
Elucidation of GPR55-Associated Signaling behind THC and LPI Reducing Effects on Ki67-Immunoreactive Nuclei in Patient-Derived Glioblastoma Cells

 , ,
, ,  ,
,
Abstract
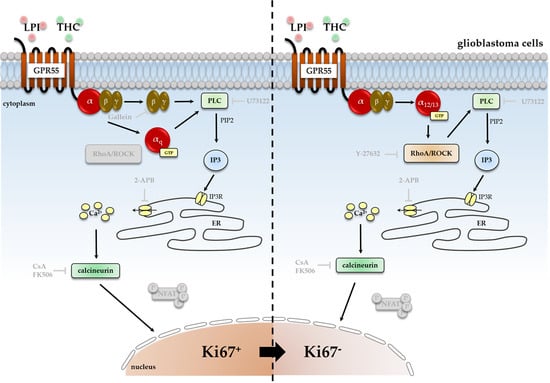
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Treatment
2.3. 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl-Tetrazolium Bromide (MTT)-Viability-Assay
2.4. PCR
2.5. Immunochemical Staining of Ki67
2.6. Immunofluorescence Labeling of NFAT
2.7. Statistics
3. Results
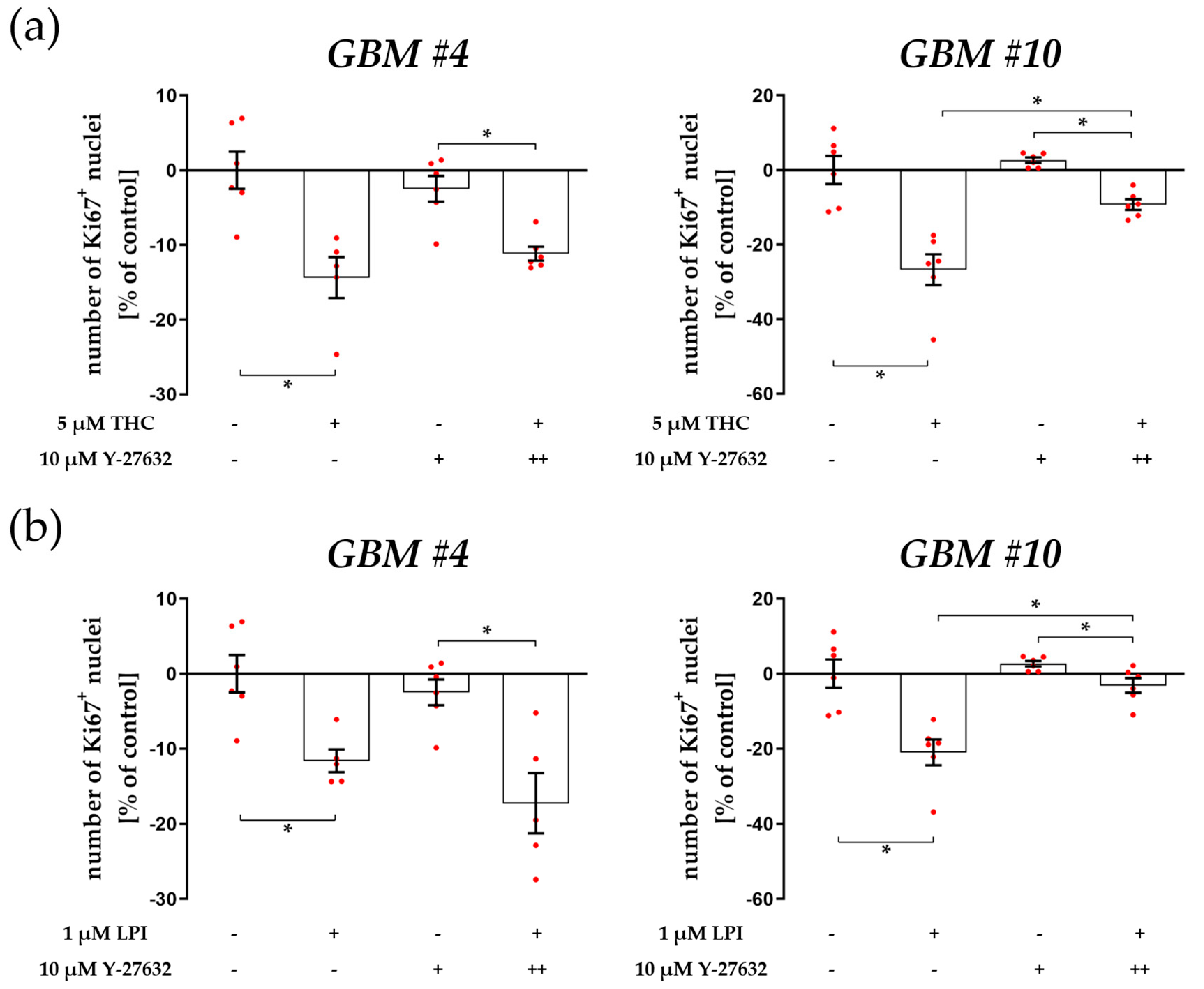
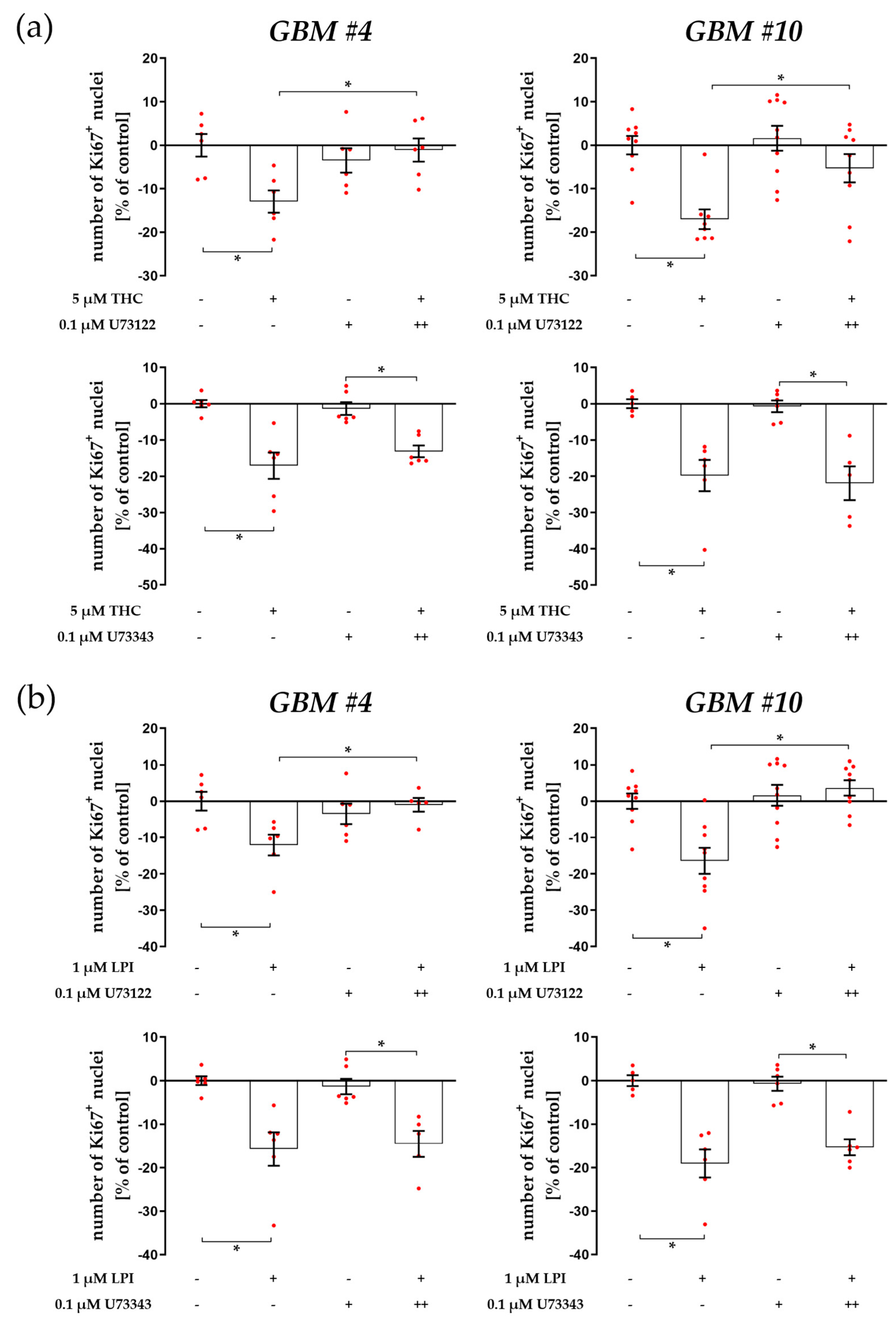
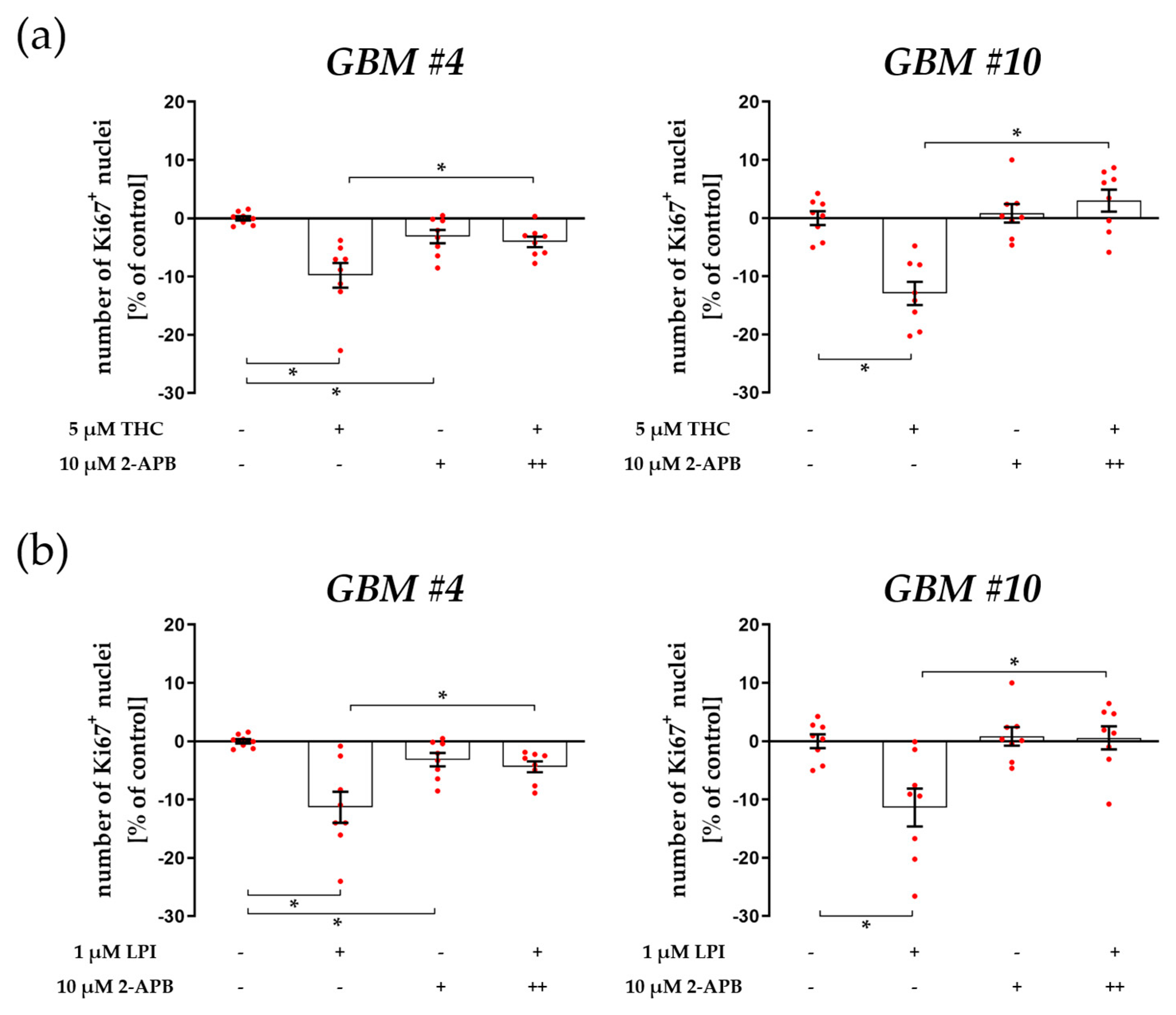
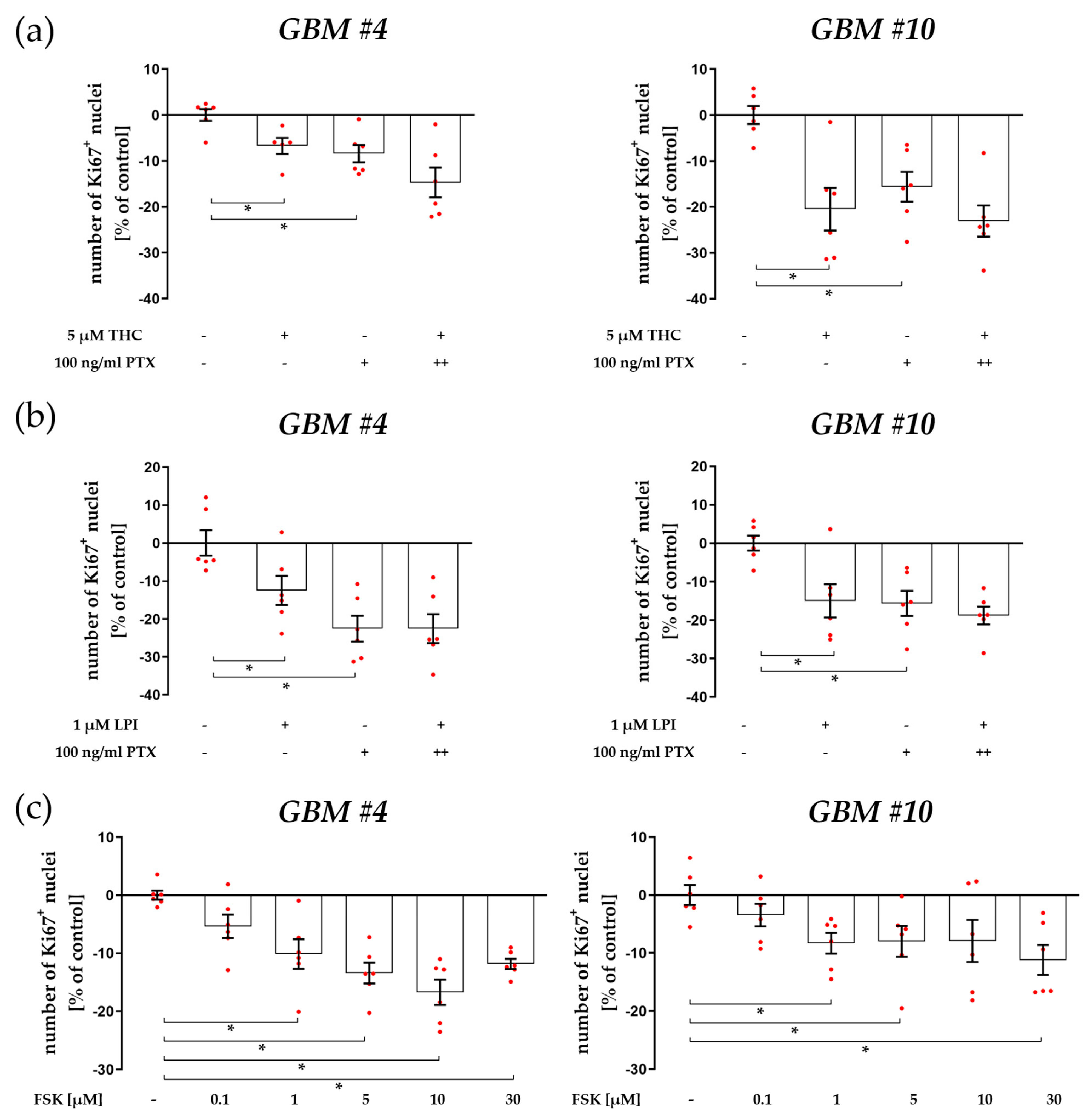
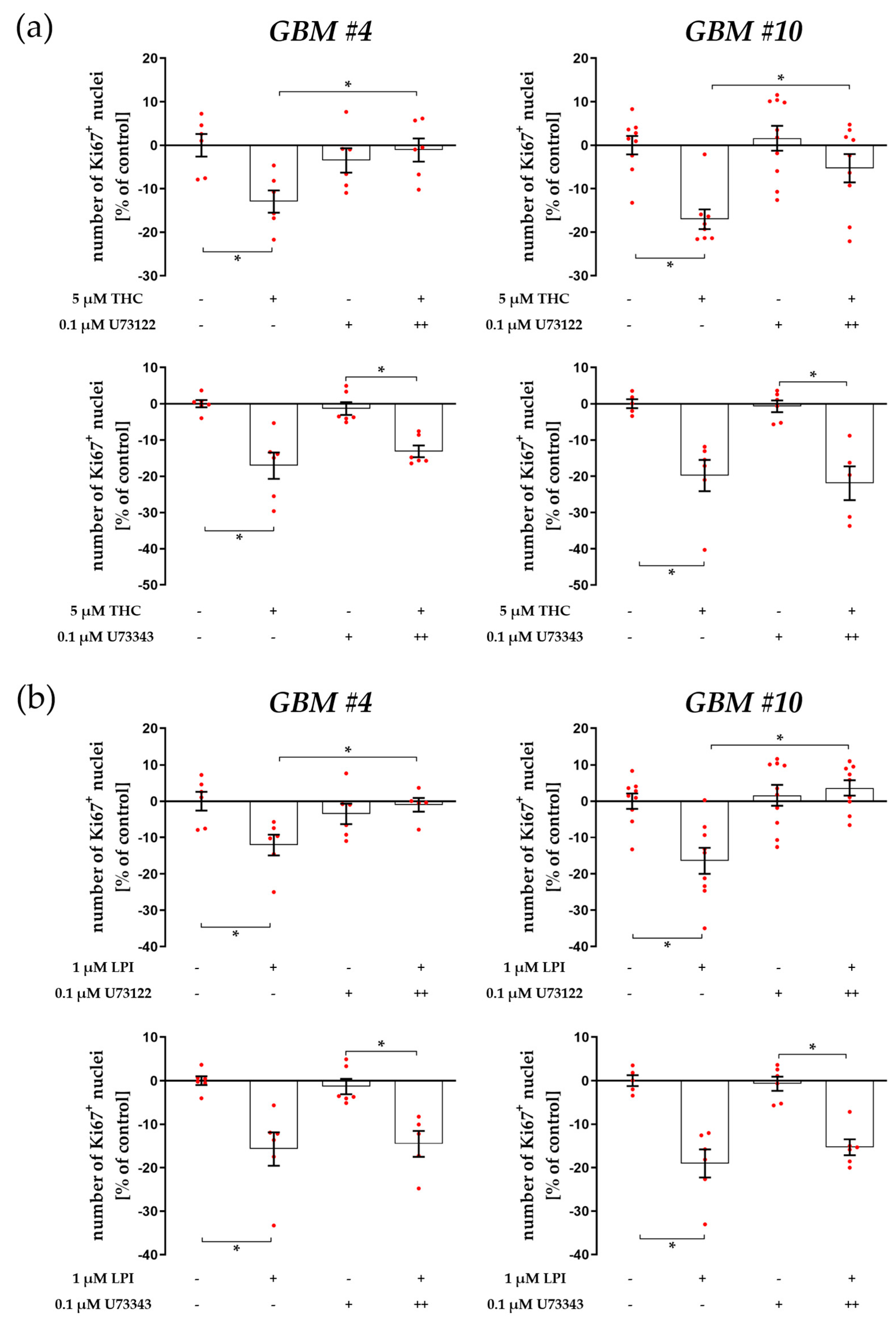
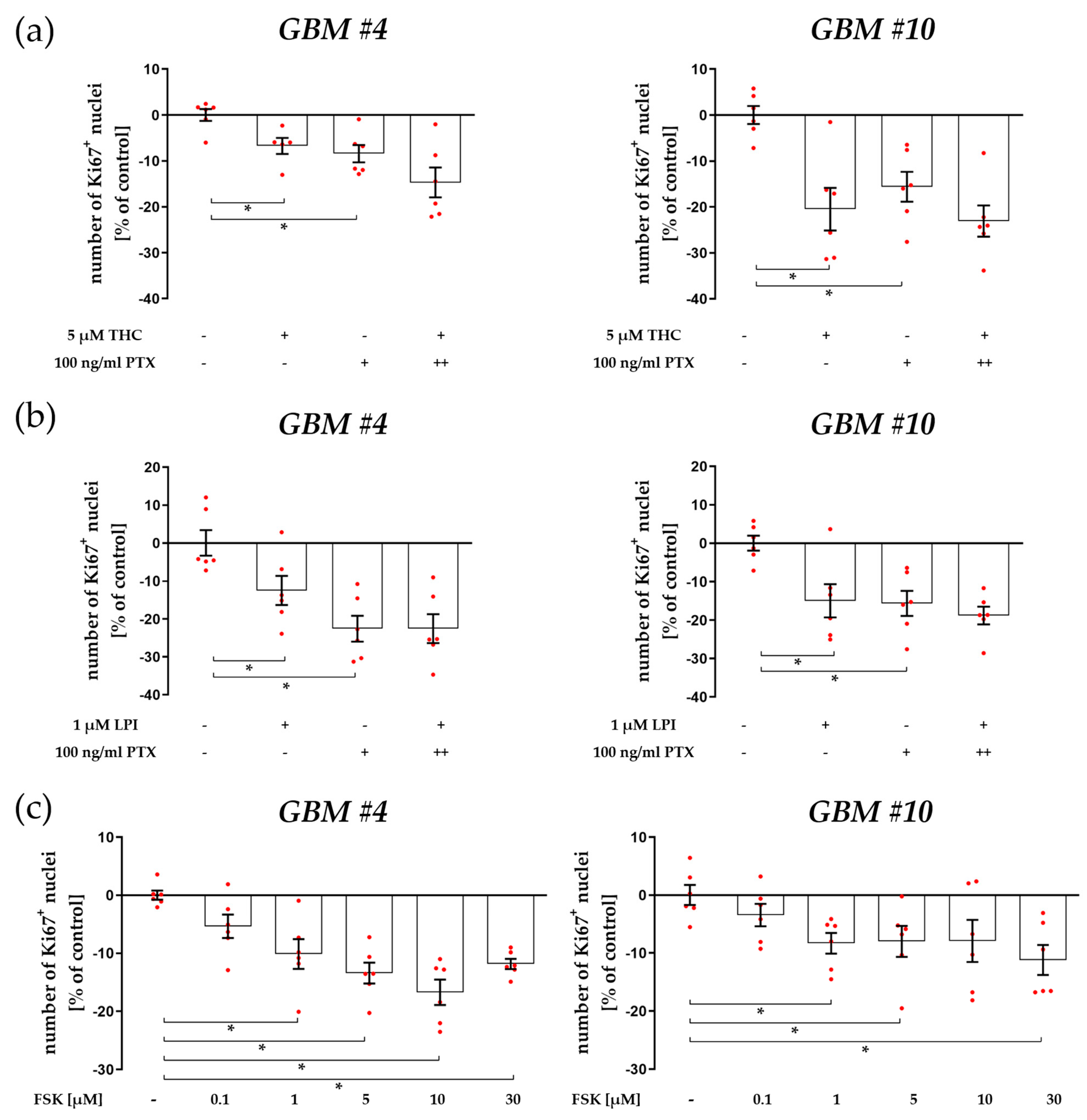
3.1. Cell-Type-Specific ROCK Signaling and a Strongly Involved PLC-IP3 Signaling
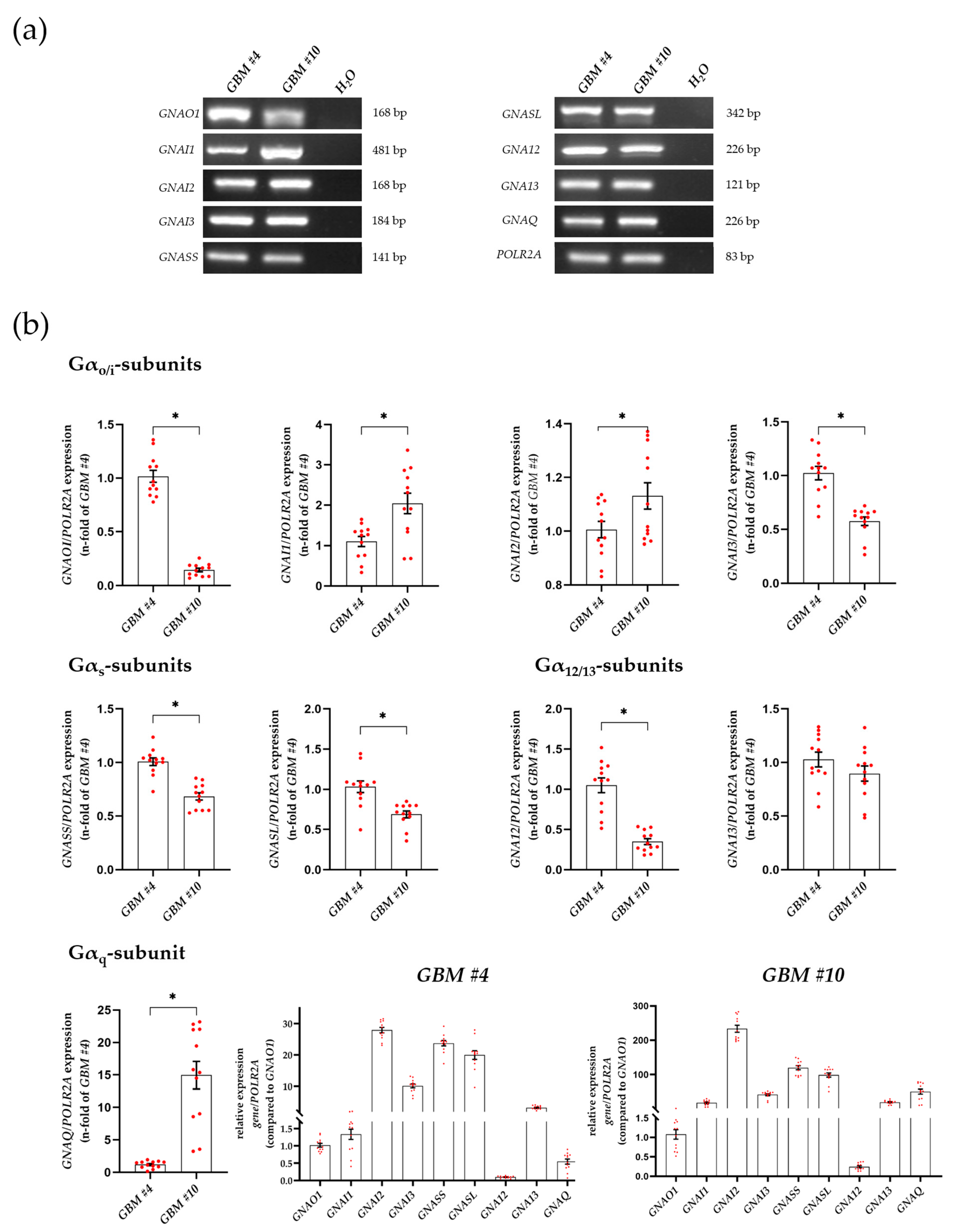
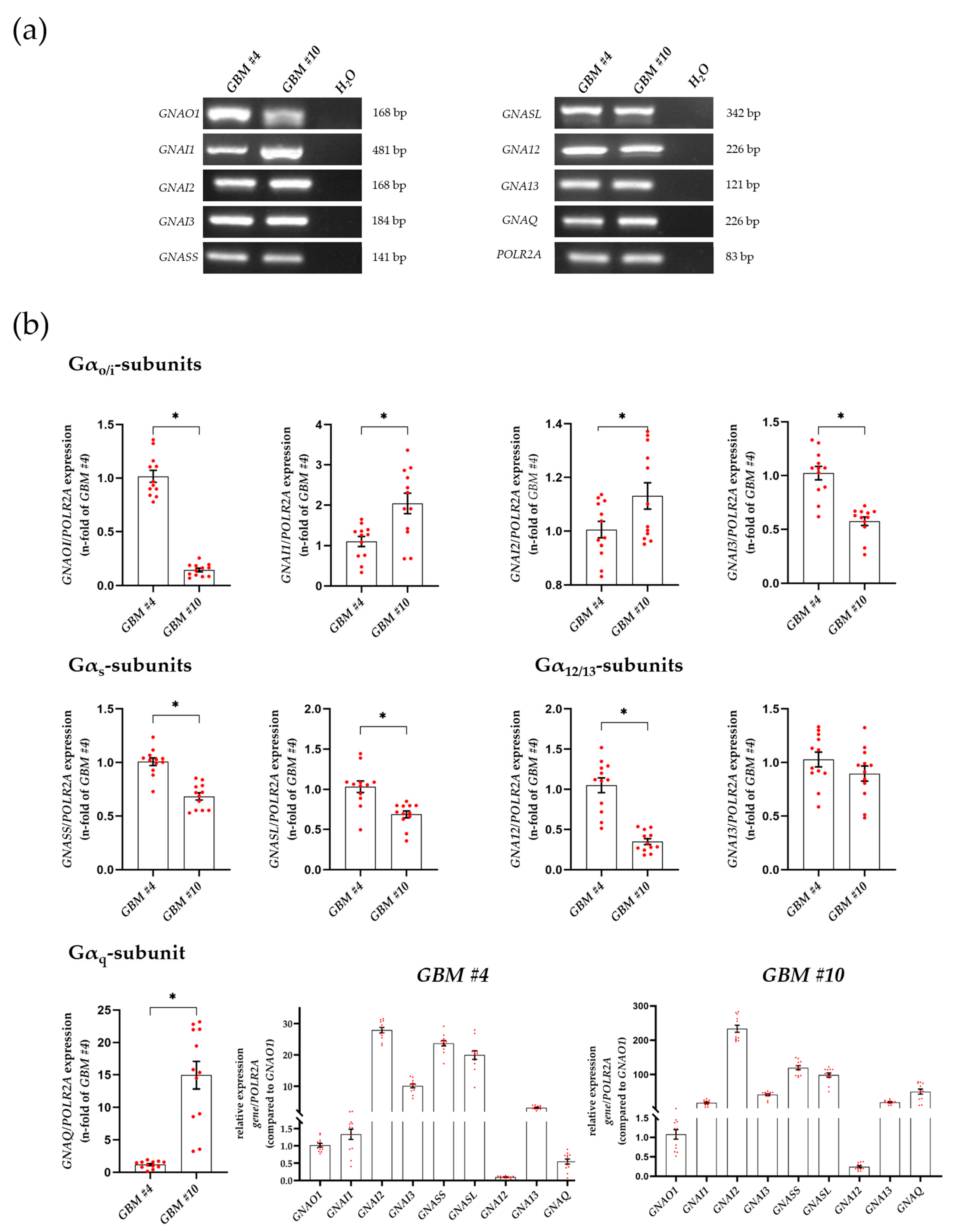
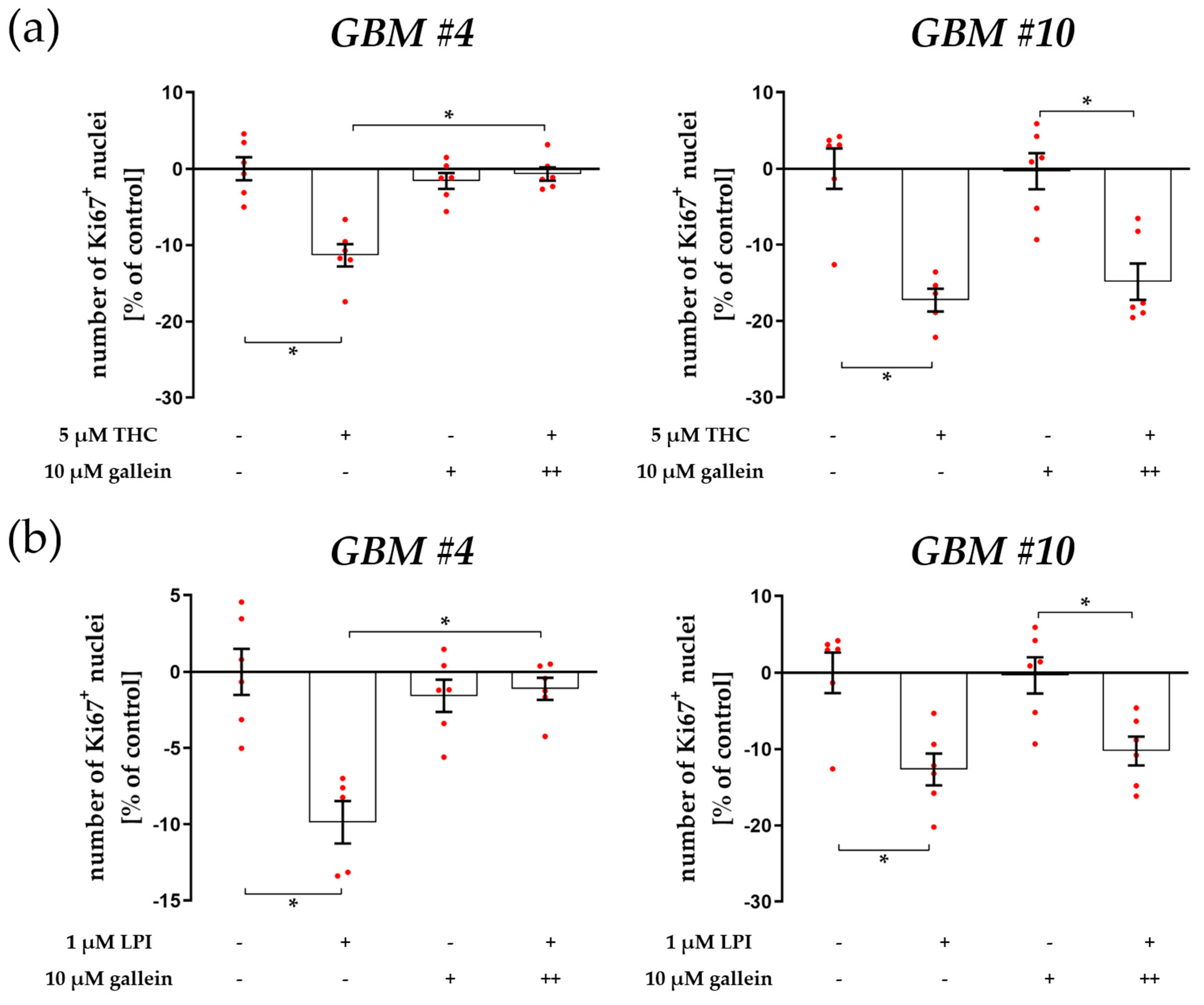
3.2. Characterization of Gα- and Gβγ-Subunits, Which Might Couple to GPR55 Signaling
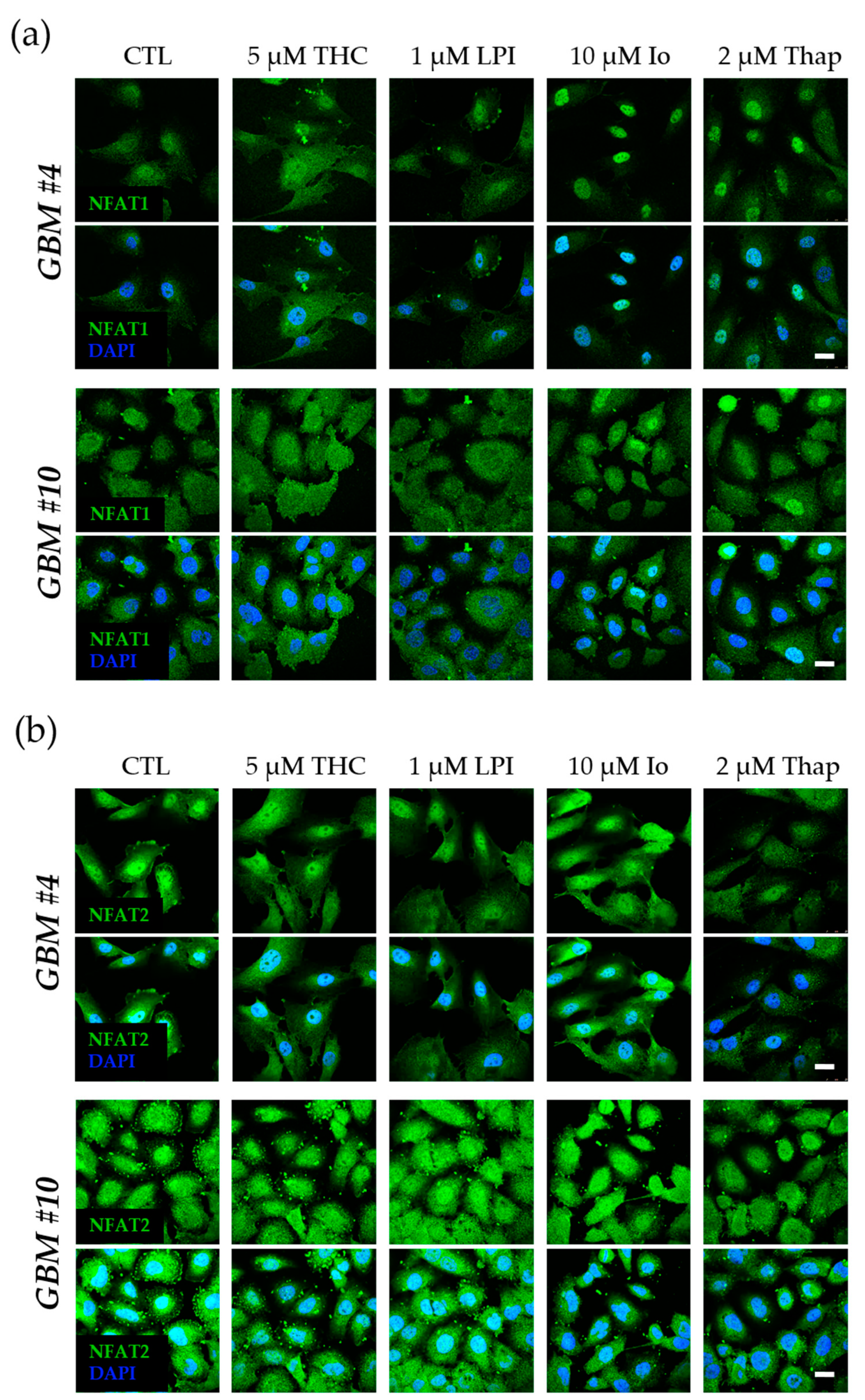
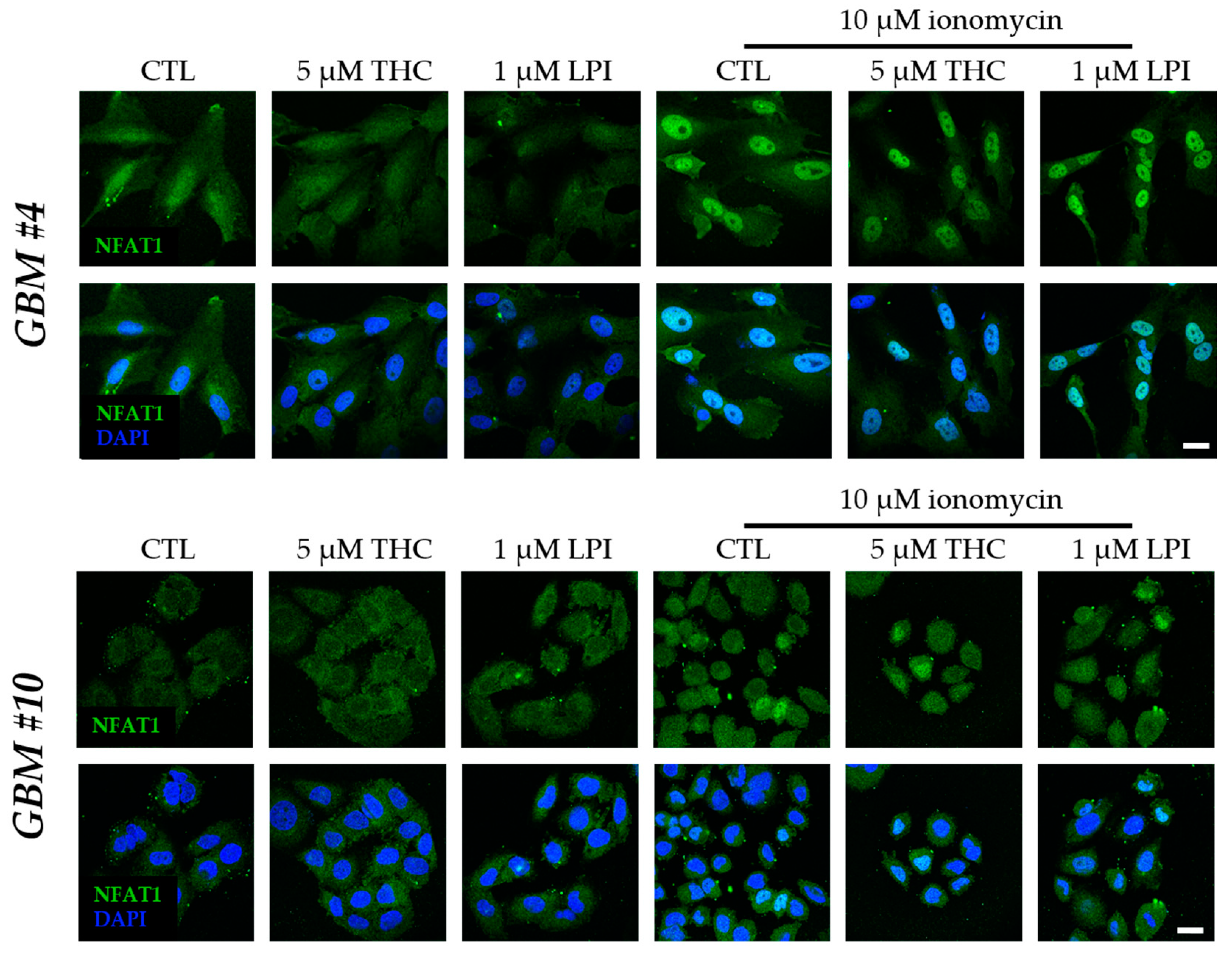
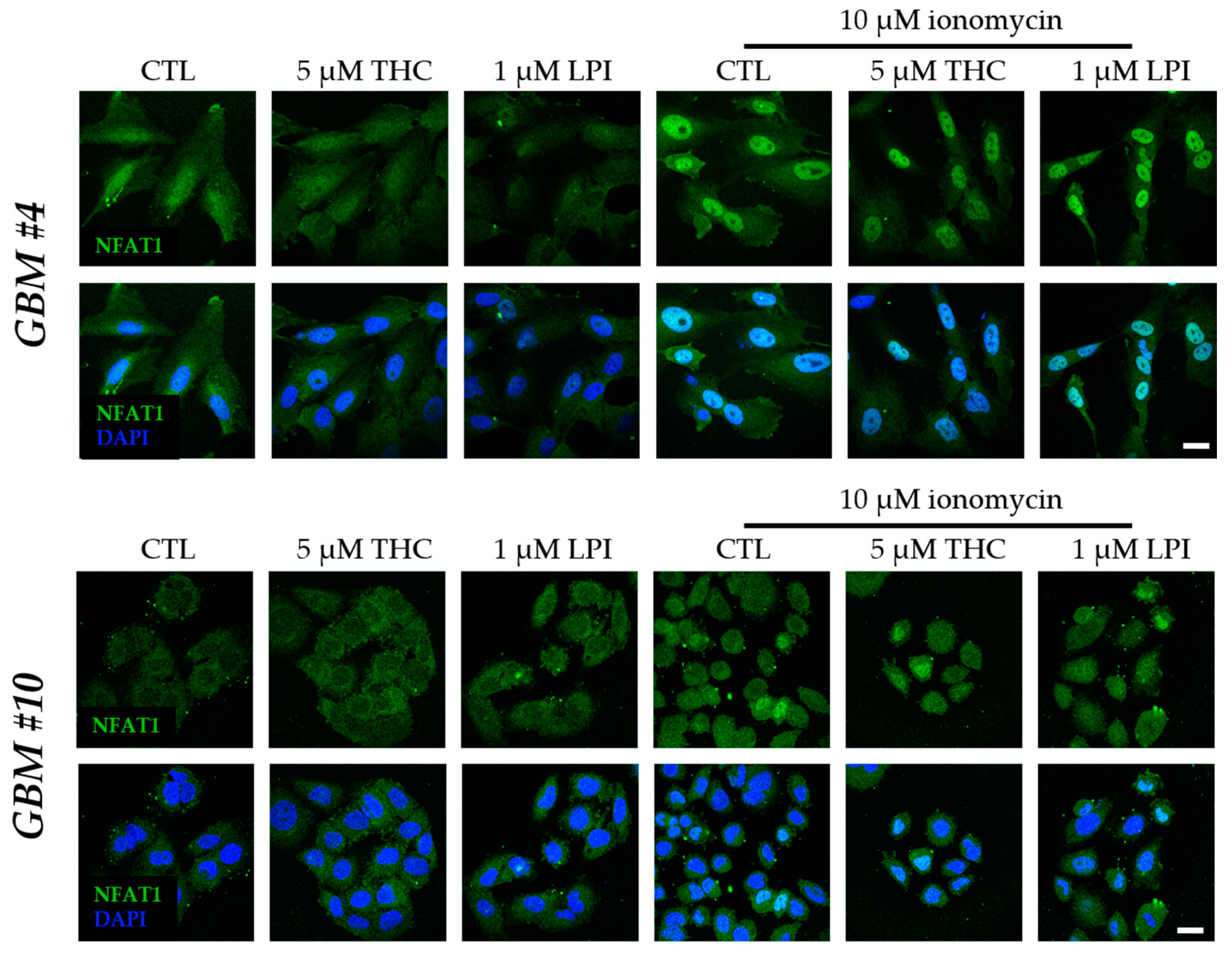
3.3. Cell-Type-Specific Calcineurin Signaling and an Unaltered Subcellular Localization of NFAT
4. Discussion
4.1. The Involvement of PLC-IP3 and RhoA-ROCK Signaling Pathways
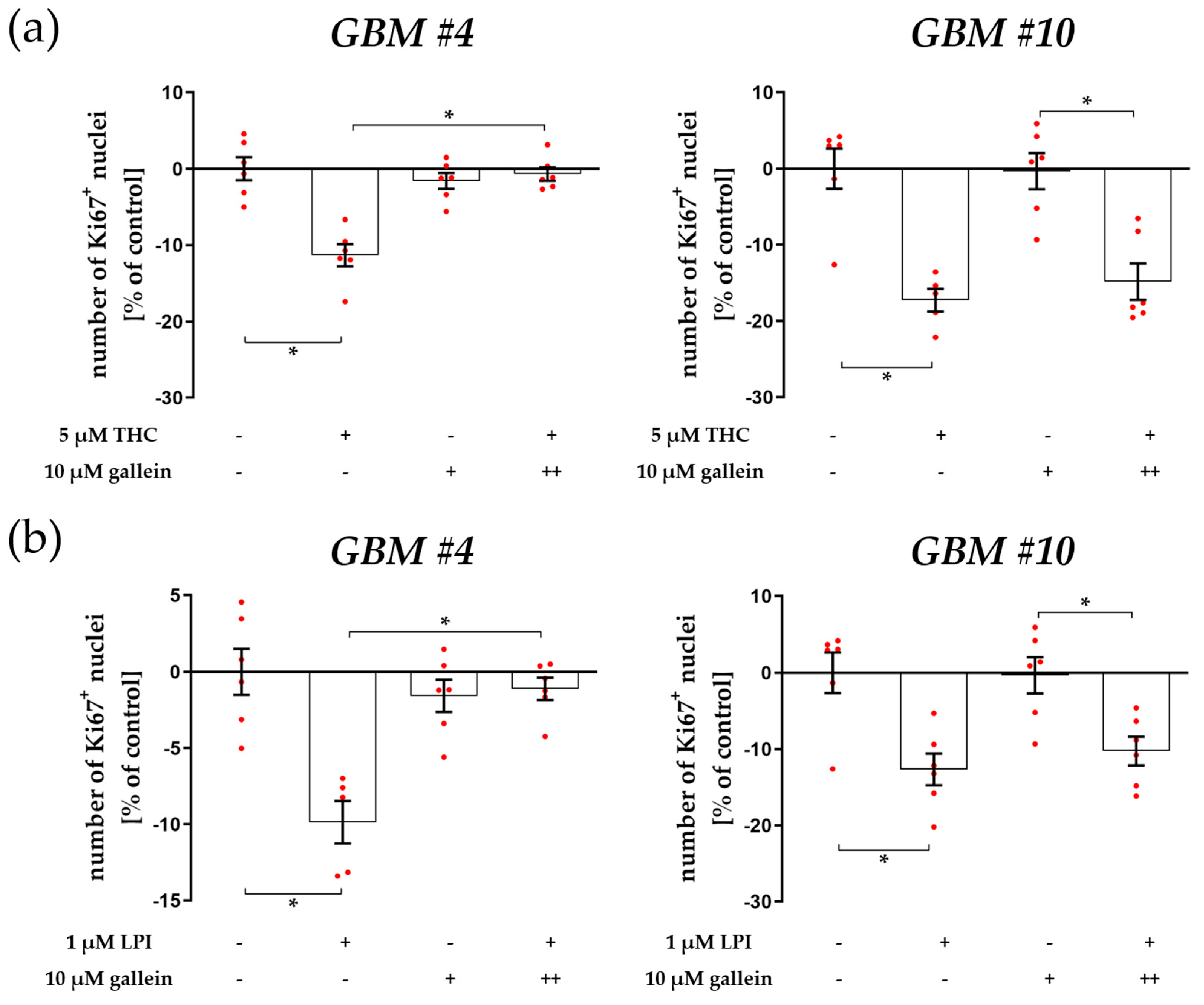
4.2. Cell-Type-Dependent Coupling of GPR55 to Gα12/13 and/or Gαq and the Role of Gβγ
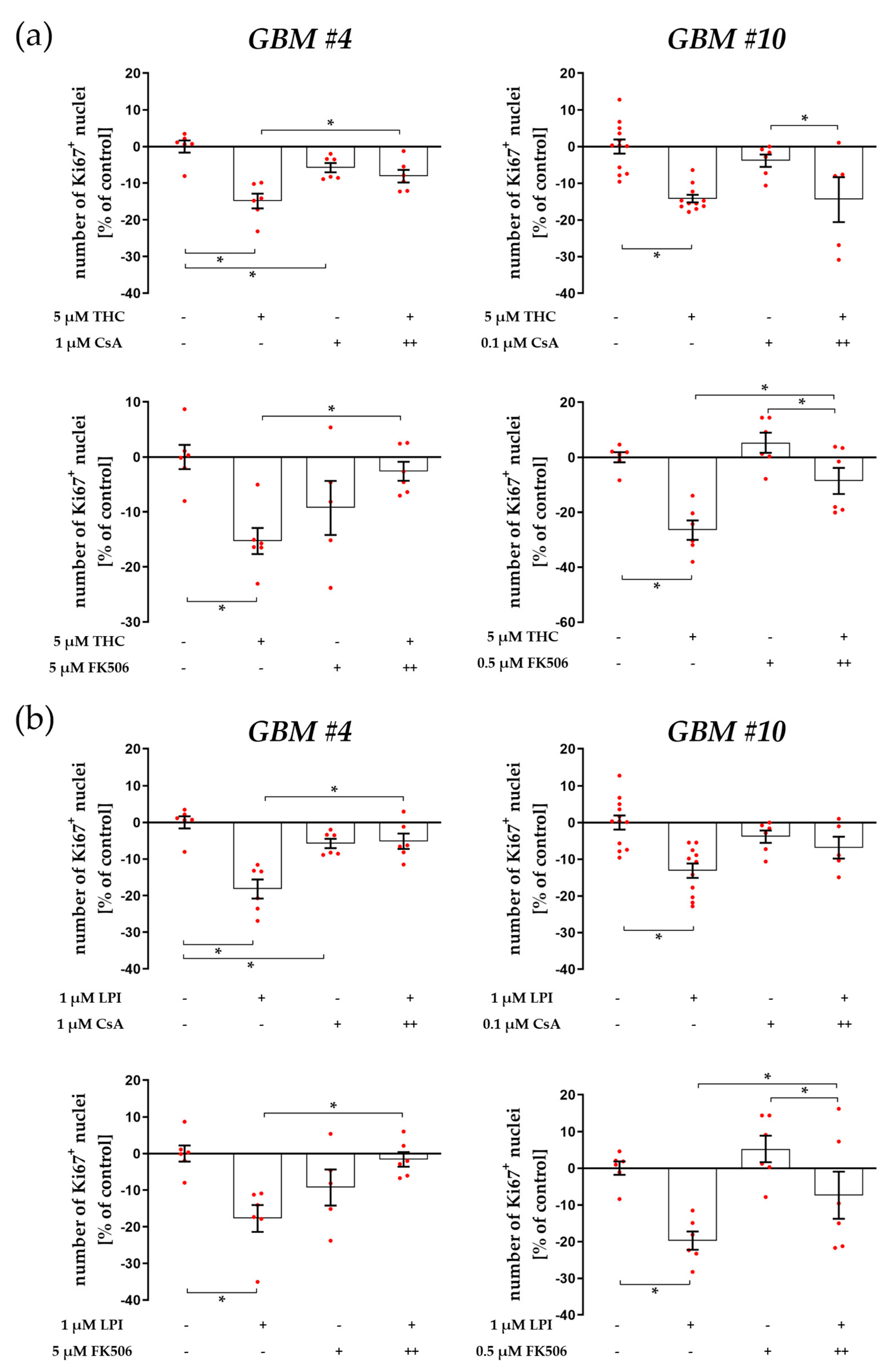
4.3. Cell-Type-Specific Calcineurin Signaling and the Role of NFAT
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The Orphan Receptor GPR55 Is a Novel Cannabinoid Receptor: GPR55, a Novel Cannabinoid Receptor. Br. J. Pharmacol. 2009, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a Cannabinoid Receptor that Increases Intracellular Calcium and Inhibits M Current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and Cloning of Three Novel Human G Protein-Coupled Receptor Genes GPR52, ΨGPR53 and GPR55: GPR55 is Extensively Expressed in Human Brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; et al. The Putative Cannabinoid Receptor GPR55 Plays a Role in Mechanical Hyperalgesia Associated with Inflammatory and Neuropathic Pain. Pain 2008, 139, 225–236. [Google Scholar] [CrossRef]
- Pietr, M.; Kozela, E.; Levy, R.; Rimmerman, N.; Lin, Y.H.; Stella, N.; Vogel, Z.; Juknat, A. Differential Changes in GPR55 during Microglial Cell Activation. FEBS Lett. 2009, 583, 2071–2076. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.B.; Kargl, J.; Andradas, C.; Brown, A.J.; Irving, A.; Sanchez, C.; Waldhoer, M. Minireview: Recent Developments in the Physiology and Pathology of the Lysophosphatidylinositol-Sensitive Receptor GPR55. Mol. Endocrinol. 2011, 25, 1835–1848. [Google Scholar] [CrossRef]
