
A Beginner’s Guide to Cell Culture: Practical Advice for Preventing Needless Problems



Abstract
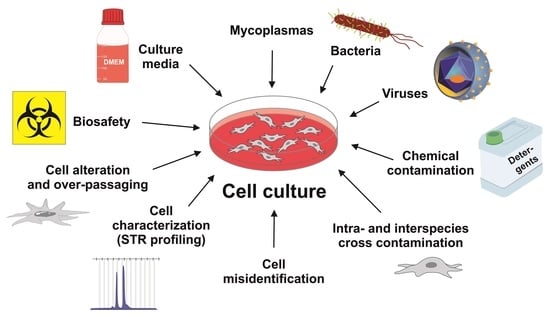
1. Introduction
2. Classification of Cell Culture Types
3. Culture Media
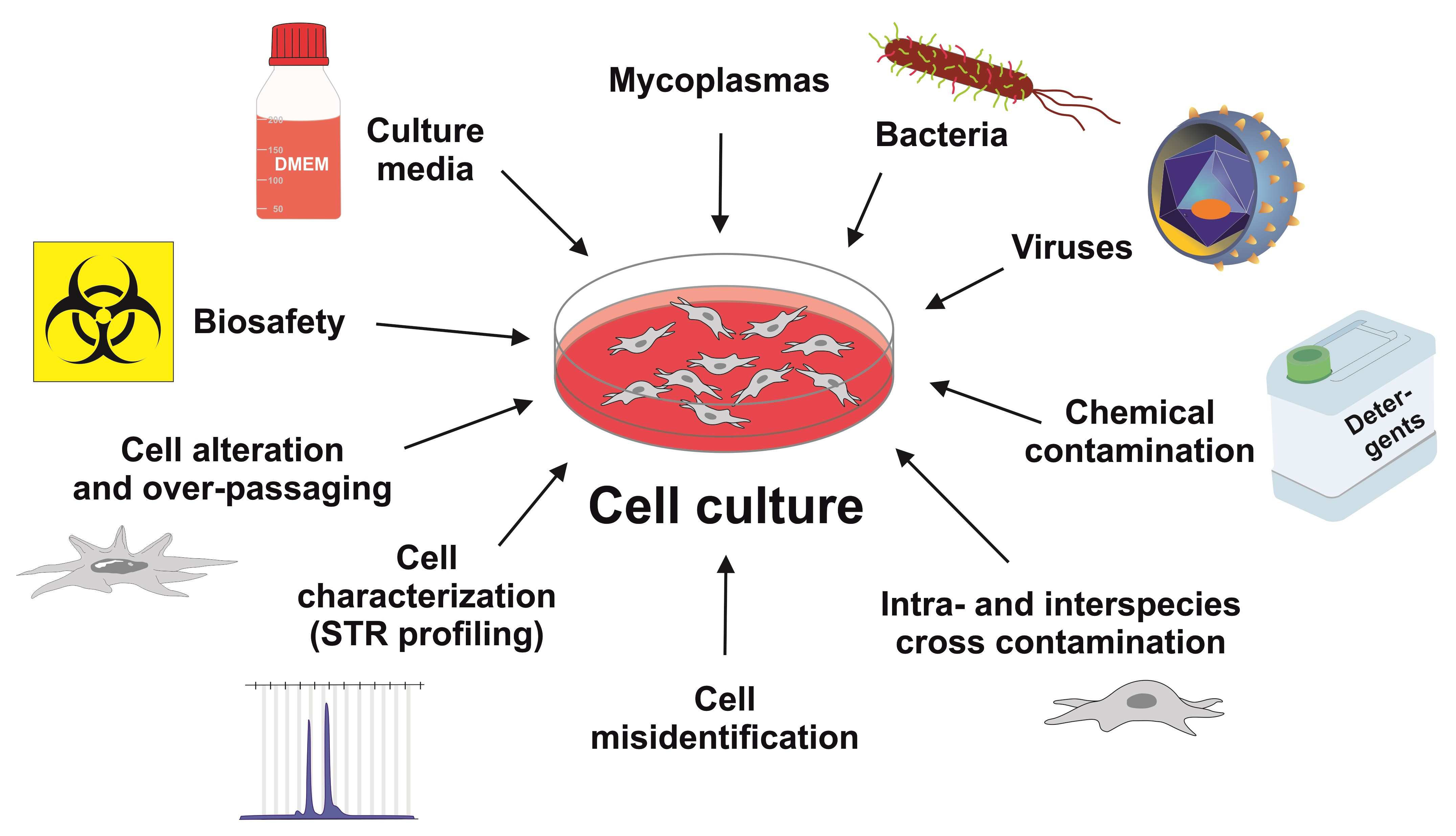
4. Phenol Red
5. Cell Contamination
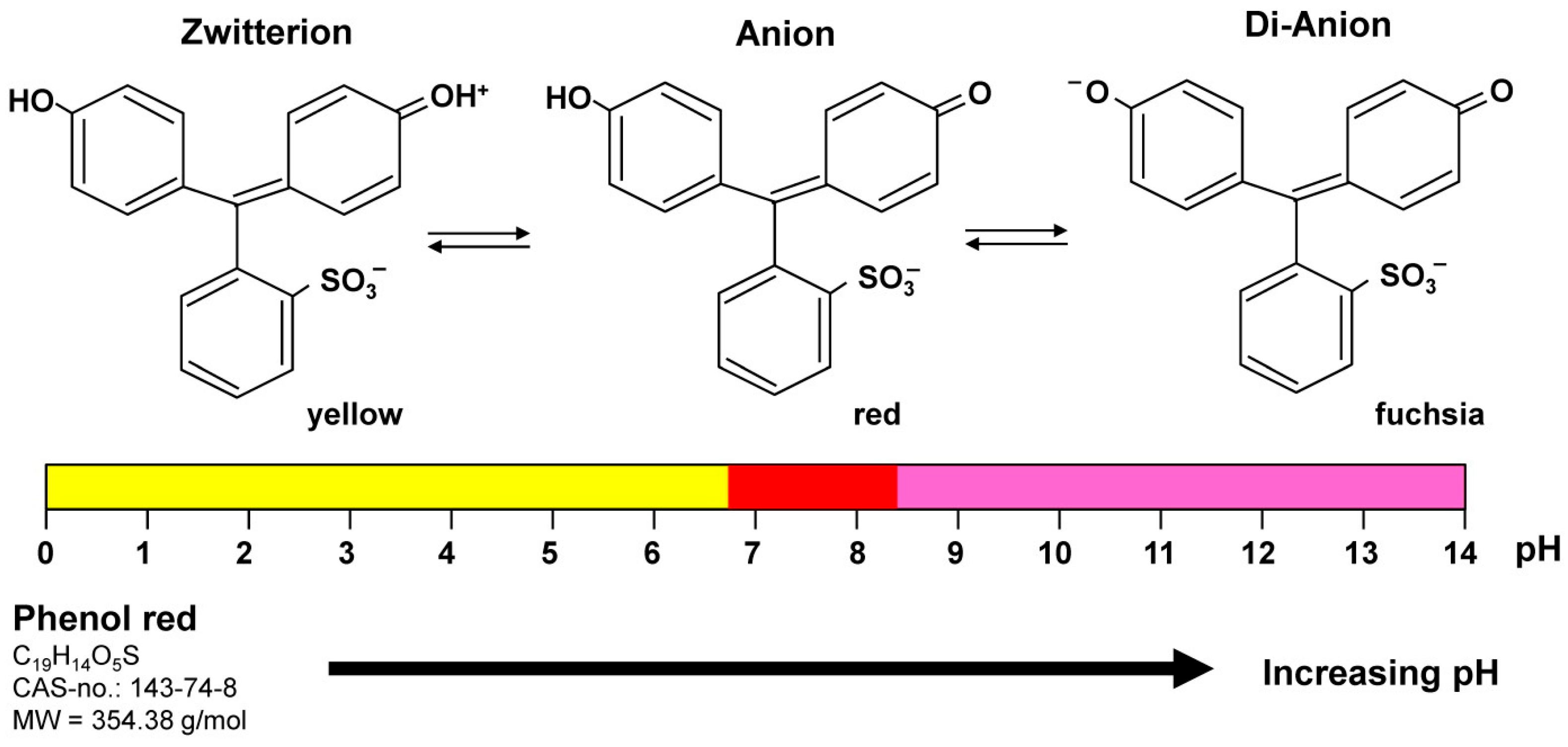
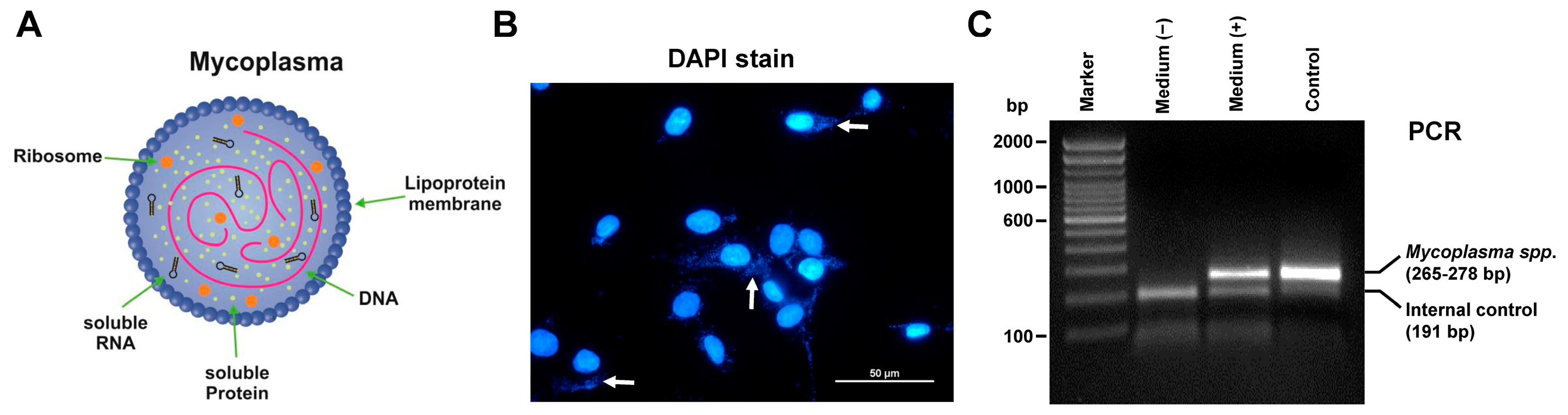
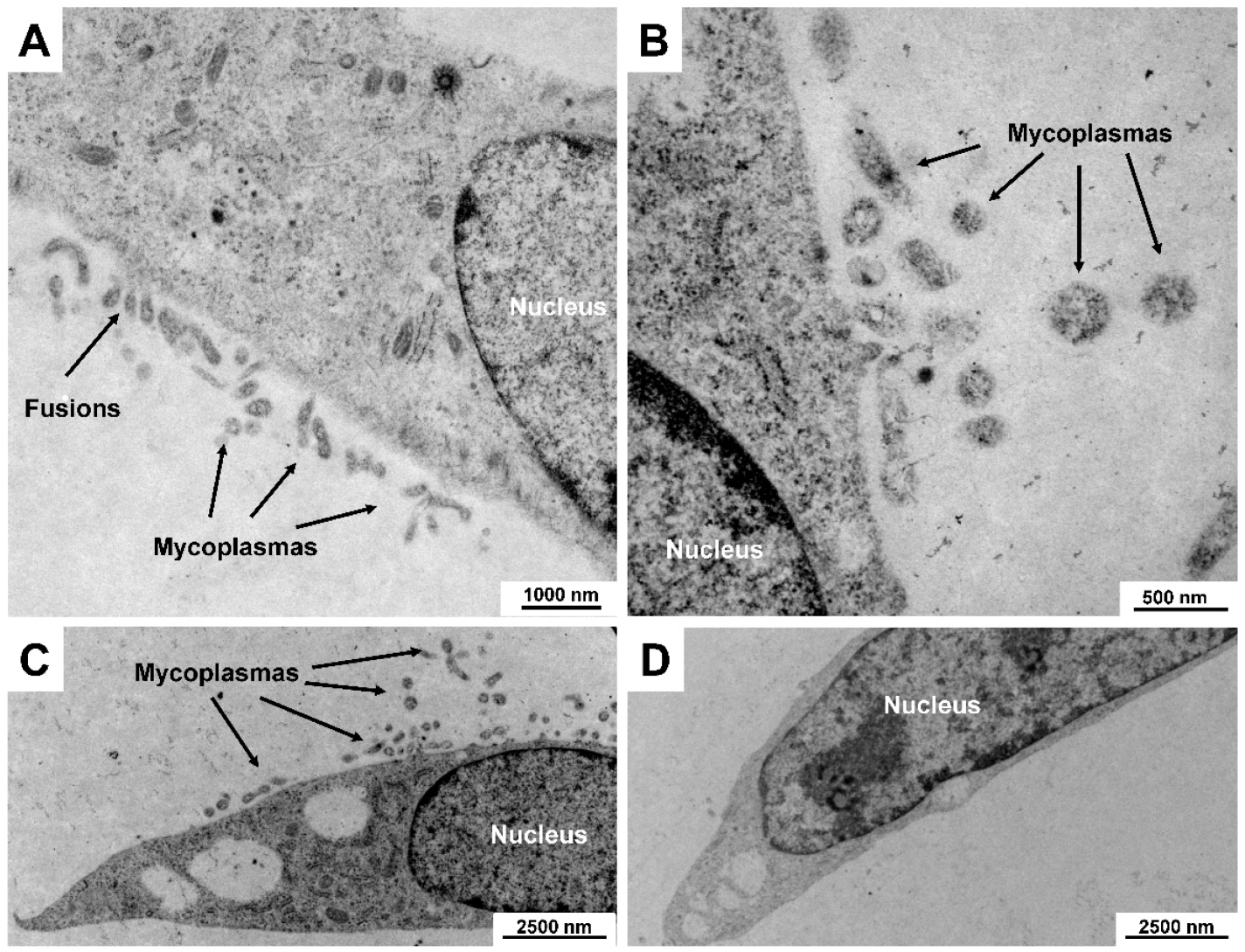
5.1. Mycoplasma Contamination
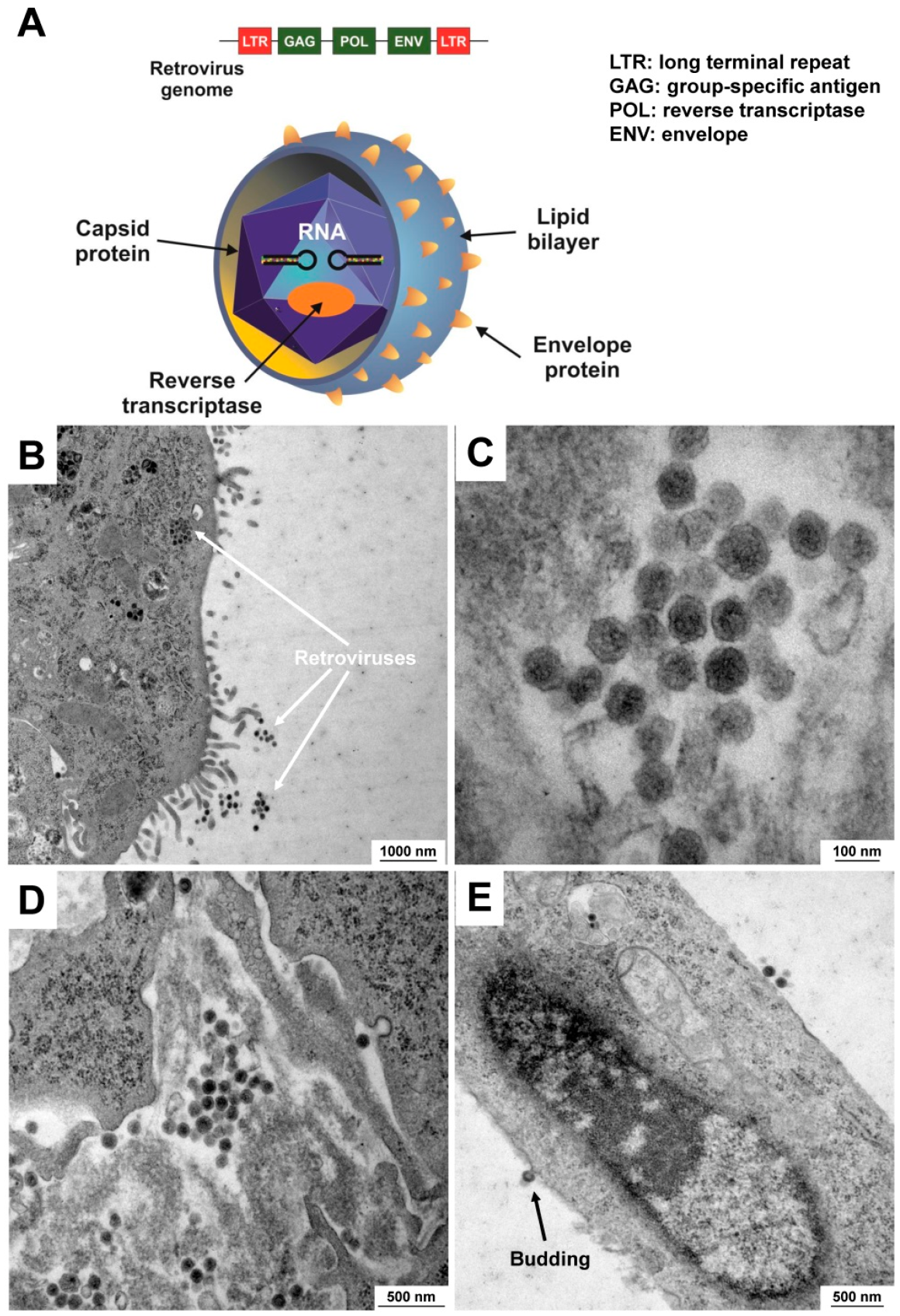
5.2. Contamination with Viruses
5.3. Chemical Contamination
5.4. Inter- and Intra-Species Cross-Contamination
5.5. Cell Misidentification
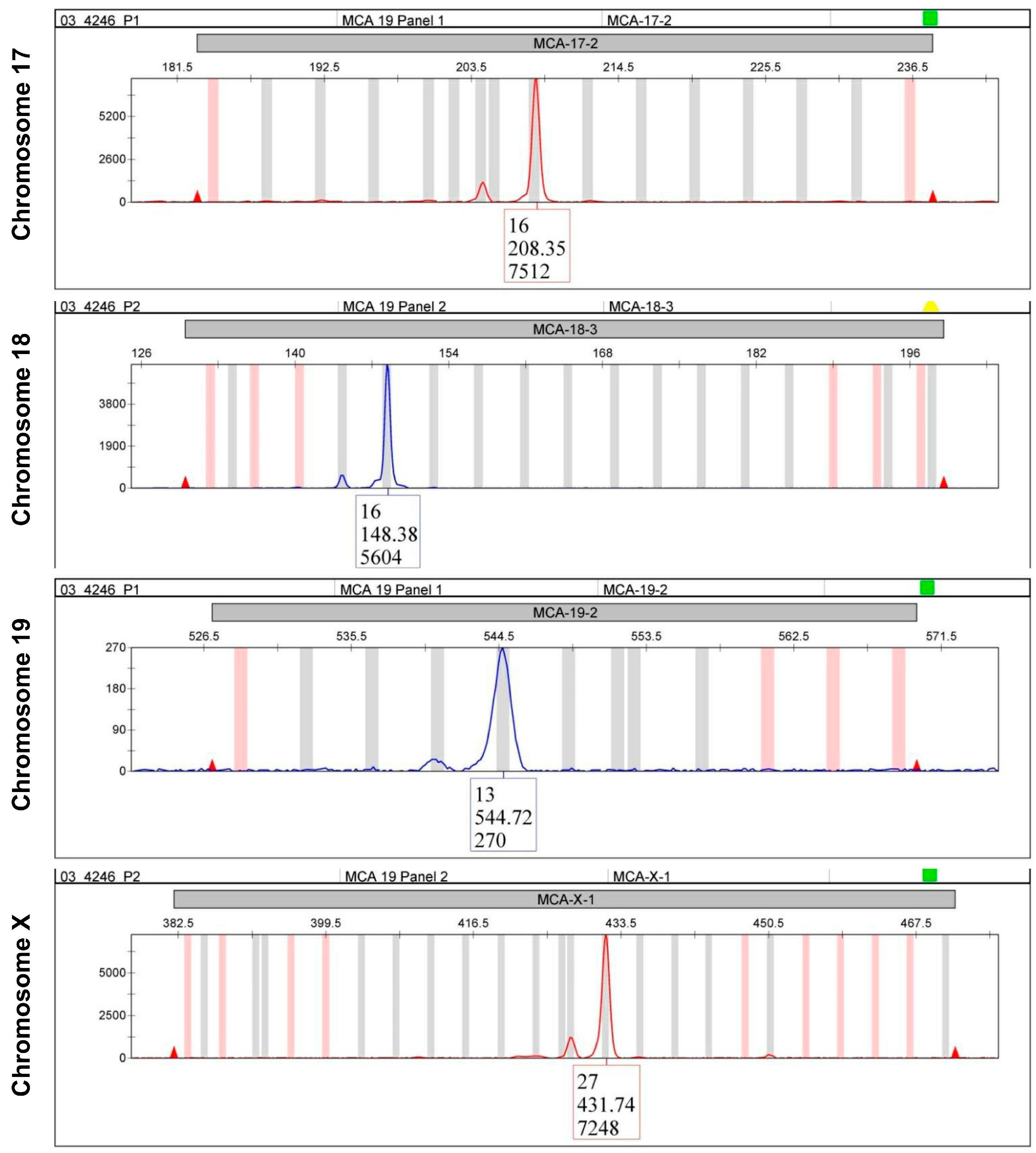
