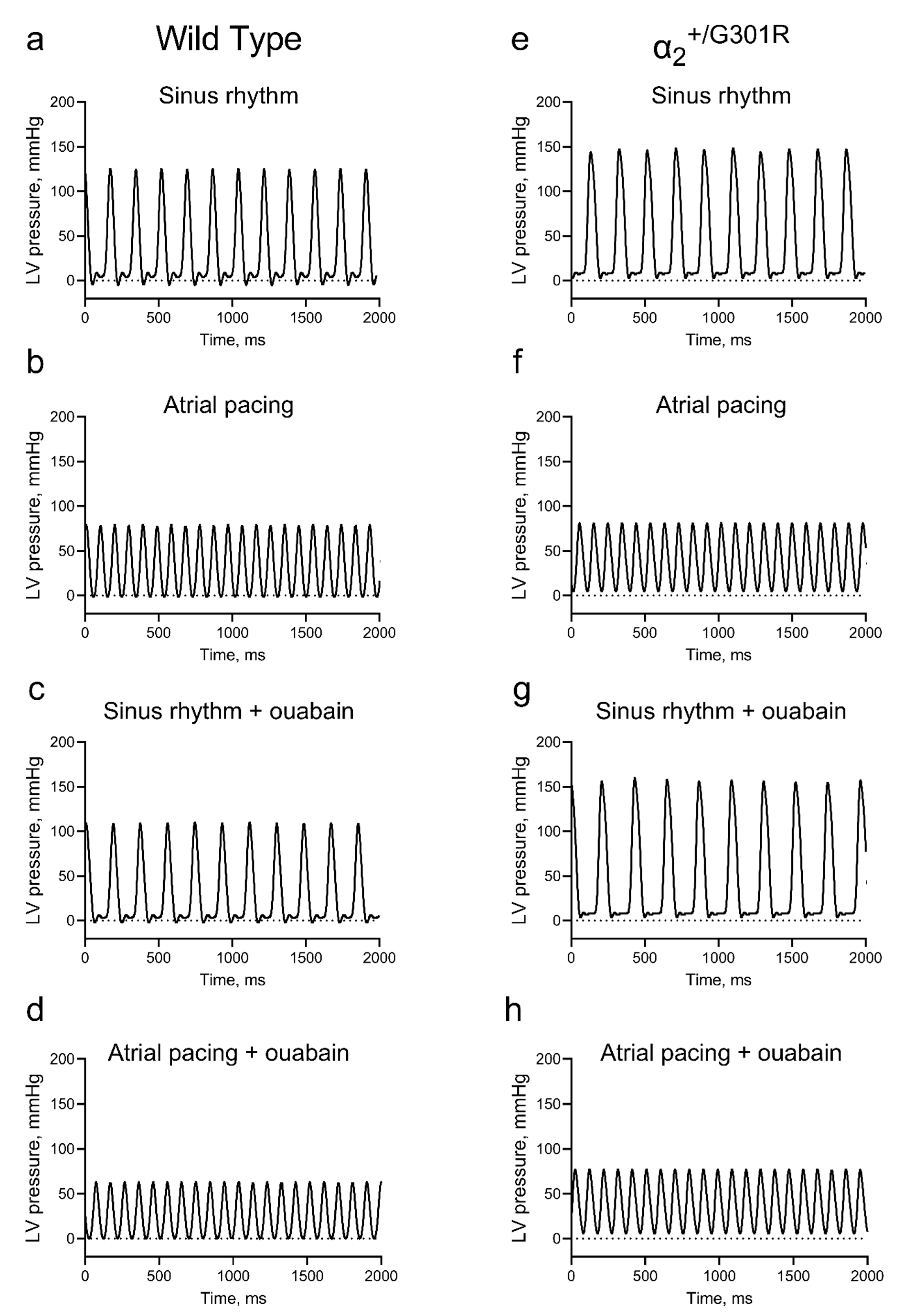
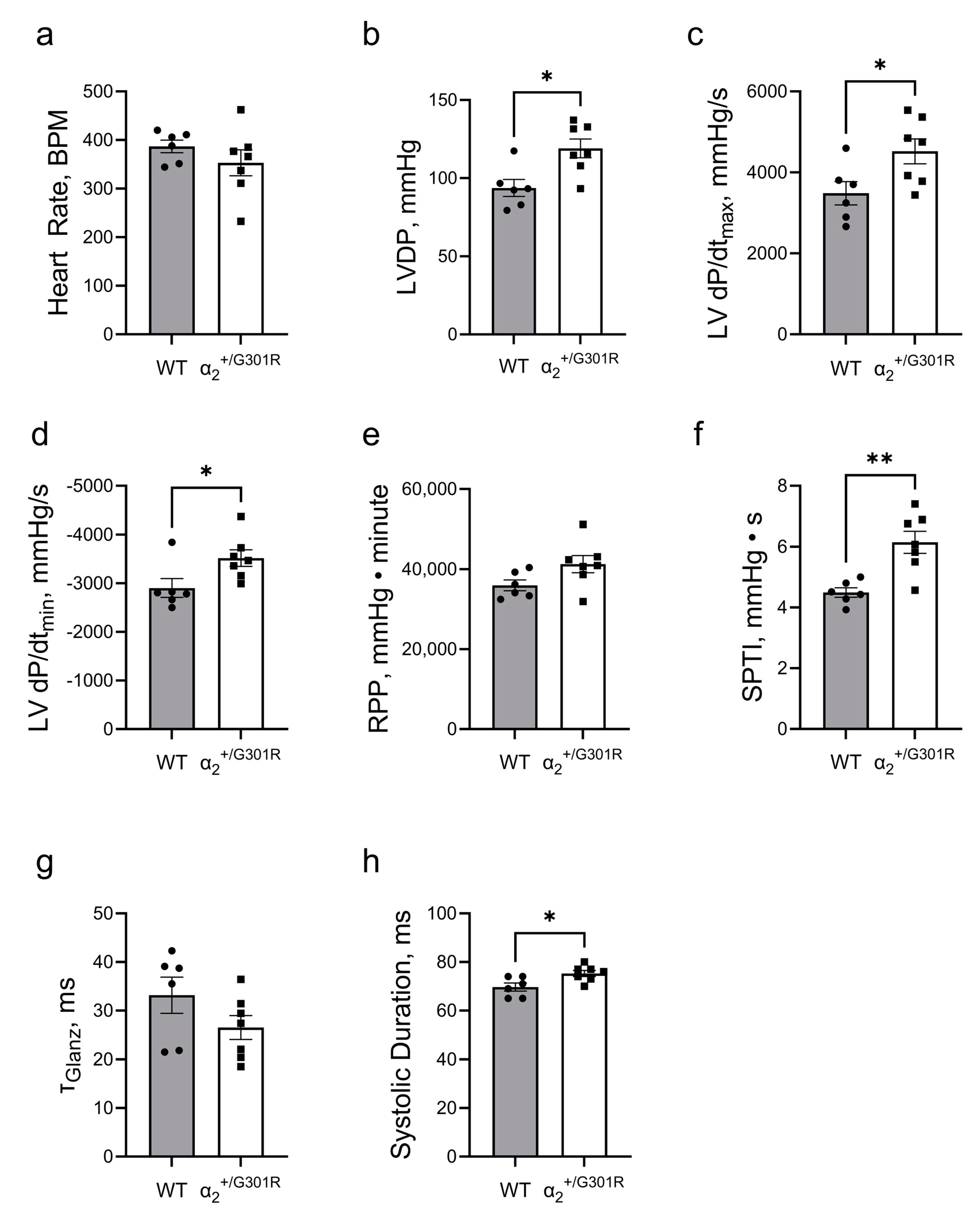
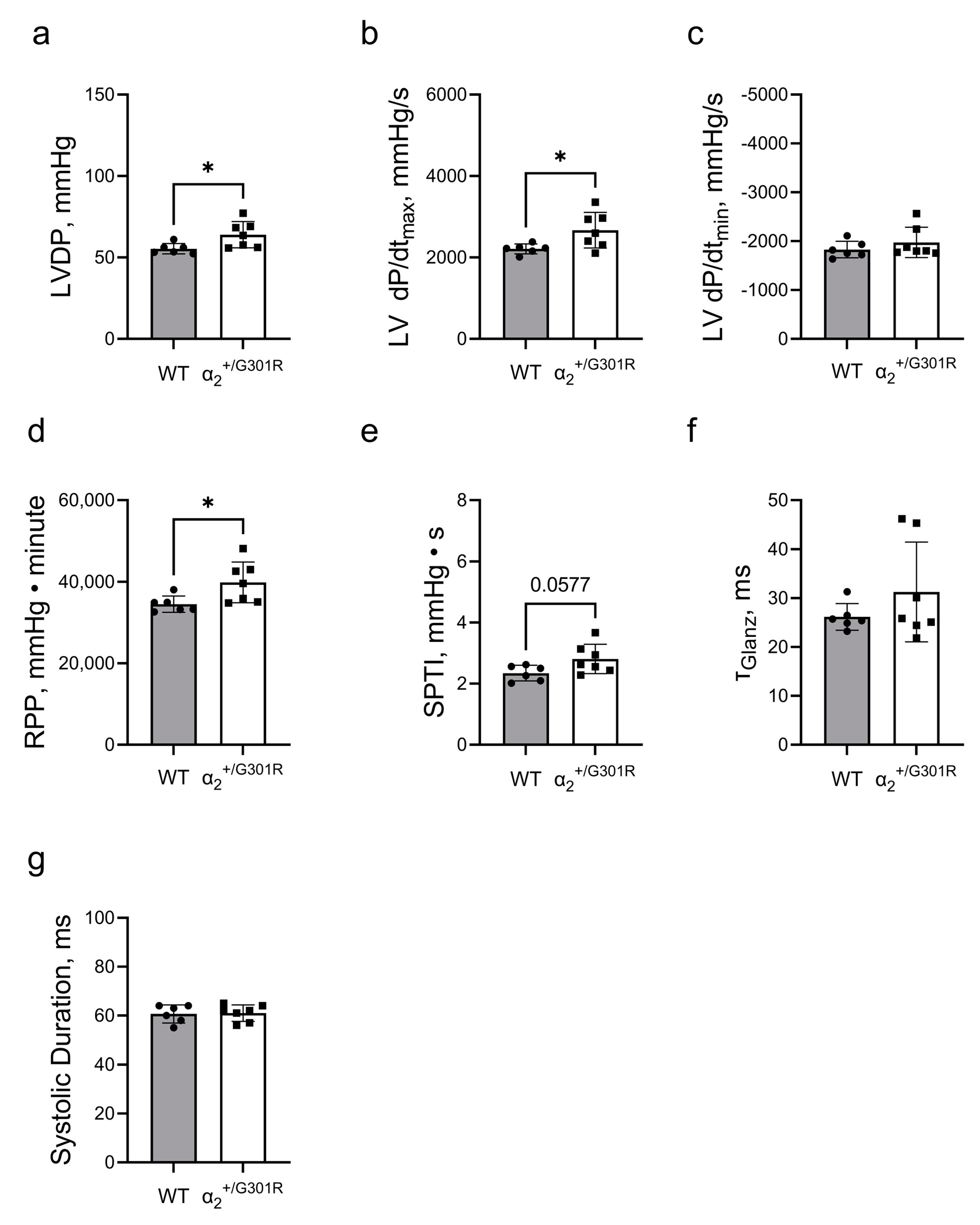
3.1. Isolated Hearts from α2+/G301R Mice Displayed Greater Contractility Than WT Hearts
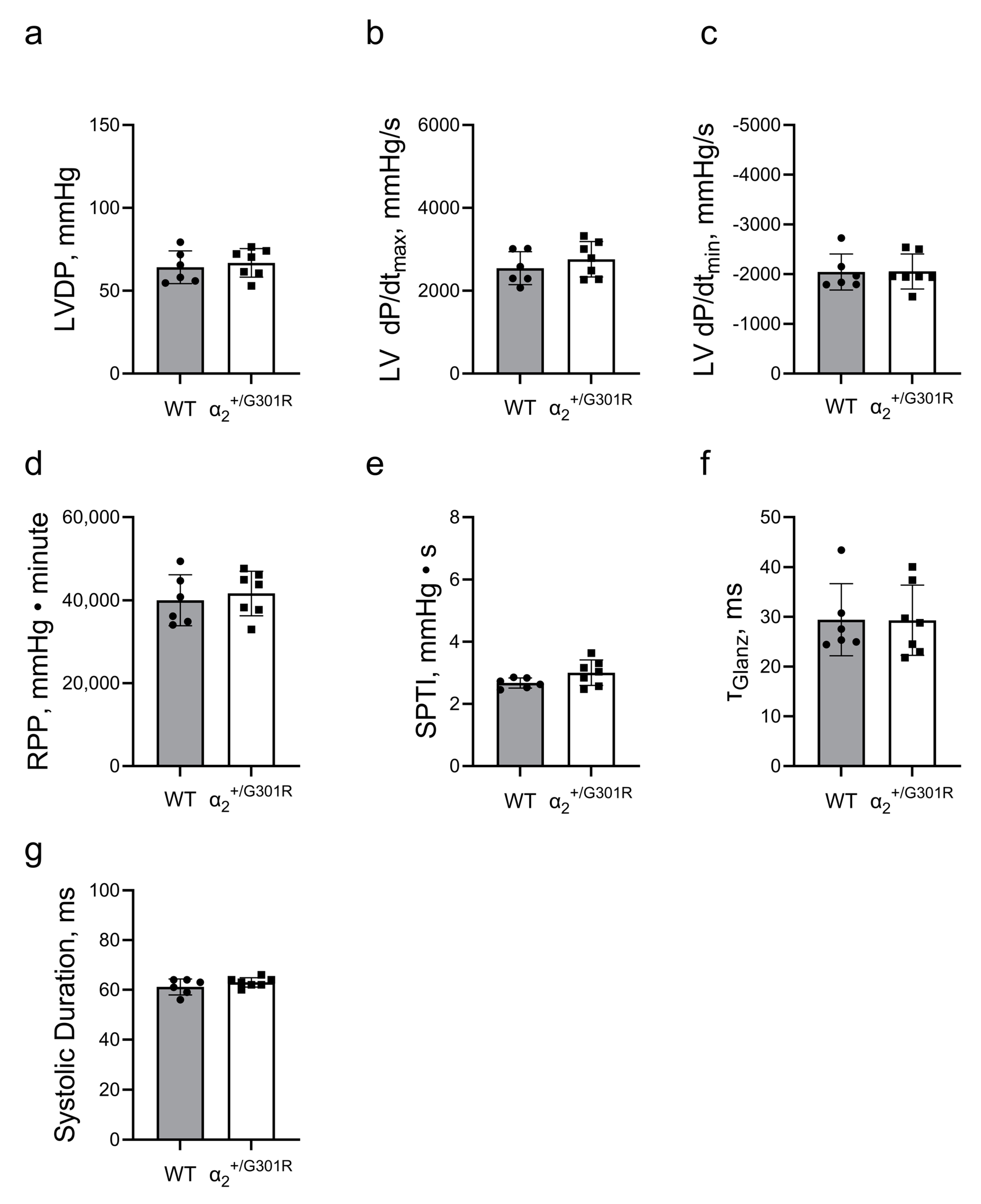
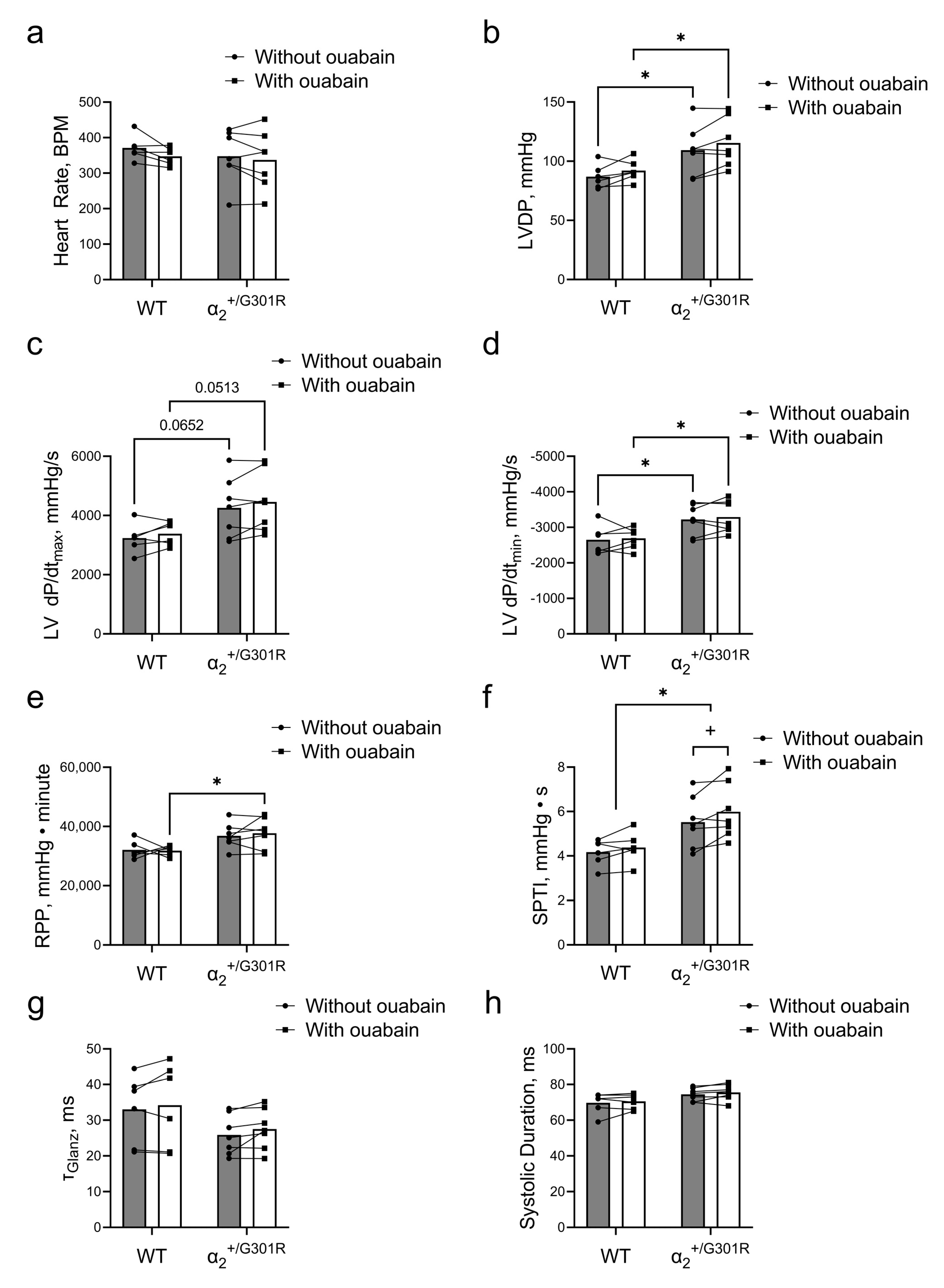
3.2. Ex Vivo Cardiac Function Was Maintained in the Presence of Ouabain during Sinus Rhythm in Both WT and α2+/G301R Hearts
3.3. Contractility during Atrial Pacing Was Augmented in α2+/G301R Hearts in the Presence of Ouabain, and This Was Associated with Increased Systolic Work
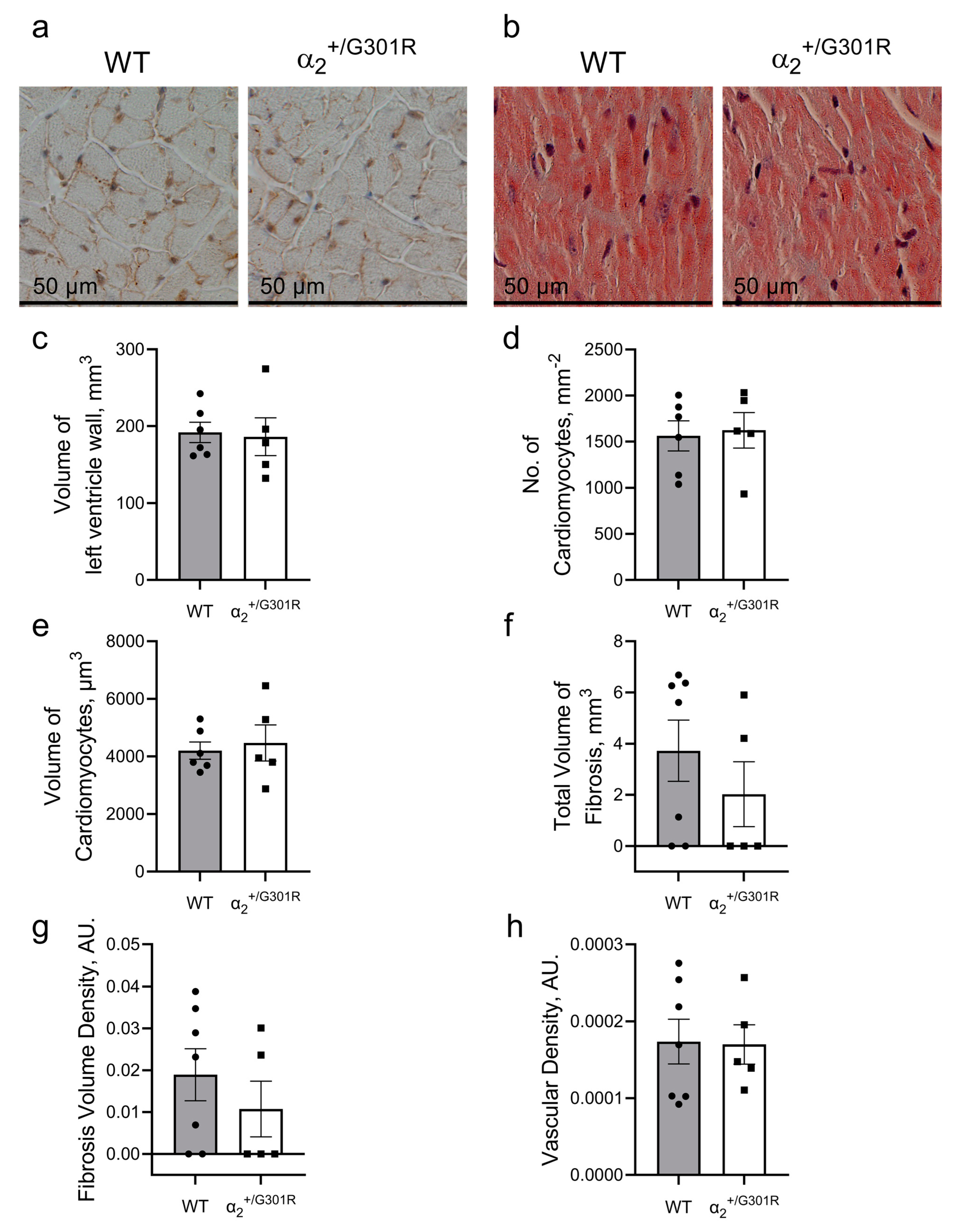
3.4. Contractile Cardiac Phenotype of α2+/G301R Mice Was Not Due to Morphological Changes in the Heart
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mensah George, A.; Roth Gregory, A.; Fuster, V. The Global Burden of Cardiovascular Diseases and Risk Factors. J. Am. Coll. Cardiol. 2019, 74, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- AIRD, W.C. Discovery of the cardiovascular system: From Galen to William Harvey. J. Thromb. Haemost. 2011, 9, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, A.P.; Han, H.C. Biomechanics of Cardiac Function. Compr. Physiol. 2015, 5, 1623–1644. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Overgaard, C.B.; Džavík, V. Inotropes and Vasopressors. Circulation 2008, 118, 1047–1056. [Google Scholar] [CrossRef]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Li, Z.; Langhans, S.A. Transcriptional regulators of Na,K-ATPase subunits. Front. Cell Dev. Biol. 2015, 3, 66. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.B.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/Calcium Exchange: Its Physiological Implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Bers, D.M.; Barry, W.H.; Despa, S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc. Res. 2003, 57, 897–912. [Google Scholar] [CrossRef]
- Weber, C.R.; Piacentino, V.; Houser, S.R.; Bers, D.M. Dynamic Regulation of Sodium/Calcium Exchange Function in Human Heart Failure. Circulation 2003, 108, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Skogestad, J.; Aronsen, J.M. Regulation of Cardiac Contractility by the Alpha 2 Subunit of the Na+/K+-ATPase. Front. Physiol. 2022, 13, 827334. [Google Scholar] [CrossRef] [PubMed]