- Oka, S.; Kimura, S.; Toshida, T.; Ota, R.; Yamashita, A.; Sugiura, T. Lysophosphatidylinositol Induces Rapid Phosphorylation of P38 Mitogen-Activated Protein Kinase and Activating Transcription Factor 2 in HEK293 Cells Expressing GPR55 and IM-9 Lymphoblastoid Cells. J. Biochem. 2010, 147, 671–678. [Google Scholar] [CrossRef]
- Kremshofer, J.; Siwetz, M.; Berghold, V.M.; Lang, I.; Huppertz, B.; Gauster, M. A Role for GPR55 in Human Placental Venous Endothelial Cells. Histochem. Cell Biol. 2015, 144, 49–58. [Google Scholar] [CrossRef]
- Simcocks, A.C.; O’Keefe, L.; Jenkin, K.A.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. A Potential Role for GPR55 in the Regulation of Energy Homeostasis. Drug Discov. Today 2014, 19, 1145–1151. [Google Scholar] [CrossRef]
- Andradas, C.; Caffarel, M.M.; Pérez-Gómez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The Orphan G Protein-Coupled Receptor GPR55 Promotes Cancer Cell Proliferation via ERK. Oncogene 2011, 30, 245–252. [Google Scholar] [CrossRef]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stančić, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 Promotes Migration and Adhesion of Colon Cancer Cells Indicating a Role in Metastasis: GPR55 in Colon Cancer. Br. J. Pharmacol. 2016, 173, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 Signalling Promotes Proliferation of Pancreatic Cancer Cells and Tumour Growth in Mice, and Its Inhibition Increases Effects of Gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Grabiec, U.; Ghadban, C.; Feese, K.; Dehghani, F. The Influence of Biomechanical Properties and Cannabinoids on Tumor Invasion. Cell Adh. Migr. 2017, 11, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Feese, K.; Greither, T.; Ghadban, C.; Jäger, V.; Dehghani, F.; Grabiec, U. Synthetic Cannabinoids Influence the Invasion of Glioblastoma Cell Lines in a Cell- and Receptor-Dependent Manner. Cancers 2019, 11, 161. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Feese, K.; Ghadban, C.; Dehghani, F.; Grabiec, U. On the Influence of Cannabinoids on Cell Morphology and Motility of Glioblastoma Cells. PLoS ONE 2019, 14, e0212037. [Google Scholar] [CrossRef]
- Kolbe, M.R.; Hohmann, T.; Hohmann, U.; Ghadban, C.; Mackie, K.; Zöller, C.; Prell, J.; Illert, J.; Strauss, C.; Dehghani, F. THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner. Cancers 2021, 13, 1064. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, Y.; Pan, Z.; Li, M.; Liu, X.; Chen, X.; Qu, G.; Zhou, L.; Xu, M.; Zheng, Q.; et al. Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells. Biomolecules 2019, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Galanti, G.; Fisher, T.; Kventsel, I.; Shoham, J.; Gallily, R.; Mechoulam, R.; Lavie, G.; Amariglio, N.; Rechavi, G.; Toren, A. Δ9-Tetrahydrocannabinol Inhibits Cell Cycle Progression by Downregulation of E2F1 in Human Glioblastoma Multiforme Cells. Acta Oncol. 2008, 47, 1062–1070. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Δ9-Tetrahydrocannabinol Inhibits Cell Cycle Progression in Human Breast Cancer Cells through Cdc2 Regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef]