6. Short Tandem Repeat Profiling
7. Cell Line Alteration and Over-Passaging
8. Biosafety Aspects in Working with Cells
9. Patient-Derived Cell Lines, Organoids, Xenograft Models, and Conditional Reprogramming
10. Cleaning and Sterilization
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hubrecht, R.C.; Carter, E. The 3Rs and humane experimental technique: Implementing change. Animals 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Pamies, D.; Leist, M.; Coecke, S.; Bowe, G.; Allen, D.G.; Gstraunthaler, G.; Bal-Price, A.; Pistollato, F.; de Vries, R.B.M.; Hogberg, H.T.; et al. Guidance document on Good Cell and Tissue Culture Practice 2.0 (GCCP 2.0). ALTEX 2022, 39, 30–70. [Google Scholar] [CrossRef] [PubMed]
- Horbach, S.P.J.M.; Halffman, W. The ghosts of HeLa: How cell line misidentification contaminates the scientific literature. PLoS ONE 2017, 12, e0186281. [Google Scholar] [CrossRef] [PubMed]
- Babic, Z.; Capes-Davis, A.; Martone, M.E.; Bairoch, A.; Ozyurt, I.B.; Gillespie, T.H.; Bandrowski, A.E. Incidences of problematic cell lines are lower in papers that use RRIDs to identify cell lines. Elife 2019, 8, e41676. [Google Scholar] [CrossRef] [PubMed]
- International Cell Line Authentication Committee (ICLAC). Available online: https://iclac.org/ (accessed on 18 February 2023).