- Swift, F.; Tovsrud, N.; Enger, U.H.; Sjaastad, I.; Sejersted, O.M. The Na+/K+-ATPase α2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc. Res. 2007, 75, 109–117. [Google Scholar] [CrossRef]
- Despa, S.; Bers, D.M. Functional analysis of Na+/K+-ATPase isoform distribution in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 2007, 293, C321–C327. [Google Scholar] [CrossRef]
- Aronsen, J.M.; Swift, F.; Sejersted, O.M. Cardiac sodium transport and excitation–contraction coupling. J. Mol. Cell. Cardiol. 2013, 61, 11–19. [Google Scholar] [CrossRef]
- Dostanic, I.; Schultz, J.E.J.; Lorenz, J.N.; Lingrel, J.B. The α1 Isoform of Na,K-ATPase Regulates Cardiac Contractility and Functionally Interacts and Co-localizes with the Na/Ca Exchanger in Heart. J. Biol. Chem. 2004, 279, 54053–54061. [Google Scholar] [CrossRef]
- Botelho, A.F.M.; Pierezan, F.; Soto-Blanco, B.; Melo, M.M. A review of cardiac glycosides: Structure, toxicokinetics, clinical signs, diagnosis and antineoplastic potential. Toxicon 2019, 158, 63–68. [Google Scholar] [CrossRef]
- Altamirano, J.; Li, Y.; DeSantiago, J.; Piacentino, V., 3rd; Houser, S.R.; Bers, D.M. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 2006, 575, 845–854. [Google Scholar] [CrossRef]
- Price, E.M.; Lingrel, J.B. Structure-function relationships in the Na,K-ATPase alpha subunit: Site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry 1988, 27, 8400–8408. [Google Scholar] [CrossRef]
- Obrien, W.J.; Lingrel, J.B.; Wallick, E.T. Ouabain Binding Kinetics of the Rat Alpha Two and Alpha Three Isoforms of the Sodium-Potassium Adenosine Triphosphate. Arch. Biochem. Biophys. 1994, 310, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Lingrel, J.B.; Bers, D.M. Na+/K+-ATPase α2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc. Res. 2012, 95, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, M.; Ursu, S.; Lehmann-Horn, F.; Liana, V.; Giovanni, A.; Paola, G.; Frontali, M.; Jurkat-Rott, K. A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 