- Sánchez, C.; Galve-Roperh, I.; Canova, C.; Brachet, P.; Guzmán, M. Delta9-Tetrahydrocannabinol Induces Apoptosis in C6 Glioma Cells. FEBS Lett. 1998, 436, 6–10. [Google Scholar] [CrossRef]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L.; et al. The Stress-Regulated Protein P8 Mediates Cannabinoid-Induced Apoptosis of Tumor Cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. In Phytocannabinoids; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 103, pp. 103–131. ISBN 978-3-319-45539-6. [Google Scholar]
- Huang, L.; Ramirez, J.C.; Frampton, G.A.; Golden, L.E.; Quinn, M.A.; Pae, H.Y.; Horvat, D.; Liang, L.; DeMorrow, S. Anandamide Exerts Its Antiproliferative Actions on Cholangiocarcinoma by Activation of the GPR55 Receptor. Lab. Investig. 2011, 91, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Akimov, M.G.; Gretskaya, N.M.; Dudina, P.V.; Sherstyanykh, G.D.; Zinchenko, G.N.; Serova, O.V.; Degtyaryova, K.O.; Deyev, I.E.; Bezuglov, V.V. The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells. Int. J. Mol. Sci. 2023, 24, 5524. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Balenga, N.A.B.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 Ligand L-α-lysophosphatidylinositol Promotes RhoA-dependent Ca2+ Signaling and NFAT Activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Ueno, S.; Yanagihata, Y.; Nakahata, N. Lysophosphatidylinositol Causes Neurite Retraction via GPR55, G13 and RhoA in PC12 Cells. PLoS ONE 2011, 6, e24284. [Google Scholar] [CrossRef]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The Putative Cannabinoid Receptor GPR55 Defines a Novel Autocrine Loop in Cancer Cell Proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef]
- Vong, C.T.; Tseng, H.H.L.; Kwan, Y.W.; Lee, S.M.-Y.; Hoi, M.P.M. G-Protein Coupled Receptor 55 Agonists Increase Insulin Secretion through Inositol Trisphosphate-Mediated Calcium Release in Pancreatic β-Cells. Eur. J. Pharmacol. 2019, 854, 372–379. [Google Scholar] [CrossRef]
- Waldeck-Weiermair, M.; Zoratti, C.; Osibow, K.; Balenga, N.; Goessnitzer, E.; Waldhoer, M.; Malli, R.; Graier, W.F. Integrin Clustering Enables Anandamide-Induced Ca2+ Signaling in Endothelial Cells via GPR55 by Protection against CB1-Receptor-Triggered Repression. J. Cell Sci. 2008, 121, 1704–1717. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.; Schröder, R.; Kargl, J.K.; Platzer, W.; Martini, L.; Arthur, S.; Penman, J.; Whistler, J.L.; Kostenis, E.; et al. GPR55 Ligands Promote Receptor Coupling to Multiple Signalling Pathways: GPR55 Signalling. Br. J. Pharmacol. 2010, 160, 604–614. [Google Scholar] [CrossRef]
- Shen, X.; Jiang, H.; Ying, M.; Xie, Z.; Li, X.; Wang, H.; Zhao, J.; Lin, C.; Wang, Y.; Feng, S.; et al. Calcineurin Inhibitors Cyclosporin A and Tacrolimus Protect against Podocyte Injury Induced by Puromycin Aminonucleoside in Rodent Models. Sci. Rep. 2016, 6, 32087. [Google Scholar] [CrossRef]