- Wang, Y.; Chen, S.; Yan, Z.; Pei, M. A prospect of cell immortalization combined with matrix microenvironmental optimization strategy for tissue engineering and regeneration. Cell Biosci. 2019, 9, 7. [Google Scholar] [CrossRef]
- Khurana, A.; Sayed, N.; Singh, V.; Khurana, I.; Allawadhi, P.; Rawat, P.S.; Navik, U.; Pasumarthi, S.K.; Bharani, K.K.; Weiskirchen, R. A comprehensive overview of CRISPR/Cas 9 technology and application thereof in drug discovery. J. Cell. Biochem. 2022, 123, 1674–1698. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Cho, J.; Kwak, J.; Sung, Y.H.; Kang, B.C. Immortalization of primary marmoset skin fibroblasts by CRISPR-Cas9-mediated gene targeting. Anim. Cells Syst. 2022, 26, 266–274. [Google Scholar] [CrossRef]
- Coecke, S.; Balls, M.; Bowe, G.; Davis, J.; Gstraunthaler, G.; Hartung, T.; Hay, R.; Merten, O.W.; Price, A.; Schechtman, L.; et al. Guidance on good cell culture practice. A report of the second ECVAM task force on good cell culture practice. Altern. Lab. Anim. 2005, 33, 261–287. [Google Scholar] [CrossRef]
- Mao, Z.; Ke, Z.; Gorbunova, V.; Seluanov, A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging 2012, 4, 431–435. [Google Scholar] [CrossRef]
- Shen, C.F.; Guilbault, C.; Li, X.; Elahi, S.M.; Ansorge, S.; Kamen, A.; Gilbert, R. Development of suspension adapted Vero cell culture process technology for production of viral vaccines. Vaccine 2019, 37, 6996–7002. [Google Scholar] [CrossRef]
- Moreira, A.S.; Silva, A.C.; Sousa, M.F.Q.; Hagner-McWhirterc, Å.; Ahlénc, G.; Lundgren, M.; Coroadinha, A.S.; Alves, P.M.; Peixoto, C.; Carrondo, M.J.T. Establishing suspension cell cultures for improved manufacturing of oncolytic adenovirus. Biotechnol. J. 2020, 15, e1900411. [Google Scholar] [CrossRef]
- Drescher, H.; Weiskirchen, S.; Weiskirchen, R. Flow cytometry: A blessing and a curse. Biomedicines 2021, 9, 1613. [Google Scholar] [CrossRef]
- Lai, T.Y.; Cao, J.; Ou-Yang, P.; Tsai, C.Y.; Lin, C.W.; Chen, C.C.; Tsai, M.K.; Lee, C.Y. Different methods of detaching adherent cells and their effects on the cell surface expression of Fas receptor and Fas ligand. Sci. Rep. 2022, 12, 5713. [Google Scholar] [CrossRef]
- Schellenberger, V.; Schellenberger, U.; Mitin, Y.V.; Jakubke, H.D. Characterization of the S’-subsite specificity of porcine pancreatic elastase. Eur. J. Biochem. 1989, 179, 161–163. [Google Scholar] [CrossRef]
- Stenn, K.S.; Link, R.; Moellmann, G.; Madri, J.; Kuklinska, E. Dispase, a neutral protease from Bacillus polymyxa, is a powerful fibronectinase and type IV collagenase. J. Investig. Dermatol. 1989, 93, 287–290. [Google Scholar] [CrossRef]
- Tsuji, K.; Ojima, M.; Otabe, K.; Horie, M.; Koga, H.; Sekiya, I.; Muneta, T. Effects of different cell-detaching methods on the viability and cell surface antigen expression of synovial mesenchymal stem cells. Cell Transplant. 2017, 26, 1089–1102. [Google Scholar] [CrossRef]
- Nowak-Terpiłowska, A.; Śledziński, P.; Zeyland, J. Impact of cell harvesting methods on detection of cell surface proteins and apoptotic markers. Braz. J. Med. Biol. Res. 2021, 54, e10197. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Berthois, Y.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Phenol red in tissue culture media is a weak estrogen: Implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef]
- Li, W.; Fan, Z.; Lin, Y.; Wang, T.Y. Serum-free medium for recombinant protein expression in Chinese hamster ovary cells. Front. Bioeng. Biotechnol. 2021, 9, 646363. [Google Scholar] [CrossRef]
- Brunner, D.; Frank, J.; Appl, H.; Schöffl, H.; Pfaller, W.; Gstraunthaler, G. Serum-free cell culture: The serum-free media interactive online database. ALTEX 2010, 27, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Asayama, Y. Animal-cell culture media: History, characteristics, and current issues. Reprod. Med. Biol. 2017, 16, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Llobet, L.; Montoya, J.; López-Gallardo, E.; Ruiz-Pesini, E. Side effects of culture media antibiotics on cell differentiation. Tissue Eng. Part C Methods 2015, 21, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Ryu, A.H.; Eckalbar, W.L.; Kreimer, A.; Yosef, N.; Ahituv, N. Use antibiotics in cell culture with caution: Genome-wide identification of antibiotic-induced changes in gene expression and regulation. Sci. Rep. 2017, 7, 7533. [Google Scholar] [CrossRef] [PubMed]
- Varghese, D.S.; Parween, S.; Ardah, M.T.; Emerald, B.S.; Ansari, S.A. Effects of aminoglycoside antibiotics on human embryonic stem cell viability during differentiation in vitro. Stem Cells Int. 2017, 2017, 2451927. [Google Scholar] [CrossRef]
- Farzaneh, M. Concise Review; Effects of antibiotics and antimycotics on the biological properties of human pluripotent and multipotent stem cells. Curr. Stem Cell Res. Ther. 2021, 16, 400–405. [Google Scholar] [CrossRef]
- Hassan, S.N.; Ahmad, F. The relevance of antibiotic supplements in mammalian cell cultures: Towards a paradigm shift. Gulhane Med. J. 2020, 62, 224–230. [Google Scholar] [CrossRef]
- Herwaldt, B.L. Laboratory-acquired parasitic infections from accidental exposures. Clin. Microbiol. Rev. 2001, 14, 659–688. [Google Scholar] [CrossRef]
- Avar, M.; Heinzer, D.; Steinke, N.; Doğançay, B.; Moos, R.; Lugan, S.; Cosenza, C.; Hornemann, S.; Andréoletti, O.; Aguzzi, A. Prion infection, transmission, and cytopathology modeled in a low-biohazard human cell line. Life Sci. Alliance 2020, 3, e202000814. [Google Scholar] [CrossRef]
- Chou, M.L.; Bailey, A.; Avory, T.; Tanimoto, J.; Burnouf, T. Removal of transmissible spongiform encephalopathy prion from large volumes of cell culture media supplemented with fetal bovine serum by using hollow fiber anion-exchange membrane chromatography. PLoS ONE 2015, 10, e0122300. [Google Scholar] [CrossRef]
- Zigler, J.S., Jr.; Lepe-Zuniga, J.L.; Vistica, B.; Gery, I. Analysis of the cytotoxic effects of light-exposed HEPES-containing culture medium. Vitr. Cell. Dev. Biol. 1985, 21, 282–287. [Google Scholar] [CrossRef]
- Capes-Davis, A.; Theodosopoulos, G.; Atkin, I.; Drexler, H.G.; Kohara, A.; MacLeod, R.A.; Masters, J.R.; Nakamura, Y.; Reid, Y.A.; Reddel, R.R.; et al. Check your cultures! A list of cross-contaminated or misidentified cell lines. Int. J. Cancer 2010, 127, 1–8. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Zheng, C.; Shen, C. Investigation of cross-contamination and misidentification of 278 widely used tumor cell lines. PLoS ONE 2017, 12, e0170384. [Google Scholar] [CrossRef]
- Weiskirchen, R. Established liver cell lines: Are you sure to have the right ones? Livers 2022, 2, 171–177. [Google Scholar] [CrossRef]
- Rottem, S. Interaction of mycoplasmas with host cells. Physiol. Rev. 2003, 83, 417–432. [Google Scholar] [CrossRef]
- Nikfarjam, L.; Farzaneh, P. Prevention and detection of Mycoplasma contamination in cell culture. Cell J. 2012, 13, 203–212. [Google Scholar] [PubMed]
- Volokhov, D.V.; Graham, L.J.; Brorson, K.A.; Chizhikov, V.E. Mycoplasma testing of cell substrates and biologics: Review of alternative non-microbiological techniques. Mol. Cell. Probes 2011, 25, 69–77. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Drexler, H.G. Detection of Mycoplasma contamination in cell cultures. Curr. Protoc. Mol. Biol. 2014, 106, 28.4.1–28.4.14. [Google Scholar] [CrossRef]
- Lawson-Ferreira, R.; Santiago, M.A.; Chometon, T.Q.; Costa, V.A.; Silva, S.A.; Bertho, A.L.; de Filippis, I. Flow-cytometric method for viability analysis of mycoplasma gallisepticum and other cell-culture-contaminant Mollicutes. Curr. Microbiol. 2021, 78, 67–77. [Google Scholar] [CrossRef]