2004, 5, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Santoro, L.; Manganelli, F.; Fortunato, M.R.; Soldovieri, M.V.; Ambrosino, P.; Iodice, R.; Pisciotta, C.; Tessa, A.; Santorelli, F.; Taglialatela, M. A new Italian FHM2 family: Clinical aspects and functional analysis of the disease-associated mutation. Cephalalgia 2011, 31, 808–819. [Google Scholar] [CrossRef]
- Stoica, A.; Larsen, B.R.; Assentoft, M.; Holm, R.; Holt, L.M.; Vilhardt, F.; Vilsen, B.; Lykke-Hartmann, K.; Olsen, M.L.; MacAulay, N. The α2β2 isoform combination dominates the astrocytic Na+/K+-ATPase activity and is rendered nonfunctional by the α2.G301R familial hemiplegic migraine type 2-associated mutation. Glia 2017, 65, 1777–1793. [Google Scholar] [CrossRef]
- Staehr, C.; Rohde, P.D.; Krarup, N.T.; Ringgaard, S.; Laustsen, C.; Johnsen, J.; Nielsen, R.; Beck, H.C.; Morth, J.P.; Lykke-Hartmann, K.; et al. Migraine Associated Mutation in the Na,K-ATPase Leads to Disturbances in Cardiac Metabolism and Reduced Cardiac Function. J. Am. Heart Assoc. 2022, 11, e021814. [Google Scholar] [CrossRef]
- Rajanathan, R.; Pedersen, T.M.; Guldbrandsen, H.O.; Olesen, L.F.; Thomsen, M.B.; Bøtker, H.E.; Matchkov, V.V. Augmented Ouabain-Induced Vascular Response Reduces Cardiac Efficiency in Mice with Migraine-Associated Mutation in the Na+, K+-ATPase α2-Isoform. Biomedicines 2023, 11, 344. [Google Scholar]
- Salahudeen, M.S.; Nishtala, P.S. An overview of pharmacodynamic modelling, ligand-binding approach and its application in clinical practice. Saudi Pharm. J. 2017, 25, 165–175. [Google Scholar] [CrossRef]
- Buchwald, P. A Receptor Model With Binding Affinity, Activation Efficacy, and Signal Amplification Parameters for Complex Fractional Response Versus Occupancy Data. Front. Pharm. 2019, 10, 605. [Google Scholar] [CrossRef]
- Bøttger, P.; Glerup, S.; Gesslein, B.; Illarionova, N.B.; Isaksen, T.J.; Heuck, A.; Clausen, B.H.; Füchtbauer, E.M.; Gramsbergen, J.B.; Gunnarson, E.; et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016, 6, 22047. [Google Scholar] [CrossRef]