- Maguire, O.; Tornatore, K.M.; O’Loughlin, K.L.; Venuto, R.C.; Minderman, H. Nuclear Translocation of Nuclear Factor of Activated T Cells (NFAT) as a Quantitative Pharmacodynamic Parameter for Tacrolimus. Cytom. Part A 2013, 83, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Surve, C.R.; Lehmann, D.; Smrcka, A.V. A Chemical Biology Approach Demonstrates G Protein Βγ Subunits Are Sufficient to Mediate Directional Neutrophil Chemotaxis. J. Biol. Chem. 2014, 289, 17791–17801. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a Lysophosphatidylinositol Receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Small-Howard, A.L.; Shimoda, L.M.N.; Adra, C.N.; Turner, H. Anti-Inflammatory Potential of CB1-Mediated cAMP Elevation in Mast Cells. Biochem. J. 2005, 388, 465–473. [Google Scholar] [CrossRef] [PubMed]
- AlSuleimani, Y.M.; Hiley, C.R. The GPR55 Agonist Lysophosphatidylinositol Relaxes Rat Mesenteric Resistance Artery and Induces Ca2+ Release in Rat Mesenteric Artery Endothelial Cells: The GPR55 Agonist Lysophosphatidylinositol. Br. J. Pharmacol. 2015, 172, 3043–3057. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Oka, S.; Tanikawa, T.; Nemoto-Sasaki, Y.; Matsumoto, N.; Ishiguro, H.; Arata, Y.; Sugiura, T.; Yamashita, A. Lysophosphatidylinositol Induced Morphological Changes and Stress Fiber Formation through the GPR55-RhoA-ROCK Pathway. Int. J. Mol. Sci. 2022, 23, 10932. [Google Scholar] [CrossRef]
- Akimov, M.G.; Gamisonia, A.M.; Dudina, P.V.; Gretskaya, N.M.; Gaydaryova, A.A.; Kuznetsov, A.S.; Zinchenko, G.N.; Bezuglov, V.V. GPR55 Receptor Activation by the N-Acyl Dopamine Family Lipids Induces Apoptosis in Cancer Cells via the Nitric Oxide Synthase (nNOS) Over-Stimulation. Int. J. Mol. Sci. 2021, 22, 622. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Hille, B.; Mackie, K. The Cannabinoid Agonist WIN55,212-2 Increases Intracellular Calcium via CB1 Receptor Coupling to Gq/11 G Proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef]
- Marzban, H.; Kong, J.; Mehr, S.; Vriend, J.; Li, J.; Guan, T.; Chung, S.; Mirzaei, N.; Marzban, A.; Shojaei, S.; et al. Mevalonate Cascade and Neurodevelopmental and Neurodegenerative Diseases: Future Targets for Therapeutic Application. Curr. Mol. Pharmacol. 2017, 10, 115–140. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Reece, J.; Putney, J.W. Role of the Cytoskeleton in Calcium Signaling in NIH 3T3 Cells. J. Biol. Chem. 1997, 272, 26555–26561. [Google Scholar] [CrossRef]