- Wehbe, K.; Vezzalini, M.; Cinque, G. Detection of mycoplasma in contaminated mammalian cell culture using FTIR microspectroscopy. Anal. Bioanal. Chem. 2018, 410, 3003–3016. [Google Scholar] [CrossRef]
- Göbel, U.B.; Stanbridge, E.J. Cloned mycoplasma ribosomal RNA genes for the detection of mycoplasma contamination in tissue cultures. Science 1984, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G.; Uphoff, C.C. Mycoplasma contamination of cell cultures: Incidence, sources, effects, detection, elimination, prevention. Cytotechnology 2002, 39, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Bøsling, J.; Poulsen, S.M.; Vester, B.; Long, K.S. Resistance to the peptidyl transferase inhibitor tiamulin caused by mutation of ribosomal protein l3. Antimicrob. Agents Chemother. 2003, 47, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Garmyn, A.; Vereecken, M.; Degussem, K.; Depondt, W.; Haesebrouck, F.; Martel, A. Efficacy of tiamulin alone or in combination with chlortetracycline against experimental Mycoplasma gallisepticum infection in chickens. Poult. Sci. 2017, 96, 3367–3374. [Google Scholar] [CrossRef] [PubMed]
- Uphoff, C.C.; Denkmann, S.A.; Drexler, H.G. Treatment of mycoplasma contamination in cell cultures with Plasmocin. J. Biomed. Biotechnol. 2012, 2012, 267678. [Google Scholar] [CrossRef]
- Puty, B.; Nogueira, I.C.D.C.; Nogueira, L.S.; Vasconcelos, C.P.; Araújo, T.M.C.; Bittencourt, L.O.; Ferreira, R.O.; Oliveira, E.H.C.; Leal, W.G.; Lima, R.R. Genotoxic effect of non-lethal concentrations of minocycline in human glial cell culture. Biomed. Pharmacother. 2020, 128, 110285. [Google Scholar] [CrossRef]
- Wang, J.; Wei, Q.; Wang, C.Y.; Hill, W.D.; Hess, D.C.; Dong, Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J. Biol. Chem. 2004, 279, 19948–19954. [Google Scholar] [CrossRef]
- Yang, X.; Pei, S.; Wang, H.; Jin, Y.; Yu, F.; Zhou, B.; Zhang, H.; Zhang, D.; Lin, D. Tiamulin inhibits breast cancer growth and pulmonary metastasis by decreasing the activity of CD73. BMC Cancer 2017, 17, 255. [Google Scholar] [CrossRef]
- Merten, O.W. Virus contaminations of cell cultures—A biotechnological view. Cytotechnology 2002, 39, 91–116. [Google Scholar] [CrossRef]
- Gombold, J.; Karakasidis, S.; Niksa, P.; Podczasy, J.; Neumann, K.; Richardson, J.; Sane, N.; Johnson-Leva, R.; Randolph, V.; Sadoff, J.; et al. Systematic evaluation of in vitro and in vivo adventitious virus assays for the detection of viral contamination of cell banks and biological products. Vaccine 2014, 32, 2916–2926. [Google Scholar] [CrossRef]
- Schröder, S.K.; Schüler, H.M.; Petersen, K.V.; Tesauro, C.; Knudsen, B.R.; Pedersen, F.S.; Krus, F.; Buhl, E.M.; Roeb, E.; Roderfeld, M.; et al. Genetic and molecular characterization of the immortalized murine hepatic stellate cell line GRX. Cells 2022, 11, 1504. [Google Scholar] [CrossRef]
- Richert-Pöggeler, K.R.; Franzke, K.; Hipp, K.; Kleespies, R.G. Electron microscopy methods for virus diagnosis and high resolution analysis of viruses. Front. Microbiol. 2019, 9, 3255. [Google Scholar] [CrossRef]
- Roingeard, P.; Raynal, P.I.; Eymieux, S.; Blanchard, E. Virus detection by transmission electron microscopy: Still useful for diagnosis and a plus for biosafety. Rev. Med. Virol. 2019, 29, e2019. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, S.; Martin, J.L.; Mueller, J.D.; Mansky, L.M. Morphology and ultrastructure of retrovirus particles. AIMS Biophys. 2015, 2, 343–369. [Google Scholar] [CrossRef]
- Borojevic, R.; Monteiro, A.N.; Vinhas, S.A.; Domont, G.B.; Mourão, P.A.; Emonard, H.; Grimaldi, G., Jr.; Grimaud, J.A. Establishment of a continuous cell line from fibrotic schistosomal granulomas in mice livers. Vitr. Cell. Dev. Biol. 1985, 21, 382–390. [Google Scholar] [CrossRef]
- Nims, R.W.; Price, P.J. Best practices for detecting and mitigating the risk of cell culture contaminants. Vitr. Cell. Dev. Biol. Anim. 2017, 53, 872–879. [Google Scholar] [CrossRef]
- Grzelak, A.; Rychlik, B.; Bartosz, G. Light-dependent generation of reactive oxygen species in cell culture media. Free Radic. Biol. Med. 2001, 30, 1418–1425. [Google Scholar] [CrossRef]
- Shaban, S.; El-Husseny, M.W.A.; Abushouk, A.I.; Salem, A.M.A.; Mamdouh, M.; Abdel-Daim, M.M. Effects of antioxidant supplements on the survival and differentiation of stem cells. Oxidative Med. Cell. Longev. 2017, 2017, 5032102. [Google Scholar] [CrossRef]
- Saito, Y. Diverse cytoprotective actions of vitamin E isoforms- role as peroxyl radical scavengers and complementary functions with selenoproteins. Free Radic. Biol. Med. 2021, 175, 121–129. [Google Scholar] [CrossRef]
- Stolwijk, J.M.; Falls-Hubert, K.C.; Searby, C.C.; Wagner, B.A.; Buettner, G.R. Simultaneous detection of the enzyme activities of GPx1 and GPx4 guide optimization of selenium in cell biological experiments. Redox Biol. 2020, 32, 101518. [Google Scholar] [CrossRef] [PubMed]
- Gartler, S.M. Apparent Hela cell contamination of human heteroploid cell lines. Nature 1968, 217, 750–751. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.M.; Cobo, F.; Nieto, A.; Cortés, J.L.; Montes, R.M.; Catalina, P.; Concha, A. Identity tests: Determination of cell line cross-contamination. Cytotechnology 2006, 51, 45–50. [Google Scholar] [CrossRef] [PubMed]
- American Type Culture Collection Standards Development Organization Workgroup ASN-0002. Cell line misidentification: The beginning of the end. Nat. Rev. Cancer 2010, 10, 441–448. [Google Scholar] [CrossRef]
- Almeida, J.L.; Cole, K.D.; Plant, A.L. Standards for cell line authentication and beyond. PLoS Biol. 2016, 14, e1002476. [Google Scholar] [CrossRef]
- Almeida, J.L.; Dakic, A.; Kindig, K.; Kone, M.; Letham, D.L.D.; Langdon, S.; Peat, R.; Holding-Pillai, J.; Hall, E.M.; Ladd, M.; et al. Interlaboratory study to validate a STR profiling method for intraspecies identification of mouse cell lines. PLoS ONE 2019, 14, e0218412. [Google Scholar] [CrossRef]
- Kroh, A.; Walter, J.; Schüler, H.; Nolting, J.; Eickhoff, R.; Heise, D.; Neumann, U.P.; Cramer, T.; Ulmer, T.F.; Fragoulis, A. A newly established murine cell line as a model for hepatocellular cancer in non-alcoholic steatohepatitis. Int. J. Mol. Sci. 2019, 20, 5658. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology (NIST). STRBase Standard Reference Database SRD-130. Available online: https://strbase.nist.gov (accessed on 18 February 2023).
- Cellosaurus. CLASTR 1.4.4. Available online: https://www.cellosaurus.org/str-search/ (accessed on 18 February 2023).
- Cao, J.; Wu, X.; Qin, X.; Li, Z. Uncovering the effect of passage number on HT29 cell line based on the cell metabolomic approach. J. Proteome Res. 2021, 20, 1582–1590. [Google Scholar] [CrossRef]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575–586. [Google Scholar] [CrossRef]
- Jensen, H.L.; Norrild, B. The effects of cell passages on the cell morphology and the outcome of herpes simplex virus type 1 infection. J. Virol. Methods 2000, 84, 139–152. [Google Scholar] [CrossRef]
- Wenger, S.L.; Senft, J.R.; Sargent, L.M.; Bamezai, R.; Bairwa, N.; Grant, S.G. Comparison of established cell lines at different passages by karyotype and comparative genomic hybridization. Biosci. Rep. 2004, 24, 631–639. [Google Scholar] [CrossRef]
- ATCC. Animal Cell Culture Guide. Available online: https://www.atcc.org/-/media/resources/culture-guides/animal-cell-culture-guide.pdf?rev=6b6752984d6a404abbc111f893ef2f99 (accessed on 18 February 2023).