- Ikeda, K.; Onaka, T.; Yamakado, M.; Nakai, J.; Ishikawa, T.-o.; Taketo, M.M.; Kawakami, K. Degeneration of the Amygdala/Piriform Cortex and Enhanced Fear/Anxiety Behaviors in Sodium Pump α2 Subunit (Atp1a2)-Deficient Mice. J. Neurosci. 2003, 23, 4667–4676. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Onimaru, H.; Yamada, J.; Inoue, K.; Ueno, S.; Onaka, T.; Toyoda, H.; Arata, A.; Ishikawa, T.-o.; Taketo, M.M.; et al. Malfunction of Respiratory-Related Neuronal Activity in Na+, K+-ATPase α2 Subunit-Deficient Mice Is Attributable to Abnormal Cl− Homeostasis in Brainstem Neurons. J. Neurosci. 2004, 24, 10693–10701. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Okada, T. Langendorff Perfusion Method as an Ex Vivo Model to Evaluate Heart Function in Rats. Methods Mol. Biol. 2018, 1816, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Tian, R. Assessment of cardiac function and energetics in isolated mouse hearts using 31P NMR spectroscopy. J. Vis. Exp. JoVE 2010, 42, e2069. [Google Scholar] [CrossRef]
- Saupe, K.W.; Spindler, M.; Tian, R.; Ingwall, J.S. Impaired Cardiac Energetics in Mice Lacking Muscle-Specific Isoenzymes of Creatine Kinase. Circ. Res. 1998, 82, 898–907. [Google Scholar] [CrossRef]
- Dorn, G.W. Tension-Time Integrals and Genetic Cardiomyopathy: The Force Is with You. Cell 2016, 165, 1049–1050. [Google Scholar] [CrossRef]
- Klawitter, P.F.; Clanton, T.L. Tension-time index, fatigue, and energetics in isolated rat diaphragm: A new experimental model. J. Appl. Physiol. 2004, 96, 89–95. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Buckberg, G.D. The myocardial oxygen supply:demand index revisited. J. Am. Heart Assoc. 2014, 3, e000285. [Google Scholar] [CrossRef]
- Morimont, P.; Lambermont, B.; Desaive, T.; Janssen, N.; Chase, G.; D’Orio, V. Arterial dP/dtmax accurately reflects left ventricular contractility during shock when adequate vascular filling is achieved. BMC Cardiovasc. Disord. 2012, 12, 13. [Google Scholar] [CrossRef]