- Finlay, D.B.; Cawston, E.E.; Grimsey, N.L.; Hunter, M.R.; Korde, A.; Vemuri, V.K.; Makriyannis, A.; Glass, M. Gαs Signalling of the CB1 Receptor and the Influence of Receptor Number: CB1 Receptor Gαs Signalling. Br. J. Pharmacol. 2017, 174, 2545–2562. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Zavaleta, R.; Ávalos-Fuentes, J.A.; González-Hernández, A.V.; Recillas-Morales, S.; Paz-Bermúdez, F.J.; Leyva-Gómez, G.; Cortés, H.; Florán, B. Presynaptic Nigral GPR55 Receptors Stimulate [3H]-GABA Release through [3H]-cAMP Production and PKA Activation and Promote Motor Behavior. Synapse 2022, 76, e22246. [Google Scholar] [CrossRef] [PubMed]
- Pfeil, E.M.; Brands, J.; Merten, N.; Vögtle, T.; Vescovo, M.; Rick, U.; Albrecht, I.-M.; Heycke, N.; Kawakami, K.; Ono, Y.; et al. Heterotrimeric G Protein Subunit Gαq Is a Master Switch for Gβγ-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 2020, 80, 940–954.e6. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The Cannabinoid Receptor CB1 Modulates the Signaling Properties of the Lysophosphatidylinositol Receptor GPR55. J. Biol. Chem. 2012, 287, 44234–44248. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Pérez-Gómez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB2-GPR55 Receptor Heteromers Modulates Cancer Cell Signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and Cannabinoid CB 2 Receptors Modulates Signalling: Heteromerization of GPR55 and CB 2 Receptors. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [PubMed]
- Bang, G.; Ghil, S. BRET Analysis Reveals Interaction between the Lysophosphatidic Acid Receptor LPA2 and the Lysophosphatidylinositol Receptor GPR55 in Live Cells. FEBS Lett. 2021, 595, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Kurose, H. Gi/o Protein-Dependent and -Independent Actions of Pertussis Toxin (PTX). Toxins 2011, 3, 884–899. [Google Scholar] [CrossRef]
- Nishida, M.; Suda, R.; Nagamatsu, Y.; Tanabe, S.; Onohara, N.; Nakaya, M.; Kanaho, Y.; Shibata, T.; Uchida, K.; Sumimoto, H.; et al. Pertussis Toxin Up-Regulates Angiotensin Type 1 Receptors through Toll-like Receptor 4-Mediated Rac Activation. J. Biol. Chem. 2010, 285, 15268–15277. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Sternweis, P.C. Regulation of Purified Subtypes of Phosphatidylinositol-Specific Phospholipase C Beta by G Protein Alpha and Beta Gamma Subunits. J. Biol. Chem. 1993, 268, 9667–9674. [Google Scholar] [CrossRef]
- Philip, F.; Kadamur, G.; Silos, R.G.; Woodson, J.; Ross, E.M. Synergistic Activation of Phospholipase C-Β3 by Gαq and Gβγ Describes a Simple Two-State Coincidence Detector. Curr. Biol. 2010, 20, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Litosch, I. RhoA Co-Ordinates with Heterotrimeric G Proteins to Regulate Efficacy. Biochem. Biophys. Res. Commun. 2011, 415, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Rebres, R.A.; Roach, T.I.A.; Fraser, I.D.C.; Philip, F.; Moon, C.; Lin, K.-M.; Liu, J.; Santat, L.; Cheadle, L.; Ross, E.M.; et al. Synergistic Ca2+ Responses by Gαi- and Gαq-Coupled G-Protein-Coupled Receptors Require a Single PLCβ Isoform That is Sensitive to Both Gβγ and Gαq. J. Biol. Chem. 2011, 286, 942–951. [Google Scholar] [CrossRef] [PubMed]