- The Organisation for Economic Co-operation and Development OECD. Guidance Document on Good In Vitro Method Practices (GIVIMP); OECD Series on Testing and Assessment, No. 286; OECD Publishing: Paris, France, 2018. [Google Scholar] [CrossRef]
- Uzbekov, R.E. Analysis of the cell cycle and a method employing synchronized cells for study of protein expression at various stages of the cell cycle. Biochemistry 2004, 69, 485–496. [Google Scholar] [CrossRef]
- Jena, G.B.; Chavan, S. Implementation of Good Laboratory Practices (GLP) in basic scientific research: Translating the concept beyond regulatory compliance. Regul. Toxicol. Pharmacol. 2017, 89, 20–25. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Chies, J.A.B. Zoonotic spillover: Understanding basic aspects for better prevention. Genet. Mol. Biol. 2021, 44 (Suppl. 1), e20200355. [Google Scholar] [CrossRef]
- Frommer, W.; Archer, L.; Boon, B.; Brunius, G.; Collins, C.H.; Crooy, P.; Doblhoff-Dier, O.; Donikian, R.; Economidis, J.; Frontali, C.; et al. Safe biotechnology (5). Recommendations for safe work with animal and human cell cultures concerning potential human pathogens. Appl. Microbiol. Biotechnol. 1993, 39, 141–147. [Google Scholar] [CrossRef]
- Nova, N. Cross-species transmission of coronaviruses in humans and domestic mammals, what are the ecological mechanisms driving transmission, spillover, and disease emergence? Front. Public Health 2021, 9, 717941. [Google Scholar] [CrossRef]
- Herman, P.; Pauwels, K. Biosafety recommendations on the handling of animal cell cultures. In Animal Cell Culture; Al-Rubeai, M., Ed.; Springer: Cham, Switzerland, 2015; Volume 9. [Google Scholar] [CrossRef]
- Silver, A. Why the world has no universal biosafety standards. BMJ 2022, 377, o954. [Google Scholar] [CrossRef]
- Huo, K.G.; D’Arcangelo, E.; Tsao, M.S. Patient-derived cell line, xenograft and organoid models in lung cancer therapy. Transl. Lung Cancer Res. 2020, 9, 2214–2232. [Google Scholar] [CrossRef]
- Dayaram, T.; Marriott, S.J. Effect of transforming viruses on molecular mechanisms associated with cancer. J. Cell. Physiol. 2008, 216, 309–314. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.; Li, M.; Li, J.; Shen, J.; Zhao, Y.; Pang, J.; Wen, Q.; Chen, M.; Wei, B.; et al. Conditional reprogramming: Next generation cell culture. Acta Pharm. Sin. B 2020, 10, 1360–1381. [Google Scholar] [CrossRef]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Suprynowicz, F.A.; Upadhyay, G.; Krawczyk, E.; Kramer, S.C.; Hebert, J.D.; Liu, X.; Yuan, H.; Cheluvaraju, C.; Clapp, P.W.; Boucher, R.C., Jr.; et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20035–20040. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Prim. 2022, 2, 94. [Google Scholar] [CrossRef]
- Urbischek, M.; Rannikmae, H.; Foets, T.; Ravn, K.; Hyvönen, M.; de la Roche, M. Organoid culture media formulated with growth factors of defined cellular activity. Sci. Rep. 2019, 9, 6193. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Liu, M.; Mao, Y. Patient-derived organoids: A promising model for personalized cancer treatment. Gastroenterol. Rep. 2018, 6, 243–245. [Google Scholar] [CrossRef]
- Zanella, E.R.; Grassi, E.; Trusolino, L. Towards precision oncology with patient-derived xenografts. Nat. Rev. Clin. Oncol. 2022, 19, 719–732. [Google Scholar] [CrossRef]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 106. [Google Scholar] [CrossRef]
- Inoue, A.; Deem, A.K.; Kopetz, S.; Heffernan, T.P.; Draetta, G.F.; Carugo, A. Current and future horizons of patient-derived xenograft models in colorectal cancer translational research. Cancers 2019, 11, 1321. [Google Scholar] [CrossRef]
- National Cancer Institute; DCTG Division of Cancer Treatment & Diagnosis. NCI Patient-Derived Models Repository (PDMR). Available online: https://pdmr.cancer.gov/ (accessed on 18 February 2023).
- Human Cancer Models Initiative (HCMI). Available online: https://ocg.cancer.gov/programs/HCMI (accessed on 18 February 2023).
- World Health Organization (WHO). Laboratory Biosafety Manual, 4th ed.; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001131-1. [Google Scholar]
- Bentancor, M.; Vidal, S. Programmable and low-cost ultraviolet room disinfection device. HardwarX 2018, 4, e00046. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inorganic salts/buffers: CaCl2: 0.2 g/L, Fe(NO3)3 × 9 H2O: 0.0001 g/L, MgSO4: 0.09767 g/L, KCl: 0.4 g/L, NaHCO3: 3.7 g/L, NaCl: 6.4 g/L, NaH2PO4: 0.109 g/L |
| Amino acids: L-Arginine × HCl: 0.084 g/L, L-Glutamine: 0.584 g/L 1, Glycine: 0.03 g/L, L-Histidine × HCl × H2O: 0.042 g/L, L-Isoleucine: 0.105 g/L, L-Leucine: 0.105 g/L, L-Lysine × HCl: 1.46 g/L, L-Phenylalanine: 0.066 g/L, L-Serine: 0.042 g/L, L-Threonine: 0.095 g/L, L-Tryptophan: 0.016 g/L, L-Tyrosine × 2 Na × 2 H2O: 0.12037 g/L, L-Valine: 0.094 g/L |
| Vitamins: Choline chloride: 0.004 g/L, Folic acid: 0.004 g/L, myo-inositol: 0.0072 g/L, Niacinamide: 0.004 g/L, D-Pantothenic acid (hemicalcium): 0.004 g/L, Pyridoxal hydrochloride: 0.004 g/L, Riboflavin: 0.0004 g/L, Thiamine × HCl: 0.004 g/L |
| Others: D-Glucose: 4.5 g/L 2, Phenol red × Na: 0.0159 g/L 3, Pyruvic acid × Na: 0.11 g/L |
| Contaminant | Remarks |
|---|---|
| Viruses (Viridae) | Viral contamination (e.g., HIV, HBV, EBV, SHBV) is hard to detect because they do not affect cellular growth. Based on their extremely small size (~100 nm in diameter), they are not visible under a bright-field microscope. However, infections with cytopathic viruses can destroy the culture. In addition, virally infected cell cultures represent a potential health hazard for laboratory personnel. |
| Mycoplasmas (Mollicutes) | Mycoplasmas are spherical to filamentous cells with no cell walls and intracytoplasmic membranes. They are the smallest self-replicating organisms with a diameter of ~ 300 nm and small genomes (~ 500 to 1000 genes). Infection can alter the host culture’s cell functions including growth, metabolism, migration, morphology, and responsiveness towards growth factors. In addition, some mycoplasma species can provoke chromosomal aberrations and damage. |