- Hamlin, R.L.; del Rio, C. dP/dtmax—A measure of ‘baroinometry’. J. Pharmacol. Toxicol. Methods 2012, 66, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.M.; Edgett, B.A.; Huber, J.S.; Platt, M.J.; Eberl, H.J.; Lutchmedial, S.; Brunt, K.R.; Simpson, J.A. Hemodynamic assessment of diastolic function for experimental models. Am. J. Physiol.-Heart Circ. Physiol. 2020, 318, H1139–H1158. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.; Axelsen, J.B.; Ringgaard, S.; Nyengaard, J.R.; Hyldebrandt, J.A.; Bogaard, H.J.; de Man, F.S.; Nielsen-Kudsk, J.E.; Andersen, A. Effects of combined angiotensin II receptor antagonism and neprilysin inhibition in experimental pulmonary hypertension and right ventricular failure. Int. J. Cardiol. 2019, 293, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.; Nyengaard, J.R. Design-based stereological estimation of the total number of cardiac myocytes in histological sections. Basic Res. Cardiol. 2005, 100, 311–319. [Google Scholar] [CrossRef]
- Muhlfeld, C.; Nyengaard, J.R.; Mayhew, T.M. A review of state-of-the-art stereology for better quantitative 3D morphology in cardiac research. Cardiovasc. Pathol. 2010, 19, 65–82. [Google Scholar] [CrossRef]
- Bruel, A.; Oxlund, H.; Nyengaard, J.R. The total length of myocytes and capillaries, and total number of myocyte nuclei in the rat heart are time-dependently increased by growth hormone. Growth Horm. IGF Res. 2005, 15, 256–264. [Google Scholar] [CrossRef]
- Staehr, C.; Hangaard, L.; Bouzinova, E.V.; Kim, S.; Rajanathan, R.; Boegh Jessen, P.; Luque, N.; Xie, Z.; Lykke-Hartmann, K.; Sandow, S.L.; et al. Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation. J. Cereb. Blood Flow Metab. 2019, 39, 1570–1587. [Google Scholar] [CrossRef]
- Correll, R.N.; Eder, P.; Burr, A.R.; Despa, S.; Davis, J.; Bers, D.M.; Molkentin, J.D. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 2014, 114, 249–256. [Google Scholar] [CrossRef]
- Moseley, A.E.; Huddleson, J.P.; Bohanan, C.S.; James, P.F.; Lorenz, J.N.; Aronow, B.J.; Lingrel, J.B. Genetic profiling reveals global changes in multiple biological pathways in the hearts of Na, K-ATPase alpha 1 isoform haploinsufficient mice. Cell. Physiol. Biochem. 2005, 15, 145–158. [Google Scholar] [CrossRef]