- White, A.D.; Jean-Alphonse, F.G.; Fang, F.; Peña, K.A.; Liu, S.; König, G.M.; Inoue, A.; Aslanoglou, D.; Gellman, S.H.; Kostenis, E.; et al. Gq/11-Dependent Regulation of Endosomal cAMP Generation by Parathyroid Hormone Class B GPCR. Proc. Natl. Acad. Sci. USA 2020, 117, 7455–7460. [Google Scholar] [CrossRef]
- Tang, W.; Tu, Y.; Nayak, S.K.; Woodson, J.; Jehl, M.; Ross, E.M. Gβγ Inhibits Gα GTPase-Activating Proteins by Inhibition of Gα-GTP Binding during Stimulation by Receptor. J. Biol. Chem. 2006, 281, 4746–4753. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Structure and Function of IP3 Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef]
- Thiel, G.; Schmidt, T.; Rössler, O.G. Ca2+ Microdomains, Calcineurin and the Regulation of Gene Transcription. Cells 2021, 10, 875. [Google Scholar] [CrossRef]
- Park, Y.-J.; Yoo, S.-A.; Kim, M.; Kim, W.-U. The Role of Calcium–Calcineurin–NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef]
- Kissinger, C.R.; Parge, H.E.; Knighton, D.R.; Lewis, C.T.; Pelletier, L.A.; Tempczyk, A.; Kalish, V.J.; Tucker, K.D.; Showalter, R.E.; Moomaw, E.W.; et al. Crystal Structures of Human Calcineurin and the Human FKBP12–FK506–Calcineurin Complex. Nature 1995, 378, 641–644. [Google Scholar] [CrossRef]
- Jin, L.; Harrison, S.C. Crystal Structure of Human Calcineurin Complexed with Cyclosporin A and Human Cyclophilin. Proc. Natl. Acad. Sci. USA 2002, 99, 13522–13526. [Google Scholar] [CrossRef]
- Fruman, D.A.; Klee, C.B.; Bierer, B.E.; Burakoff, S.J. Calcineurin Phosphatase Activity in T Lymphocytes is Inhibited by FK 506 and Cyclosporin A. Proc. Natl. Acad. Sci. USA 1992, 89, 3686–3690. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Kohler, N.G.; Joly, A. Cyclosporine A Is an Uncompetitive Inhibitor of Proteasome Activity and Prevents NF-κB Activation. FEBS Lett. 1997, 413, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Brown, A.J.; Andersen, L.; Dorn, G.; Schicho, R.; Waldhoer, M.; Heinemann, A. A Selective Antagonist Reveals a Potential Role of G Protein–Coupled Receptor 55 in Platelet and Endothelial Cell Function. J. Pharmacol. Exp. Ther. 2013, 346, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Parekh, A.B. Distinct Spatial Ca2+ Signatures Selectively Activate Different NFAT Transcription Factor Isoforms. Mol. Cell 2015, 58, 232–243. [Google Scholar] [CrossRef]
- Morgan, A.J.; Jacob, R. Ionomycin Enhances Ca2+ Influx by Stimulating Store-Regulated Cation Entry and Not by a Direct Action at the Plasma Membrane. Biochem. J. 1994, 300, 665–672. [Google Scholar] [CrossRef]
- Razani-Boroujerdi, S.; Partridge, L.D.; Sopori, M.L. Intracellular Calcium Signaling Induced by Thapsigargin in Excitable and Inexcitable Cells. Cell Calcium 1994, 16, 467–474. [Google Scholar] [CrossRef]
- Vihma, H.; Luhakooder, M.; Pruunsild, P.; Timmusk, T. Regulation of Different Human NFAT Isoforms by Neuronal Activity. J. Neurochem. 2016, 137, 394–408. [Google Scholar] [CrossRef]