| Bacteria (Bacteriaceae) | The shape and size of bacteria can vary considerably ranging from 0.5 to 1.0 µm up to 10 to 20 µm in spiral forms. The bacterial genomes can range from about 130 kbp to over 14 Mbp and typically consist of 500–1200 genes (parasitic bacteria), 1500–1700 (free-living bacteria), and 1500–2700 genes (archaea). Most bacterial contaminants are able to quickly colonize and the flourish in cell culture media. Respective contamination can usually be readily detected by microscopy as tiny, moving granules between the cells within a few days of initial contamination. |
| Yeast and mold (Fungi) | Yeast cells are fungi that multiply faster than mammalian cells. The typical size of yeast and mold is 3–4 µm (but can be up to 40 µm). Contamination becomes clearly obvious by microscopic analysis or color change of the medium within 2–3 days. Antibiotics such as penicillin and streptomycin have no toxic effects on yeast. |
| Parasites | Different intracellular protozoan parasites (e.g., Toxoplasma gondii, Trypanosoma cruzi, Leishmania spp., Cryptosporidium parvum, Plasmodium spp.) may be included in freshly prepared primary cell cultures originating from a donor organism that is known or suspected to be infected with respective parasites. Special safety precautions should be considered and protective clothing and equipment might be necessary. Needles and other sharp objects should be omitted when working with parasite-infected cell lines [29]. |
| Prions | Prions are devoid of nucleic acids and consist primarily of protein termed PrPSc. Although most cell lines are resistant to prion infection, some cells lines are susceptible to prions and can promote stable and persistent replication of prions [30]. They can be included in cell culture media enriched with serum of bovine origin [31]. Prions are difficult to inactivate. |
| Chemical, biological, and other nonliving contaminants | Endotoxin/lipopolysaccharides, detergents, radicals, hormones, growth factors, metals, residues of disinfectants and cleaning agents, plasticizers, and other impurities can impact proper cell growth. Chemical contamination can result from contaminated reagents, water, sera or some culture additives. In addition, detergents or other deposits on storage vessels, glassware, pipettes or instruments introduced by disinfection can be sources of contamination. Plastic tubing and storage bottles can release plasticizers. Free radicals can be generated by photoactivation of tryptophan, riboflavin, or buffering agents (e.g., HEPES and PIPES) when exposed to extensive visible or fluorescent light [32]. |
| Inter- and intra-species cross-contamination | The incidence and extent of cell line cross-contamination is rather high [33,34]. The sources of inter- and intra-species cross-contamination are manifold (e.g., spreading via aerosols, usage of unplugged pipettes, sharing media and reagents among different cell lines, usage of conditioned medium, etc.) [35]. |
| Compound | Composition |
|---|---|
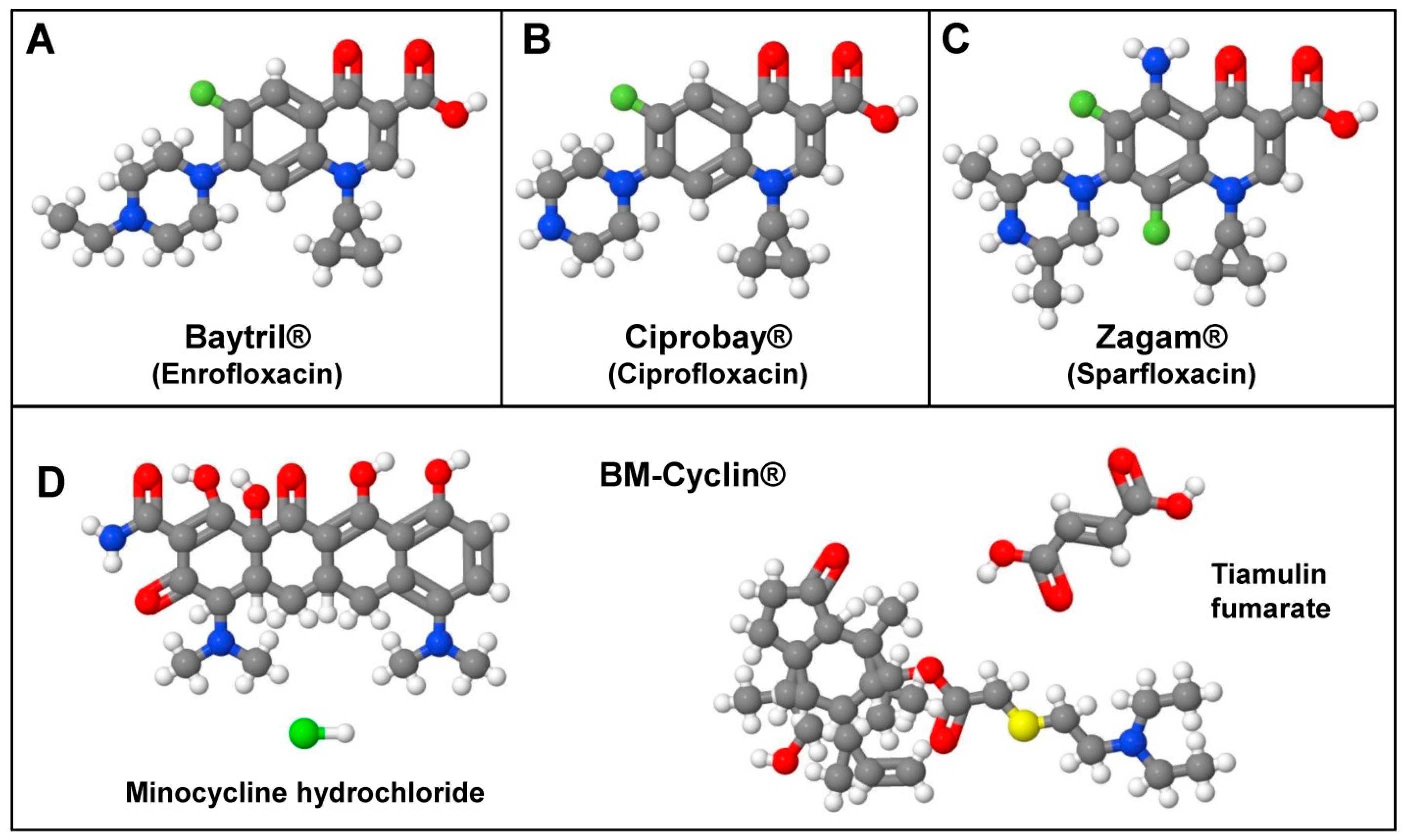
| BM-Cyclin | Tiamulin fumarate (a Macrolide) and Minocycline hydrochloride (a Tetracycline) |
| Ciprobay | Ciprofloxacin (a Quinolone) |
| Mycoplasma Removal Agent (MRA) | 4-oxo-quinoline-3-carboxylic acid derivative (a Quinolone) |
| Plasmocin | Contains two bactericidal components (a macrolide acting on the protein synthesis machinery by inhibiting the 50S ribosomal subunit and a fluoroquinolone inhibiting the DNA gyrase) |
| Baytril | Enrofloxacin (a Quinolone, inhibitor of DNA gyrase) |
| Zagam | Sparfloxacin (a Quinolone, inhibitor of DNA gyrase) |
| MycoZap | Ready-to-use combination of a not-disclosed surface-active antimicrobial peptide (MycoZap reagent 1) and a not-disclosed antibiotic (MycoZap reagent 2). |
| MycoRAZOR | Ready-to-use antibiotic mixture prepared in PBS acting against a large variety of mycoplasma by acting on the protein synthesis mechanism by interfering with the ribosome translation of the mycoplasms as well as with their transcription apparatus. |
| Normocin | Three antibiotics. Two of these compounds act on mycoplasmas, Gram-positive, and Gram-negative bacteria by blocking DNA and protein synthesis. The third compound eradicates fungi, including yeasts, by disrupting ionic exchange through the cell membrane. |
| Fungin | The soluble form of Pimarcin, a polyene that attacks yeasts, molds, and fungi by disrupting ionic exchange through the cell membrane. |
| Plasmocure | It contains two bactericidal components belonging to different antibiotic families. The first antibiotic binds to the 50S subunit of the ribosome and blocks peptidyltransferase activity, while the second antibiotic binds to isoleucyl-tRNA synthetase, thereby halting the incorporation of isoleucine into bacterial proteins. |
| Normocure | Contains three bactericidal components belonging to different antibiotic families that inhibit DNA and protein synthesis and disrupt membrane integrity by targeting structures that are absent in eukaryotic cells. |
| Human (Homo sapiens): ‘1.1B4; 1E8; 2008/C13*5.25; 222; 2474/90; 2563 (MAC-21); 28SC-ES; 2957/90; 3051/80; 3AB-OS; 41M; 5-8F; 6-10B; A172TR3 (U251-TR3); ACC2; ACC3; ACCM; ACCNS; ACCS; ADLC-5M2; AG-F; AKI; ALVA-31; ALVA-41; ALVA-55; ALVA-101; AO; ARO81-1 (ARO); AV3; AZ521; BCC1/KMC; BE-13; BEL-7402; BEL-7404; BGC-823; BHP 10-3; BHP 14-9; BHP 15-3; BHP 17-10; BHP 18-21; BHP 2-7; BHP 5-16; BHP 7-13; BIC-1; BLIN-1; BM-1604; BrCA 5; BSCC-93; C16; C-433; CAC2; CaES-17; CaMa (clone 15); CaOV; Caov-2; CaVe; CCL3; CGTH-W-1; CH1; CH1-cisR; Chang liver; CHB; CHP-234; Clom 15; Clone 1-5c-4; Clone-16; CMP; CMPII C2; CNDT2; CNE-1; CNE-2; CO (COLE); COLO-38; COLO-587; COLO-677; COLO-775; COLO-818; CoLo-TC; D18T; D-54 MG; D98/AH; D98/AH2 Clone B; DAMI; DAPT; DD; Det30A; Detroit-6 (Det6); Detroit-98; Detroit 98/AG; Detroit 98/AH-2; Detroit 98/AH-R; Detroit 98s; DM12; DM14; DRO90-1 (DRO); DuPro-1; E006AA; E006AA-hat; EB33; ECC-1; ECV-304; ED27; EH; EJ-1; Ej138; EL 1; ElCo; EPLC3-2M1; EPLC-65; ESP1; ETK-1; EU-1; EU-7; EUE; EVLC2; F2-4E5; F2-5B6; F255A4; FB2; FL; Flow 13000; Flow 5000; Flow 6000; Flow 7000; FQ; G-11; GHE; Girardi heart; GLC-82; GM1312; GOS-3; GREF-X; GR-M; GT3TKB; H-494; H7D7A; H7D7B; H7D7BD5; H7D7C; H7D7D; HAC15; HAC-84; HAG; HBC; HBL-100; HBT-3; HBT-39b; HBT-E (HBT-3 clone); HCC60; HCE; HCu-10; HCu-18; HCu-22; HCu-27; HCu-33; HCu-37; HCu-39; HCV-29Tmv; HEC-155; HEC-180; HEK; HEK/HRV; HEL-R66; HEp-2 (H.Ep.