- Bers, D.M.; Despa, S. Cardiac Myocytes Ca2+ and Na+ Regulation in Normal and Failing Hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef]
- James, P.F.; Grupp, I.L.; Grupp, G.; Woo, A.L.; Askew, G.R.; Croyle, M.L.; Walsh, R.A.; Lingrel, J.B. Identification of a Specific Role for the Na,K-ATPase α2 Isoform as a Regulator of Calcium in the Heart. Mol. Cell 1999, 3, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, N. Cardiac work and efficiency. Cardiovasc. Res. 2000, 48, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Salvi, P.; Parati, G. Aortic stiffness and myocardial ischemia. J. Hypertens. 2015, 33, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Kobe, J.; Mishra, N.; Arya, V.K.; Al-Moustadi, W.; Nates, W.; Kumar, B. Cardiac output monitoring: Technology and choice. Ann. Card. Anaesth. 2019, 22, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef]
- Antohi, E.-L.; Chioncel, O.; Mihaileanu, S. Overcoming the Limits of Ejection Fraction and Ventricular-Arterial Coupling in Heart Failure. Front. Cardiovasc. Med. 2022, 8, 2170. [Google Scholar] [CrossRef]
- Illarionava, N.B.; Brismar, H.; Aperia, A.; Gunnarson, E. Role of Na,K-ATPase α1 and α2 Isoforms in the Support of Astrocyte Glutamate Uptake. PLoS ONE 2014, 9, e98469. [Google Scholar] [CrossRef]
- Mohammadi, K.; Liu, L.; Tian, J.; Kometiani, P.; Xie, Z.; Askari, A. Positive inotropic effect of ouabain on isolated heart is accompanied by activation of signal pathways that link Na+/K+-ATPase to ERK1/2. J. Cardiovasc. Pharm. 2003, 41, 609–614. [Google Scholar] [CrossRef]
- Aksentijević, D.; Karlstaedt, A.; Basalay, M.V.; O’Brien, B.A.; Sanchez-Tatay, D.; Eminaga, S.; Thakker, A.; Tennant, D.A.; Fuller, W.; Eykyn, T.R.; et al. Intracellular sodium elevation reprograms cardiac metabolism. Nat. Commun. 2020, 11, 4337. [Google Scholar] [CrossRef]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [CrossRef]