- Yissachar, N.; Sharar Fischler, T.; Cohen, A.A.; Reich-Zeliger, S.; Russ, D.; Shifrut, E.; Porat, Z.; Friedman, N. Dynamic Response Diversity of NFAT Isoforms in Individual Living Cells. Mol. Cell 2013, 49, 322–330. [Google Scholar] [CrossRef]
- Screaton, R.A.; Conkright, M.D.; Katoh, Y.; Best, J.L.; Canettieri, G.; Jeffries, S.; Guzman, E.; Niessen, S.; Yates, J.R.; Takemori, H.; et al. The CREB Coactivator TORC2 Functions as a Calcium- and cAMP-Sensitive Coincidence Detector. Cell 2004, 119, 61–74. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The Complex Relationship between TFEB Transcription Factor Phosphorylation and Subcellular Localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Wu, H. Activation of MEF2 by Muscle Activity Is Mediated through a Calcineurin-Dependent Pathway. EMBO J. 2001, 20, 6414–6423. [Google Scholar] [CrossRef] [PubMed]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB Phosphorylation and Dephosphorylation: A Ca2+- and Stimulus Duration–Dependent Switch for Hippocampal Gene Expression. Cell 1996, 87, 1203–1214. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substances | Targets | Behavior | Solvent | Concentration | Pre-Incubation Time | Company | Article Number |
|---|---|---|---|---|---|---|---|
| 2-APB | IP3-sensitive receptors | inhibitor | DMSO | 10 µM | 30 min [28] | Tocris, Bristol, UK | 1224 |
| cyclosporine A (CsA) | calcineurin | inhibitor | DMSO | 0.1 µM 1 µM | 1 h [31] | Tocris, Bristol, UK | 1101 |
| Dronabinol (THC) | CB1 CB2 GPR18 GPR55 | agonist [22] | DMSO | 5 µM | - | THC pharm GmbH, Frankfurt am Main, Germany | THC-1016 |
| FK506 | calcineurin | inhibitor | DMSO | 0.5 µM 5 µM | 1.5 h [32] | Tocris, Bristol, UK | 3631 |
| forskolin | adenylyl cyclase | activator | DMSO | 0.1 µM 1 µM 5 µM 10 µM 30 µM | - | Sigma, Darmstadt, Germany | |
| gallein | Gβγ-subunits | inhibitor | DMSO | 10 µM | 15 min [33] | Tocris, Bristol, UK | 3090 |
| ionomycin | - | calcium ionophore | DMSO | 10 µM | - | Tocris, Bristol, UK | 1704 |
| lysophosphatidyl-inositol (LPI) | GPR55 | agonist [34] | DMSO | 1 µM | - | Sigma, Darmstadt, Germany | L7635 |
| pertussis toxine (PTX) | Gi/o-proteins | inhibitor | H2O | 100 ng/ml | 16 h [35] | Tocris, Bristol, UK | 3097 |
| U73122 | PLC | inhibitor | DMSO | 0.1 µM | 15 min [36] | Cayman Chemicals, Ann Arbor, Michigan, USA | 70740 |
| U73343 | - | inactive analogue of U73122 | DMSO | 0.1 µM | 15 min | Tocris, Bristol, UK | 4133 |
| thapsigargin | SERCA | inhibitor | DMSO | 2 µM | - | Tocris, Bristol, UK | 1138 |
| Y-27632 | ROCK | inhibitor | H2O | 10 µM | 1 h [37] | Tocris, Bristol, UK | 1254 |
| Gene | Accession Number | Forward Primer (5′-> 3′) | Reverse Primer (5′-> 3′) | Size [bp] |
|---|---|---|---|---|
| GAPDH | NM_002046 | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG | 87 |