-2); Hep-2C; Hep2 (Clone 2B); HES; HIMEG-1; HKB-1; HKMUS; HKMUS-SF; HL111783; HMV-1; HNOS; HO-8910; HO-8910PM; HONE-1; HPB-MLT; HPC-36M; hPTC; HROBML03; Hs 677.St; HSC-41; HSG; HSG-AZA1; HSG-AZA3; HSGc-C5; HS-SULTAN; HSY; hTERT-EEC; Hu1734; Hu456; Hu549; Hu609; Hu609Tmv; Hu961a, Hu961t; HuKo39; HuL-1; Hut; IMC-2; IMC-3; IMC-4; Intestine 407 (Int-407, HEI); IPDDC-A2; IPRB; IPTP/98; IST-1; J-111; J96; JCA-1; JHC; JHT; JHU012; JHU013; JHU019; JHU028; JMAR; JOSK-I; JOSK-K; JOSK-M; JOSK-S; JROECL 47 (OE47); JROECL 50 (OE50); JTC-17; JTC-3; K051; K1; K2; K5; KAK1; KAT10; KAT4; KAT5; KAT50; KAT7; KB; KB-3-1; KB-V1; KCI-MOH1; KKU-213B (KKU-M214); KKU-213C (KKU-M156); KM20; KM20L2; KM-3; KM3; KMS-21-BM; KMT-2; KOSC-3; KP-1N; KPB-M15; KPL-1; KP-P1; KSY-1; KU7; KU-YS; L-02; L-132; L-41; LC5; LC5-HIV; LED-Ti; LLC-15MB; LN-319; LN-443; LR10.6; LTEP-a2; LU; LU 106; Lu-130; M10T; M4A4; M4A4 GFP; M4A4 LM3-2 GFP; M4A4 LM3-4 CL16 GFP; MA-1; MA-160; MaTu; MC-4000; McCoy; MCF-7/AdrR (NCI/ADR-RES); MDA-MB-435; MDA-MB-435S; MDA-N; MDS; MEL-HO; MEL-WIE; MGC-803; MGH-U1 (EJ); MGH-U2 (HM); MHH-225; Minnesota EE; MKB-1; MKN28; MOBS-1; MOLT-15; MPanc-96; MRO87-1; MT-1; MT-3; MUM2C; MUTZ-1; MV522; NC-37; NCC16; NCI-H1264; NCI-H1304; NCI-H1514; NCI-H157; NCI-H1622; NCI-H1870; NCI-H249; NCI-H513; NCI-H592; NCI-H60; NCI-H630; NCI-H738; NCOL-1; NCTC 2544; NCTC 3075; ND-1; NM2C5; NM2C5 GFP; NOI-90; NOK-SI; NOSE06; NOSE07; NPA’87; NS-3; OCM-1; OCM-3; OCM-8; OCUM-6; OE; OF; ONCO-DG-1; OS 187; OST; OU-AML-1; OU-AML-2; OU-AML-3; OU-AML-4; OU-AML-5; OU-AML-6; OU-AML-7; OU-AML-8; OV2008 (A2008); Ovary1847; OVMIU; P1-1A3; P1-4D6; P39/TSUGANE (P39/TSU); Panc 01.28; Panc 06.03; PBEI; PC-93; PC-MDS; PCI-22A; PCI-22B; PCI-3; PEAZ-1; PH; PH61-N; PLB-985; PPC-1; PSV811; QGY-7701; QGY-7703; QSG-7701; RAMAK-1; RB; RBHF-1; RC-2A; RED-3; REH-6; REPC; RERF-LC-MA; RERF-LC-OK; RM-10; RMUG-L; RO-D81-1; RO-H85-1; RPMI-4788; RPMI-6666; RPTC-1; RS-1; RTSG; RY; SA4; SAM-1; SAML-1; SBC-2; SBC-7; SC (28SC); SCCTF; SCLC-16H; SCLC-24H; SEG-1; SF767; SGC-7901; SH-2; SH-3; SK-GT-5; SK-MG-1; SK-N-MC; SK-OV-4; SK-OV-6; SKW-3; SLK; SLR20; SLR24; SMMC-7721; SNB-19; SNU-1958; SPC-A1; SPI-801; SPI-802; SpR; SQ-5; SR-91; SU-DHL-7; SU-DHL-9; SUNE1; SUNE2; SW-527; SW-598; SW-608; SW-613; SW-732; SW-733; T-1; T1; T-33; T404; T406; T409; T-9; Tca8113; TCO-1; TDL-1; TDL-2; TDL-3; TDL-4; TE-12; TE-13; TE-2; TE-3; TE671; TE671 Subline No.2; TE-7; TEC61; TI-1; TK-1; TMH-1; TMM; TSCCa; TSU-Pr1; Tu-138; Tu-158LN; Tu-159; Tu-167; Tu-182; Tu-212; Tu-212LN; TuWi; U-118 MG; UM-UC-2; UM-UC-3-GFP; UPES/C; UPHHJA; UTMB-460; VC312R; WiDr; WISH; Wong-Kilbourne derivative (WKD); WRL 68; WSU-ALCL; WSU-CLL; YAA; YAP; YJ; YMB-1; YMB-1-E; Z-HL16C |
| Mouse (Mus musculus): 1-1ras1000; 1-1src; BALB/3T3 A31-1-1; BALB/3T3 A31-1-13; Bhas42; BT-B; MOC2-10 (MOC10) |
| Rat (Rattus norvegicus): HAPI; RGC-5 |
| Horse (Equus caballus): eCAS; EEK |
| Cow (Bos taurus): ECTC; LF-BK; LFBK-alphaVbeta6 |
| Mosquito (Culicidae) : Aedes aegypti, Suitor’s clone; Culiseta inornata |
| Black witch moth (Ascalapha odorata): Ao38 (BTI-Tnao38) |
| Rainbow trout (Oncorhynchus mykiss): Clone 1A; D-11; R1 |
| Carp (Cyprinus carpio): EPC |
| Dog (Canis familiaris): Fitz-HSA; UCDK9B1; UCDK9B2; UCDK9B3; UCDK9B4; UCDK9B5 |
| Mulberry tiger moth (Lemyra imparilis): FRI-SpIm-1229 |
| Guinea pig (Cavia porcellus): GPS-M; GPS-PD |
| Nile tilapia (Oreochromis niloticus): Hepa-T1 |
| White marked tussock moth (Orgyia leucostigma): IPRI-OL-7; IPRI-OL-11 |
| Cabbage moth (Mamestra brassicae): IZD-MB-0503 |
| Black-spotted frog (Pelophylax nigromaculatus): LAH1; LAH2 |
| Grass frog (Rana temporaria): LT-1 |
| Rhesus Monkey (Macaca mulatta) & Monkey (unspecified): MA-104; MS (Monkey Stable) |
| Rabbit (Oryctolagus cuniculus): MA-111 |
| Crayfish (Orconectes limosus): OLGA-PH-J/92 (OL-J/92) |
| American dog tick (Dermacentor variabilis): RML-15 [RML-RSE] |
| Pig (Sus scrofa): SJPL |
| Domestic silkmoth (Bombyx mori): SPC-BM-36 |
| STR Marker | GRX | AML12 | N-HCC25 * |
|---|---|---|---|
| STR 1-1 (MCA-1-1) | 10 | 11 | 16 |
| STR 1-2 (MCA-1-2) | 16 | 13 | 19 |
| STR 2-1 (MCA-2-1) | 9 | 9 | 16 |
| STR 3-2 (MCA-3-2) | 14 | 12 | 14 |
| STR 4-2 (MCA-4-2) | 19.3 | 20.3 | 20.3 |
| STR 5-5 (MCA-5-5) | 15 | 14, 15 | 17 |
| STR 6-4 (MCA-6-4) | 19 | 15.3 | 18 |
| STR 6-7 (MCA-6-7) | 12 | 12 | 17 |
| STR 7-1 (MCA-7-1) | 26 | 29 | 26.2 |
| STR 8-1 (MCA-8-1) | 16 | 14, 15 | 16 |
| STR 9-2 (MCA-9-2) | ND | 15 | 18 |
| STR 11-2 (MCA-11-2) | 16 | 18 | 16 |
| STR 12-1 (MCA-11-2) | 16 | 19 | 17 |
| STR 13-1 (MCA-13-1) | 17 | 15 | 17 |
| STR 15-3 (MCA-15-3) | 25.3 | 21.3 | 22.3 |
| STR 17-2 (MCA-17-2) | 16 | 13, 15 | 16 |
| STR 18-3 (MCA-18-3) | 16 | 21 | 16 |
| STR 19-2 (MCA-19-2) | 12 | 13 | 13 |
| STR X-1 (MCA-X-1) | 26, 27 | 26 | 27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskirchen, S.; Schröder, S.K.; Buhl, E.M.; Weiskirchen, R. A Beginner’s Guide to Cell Culture: Practical Advice for Preventing Needless Problems. Cells 2023, 12, 682. https://doi.org/10.3390/cells12050682
Weiskirchen S, Schröder SK, Buhl EM, Weiskirchen R. A Beginner’s Guide to Cell Culture: Practical Advice for Preventing Needless Problems. Cells. 2023; 12(5):682. https://doi.org/10.3390/cells12050682
Chicago/Turabian StyleWeiskirchen, Sabine, Sarah K. Schröder, Eva Miriam Buhl, and Ralf Weiskirchen. 2023. "A Beginner’s Guide to Cell Culture: Practical Advice for Preventing Needless Problems" Cells 12, no. 5: 682. https://doi.org/10.3390/cells12050682
APA StyleWeiskirchen, S., Schröder, S. K., Buhl, E. M., & Weiskirchen, R. (2023). A Beginner’s Guide to Cell Culture: Practical Advice for Preventing Needless Problems. Cells, 12(5), 682. https://doi.org/10.3390/cells12050682