- Xie, Z.; Askari, A. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Bouzinova, E.V.; Hangaard, L.; Staehr, C.; Mazur, A.; Ferreira, A.; Chibalin, A.V.; Sandow, S.L.; Xie, Z.; Aalkjaer, C.; Matchkov, V.V. The α2 isoform Na,K-ATPase modulates contraction of rat mesenteric small artery via cSrc-dependent Ca(2+) sensitization. Acta Physiol. 2018, 224, e13059. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.A.; Isaksen, T.J.; Heuck, A.; Scavone, C.; Lykke-Hartmann, K. The α2 Na+/K+-ATPase isoform mediates LPS-induced neuroinflammation. Sci. Rep. 2020, 10, 14180. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.M.L. Myocardial contraction-relaxation coupling. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H1741–H1749. [Google Scholar] [CrossRef]
- Borlaug, B.A.; Kass, D.A. Mechanisms of Diastolic Dysfunction in Heart Failure. Trends Cardiovasc. Med. 2006, 16, 273–279. [Google Scholar] [CrossRef]
- Chatterjee, K. Pathophysiology of Systolic and Diastolic Heart Failure. Med. Clin. N. Am. 2012, 96, 891–899. [Google Scholar] [CrossRef]
- Fazlollahi, F.; Santini Gonzalez, J.J.; Repas, S.J.; Canan, B.D.; Billman, G.E.; Janssen, P.M.L. Contraction-relaxation coupling is unaltered by exercise training and infarction in isolated canine myocardium. J. Gen. Physiol. 2021, 153, e202012829. [Google Scholar] [CrossRef]
- Hiranandani, N.; Raman, S.; Kalyanasundaram, A.; Periasamy, M.; Janssen, P.M.L. Frequency-dependent contractile strength in mice over- and underexpressing the sarco(endo)plasmic reticulum calcium-ATPase. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, R30–R36. [Google Scholar] [CrossRef]
- Ramanathan, T.; Skinner, H. Coronary blood flow. Contin. Educ. Anaesth. Crit. Care Pain 2005, 5, 61–64. [Google Scholar] [CrossRef]
- Jeong, E.M.; Dudley, S.C., Jr. Diastolic dysfunction. Circ. J. 2015, 79, 470–477. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.Q.; Liu, F.Y.; Guo, Z.; An, P.; Wang, M.Y.; Yang, Z.; Fan, D.; Tang, Q.Z. Mitochondria in Pathological Cardiac Hypertrophy Research and Therapy. Front. Cardiovasc. Med. 2021, 8, 822969. [Google Scholar] [CrossRef] [PubMed]