| GNA12 | NM_007353 | GAGCTCTGCAGGTGTGGATT | GAAGATGGGAGAGCCGTCTG | 226 |
| GNA13 | NM_006572 | CGTCGAGAATTTCAACTGGGTG | CTTTGGTGGGTCTTCTGGCA | 121 |
| GNAI1 | NM_002069 | GCTGAAGATGAAGAAATGAACCGAA | GTCCCAGATGCATTTGCCTT | 481 |
| GNAI2 | NM_002070 | CAGGCAGCTATTTGCACTGTC | AGGTCGTTCAGGTAGTAGGC | 168 |
| GNAI3 | NM_006496 | AGTTTCCGTGGTGTGAGTGA | GATTCTCCAGCACCGAGTAGC | 184 |
| GNAO1 | NM_020988 | TGGTGATAAGGAGAGAAAGGCTG | TCGTTGAGCTGATACTCCCG | 168 |
| GNAQ | NM_002072 | TGAGCACAATAAGGCTCATGC | ATCTTGTTGCGTAGGCAGGT | 226 |
| GNASL | NM_000516 | GAGCAACAGCGATGGTGAGA | TGATCGCTCGGCACATAGTC | 342 |
| GNASS | NM_080426 | GCAGAAGGACAAGCAGGTCTA | TTGGTTGCCTTCTCACTGTCTC | 141 |
| POLR2A | NM_000937 | CTTGCCCCGTGCCATGCAGA | CTCGCACCCGGCCTTCCTTG | 83 |
| TBP | NM_003194 | GAGCTGTGATGTGAAGTTTCC | TCTGGGTTTGATCATTCTGTAG | 117 |
| Antibodies | Species | Concentration (Application) | Company | Article Number |
|---|---|---|---|---|
| anti-Ki67 | rabbit | 1:200 (ICC) | DSC innovative Diagnostic-System, Hamburg, Germany | KI681C002 |
| anti-NFAT1 | rabbit | 1.200 (IF) | Cell Signaling, Danvers, MA, USA | 5861 |
| anti-NFAT2 | rabbit | 1:200 (IF) | Invitrogen, Schwerte, Germany | PA5-90432 |
| anti-NFAT3 | rabbit | 1:200 (IF) | Invitrogen, Schwerte, Germany | PA1-021 |
| anti-NFAT4 | rabbit | 1:200 (IF) | Invitrogen, Schwerte, Germany | PA5-99546 |
| anti-rabbit Alexa488 conjugated | goat | 1:200 (IF) | Invitrogen, Schwerte, Germany | A11034 |
| anti-rabbit IgG, biotin conjugated | goat | 1:100 (ICC) | Sigma, Darmstadt, Germany | B7389 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolbe, M.R.; Hohmann, T.; Hohmann, U.; Maronde, E.; Golbik, R.; Prell, J.; Illert, J.; Strauss, C.; Dehghani, F. Elucidation of GPR55-Associated Signaling behind THC and LPI Reducing Effects on Ki67-Immunoreactive Nuclei in Patient-Derived Glioblastoma Cells. Cells 2023, 12, 2646. https://doi.org/10.3390/cells12222646
Kolbe MR, Hohmann T, Hohmann U, Maronde E, Golbik R, Prell J, Illert J, Strauss C, Dehghani F. Elucidation of GPR55-Associated Signaling behind THC and LPI Reducing Effects on Ki67-Immunoreactive Nuclei in Patient-Derived Glioblastoma Cells. Cells. 2023; 12(22):2646. https://doi.org/10.3390/cells12222646
Chicago/Turabian StyleKolbe, Marc Richard, Tim Hohmann, Urszula Hohmann, Erik Maronde, Ralph Golbik, Julian Prell, Jörg Illert, Christian Strauss, and Faramarz Dehghani. 2023. "Elucidation of GPR55-Associated Signaling behind THC and LPI Reducing Effects on Ki67-Immunoreactive Nuclei in Patient-Derived Glioblastoma Cells" Cells 12, no. 22: 2646. https://doi.org/10.3390/cells12222646
APA StyleKolbe, M. R., Hohmann, T., Hohmann, U., Maronde, E., Golbik, R., Prell, J., Illert, J., Strauss, C., & Dehghani, F. (2023). Elucidation of GPR55-Associated Signaling behind THC and LPI Reducing Effects on Ki67-Immunoreactive Nuclei in Patient-Derived Glioblastoma Cells. Cells, 12(22), 2646. https://doi.org/10.3390/cells12222646