- How, O.-J.; Aasum, E.; Severson, D.L.; Chan, W.Y.A.; Essop, M.F.; Larsen, T.S. Increased Myocardial Oxygen Consumption Reduces Cardiac Efficiency in Diabetic Mice. Diabetes 2006, 55, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Biondo, E.D.; Spontarelli, K.; Ababioh, G.; Méndez, L.; Artigas, P. Diseases caused by mutations in the Na+/K+ pump α1 gene ATP1A1. Am. J. Physiol.-Cell Physiol. 2021, 321, C394–C408. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J.; Arystarkhova, E.; Penniston, J.T.; Swoboda, K.J.; Brashear, A.; Ozelius, L.J. Genotype-structure-phenotype relationships diverge in paralogs ATP1A1, ATP1A2, and ATP1A3. Neurol. Genet. 2019, 5, e303. [Google Scholar] [CrossRef]
- Pietrobon, D. Familial hemiplegic migraine. Neurotherapeutics 2007, 4, 274–284. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef]
- Sabbah, H.N. Silent disease progression in clinically stable heart failure. Eur. J. Heart Fail. 2017, 19, 469–478. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajanathan, R.; Riera, C.V.i.; Pedersen, T.M.; Staehr, C.; Bouzinova, E.V.; Nyengaard, J.R.; Thomsen, M.B.; Bøtker, H.E.; Matchkov, V.V. Hypercontractile Cardiac Phenotype in Mice with Migraine-Associated Mutation in the Na+,K+-ATPase α2-Isoform. Cells 2023, 12, 1108. https://doi.org/10.3390/cells12081108
Rajanathan R, Riera CVi, Pedersen TM, Staehr C, Bouzinova EV, Nyengaard JR, Thomsen MB, Bøtker HE, Matchkov VV. Hypercontractile Cardiac Phenotype in Mice with Migraine-Associated Mutation in the Na+,K+-ATPase α2-Isoform. Cells. 2023; 12(8):1108. https://doi.org/10.3390/cells12081108
Chicago/Turabian StyleRajanathan, Rajkumar, Clàudia Vilaseca i Riera, Tina Myhre Pedersen, Christian Staehr, Elena V. Bouzinova, Jens Randel Nyengaard, Morten B. Thomsen, Hans Erik Bøtker, and Vladimir V. Matchkov. 2023. "Hypercontractile Cardiac Phenotype in Mice with Migraine-Associated Mutation in the Na+,K+-ATPase α2-Isoform" Cells 12, no. 8: 1108. https://doi.org/10.3390/cells12081108
APA StyleRajanathan, R., Riera, C. V. i., Pedersen, T. M., Staehr, C., Bouzinova, E. V., Nyengaard, J. R., Thomsen, M. B., Bøtker, H. E., & Matchkov, V. V. (2023). Hypercontractile Cardiac Phenotype in Mice with Migraine-Associated Mutation in the Na+,K+-ATPase α2-Isoform. Cells, 12(8), 1108. https://doi.org/10.3390/cells12081108