
Evaluating the Mechanism of Cell Death in Melanoma Induced by the Cannabis Extract PHEC-66

 , ,
, ,  and
and
Abstract
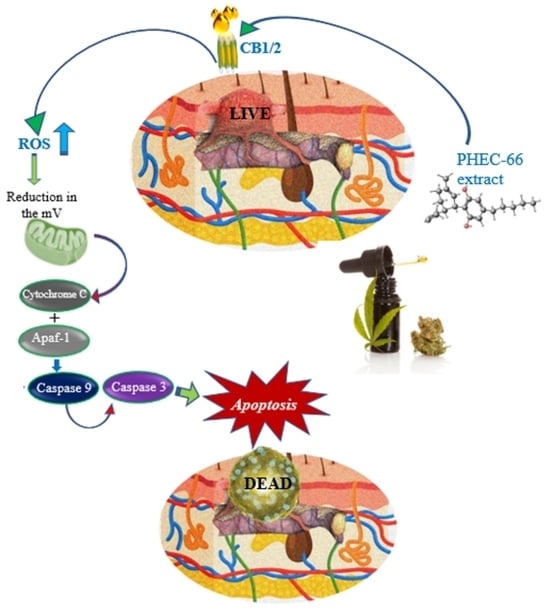
1. Introduction
2. Materials and Methods
2.1. Materials
Cell Culture
2.2. Methods
2.2.1. Quantitative Polymerase Chain Reaction (qPCR)
2.2.2. CB1/CB2 Receptors Antagonism Assay
2.2.3. Reactive Oxygen Species (ROS)
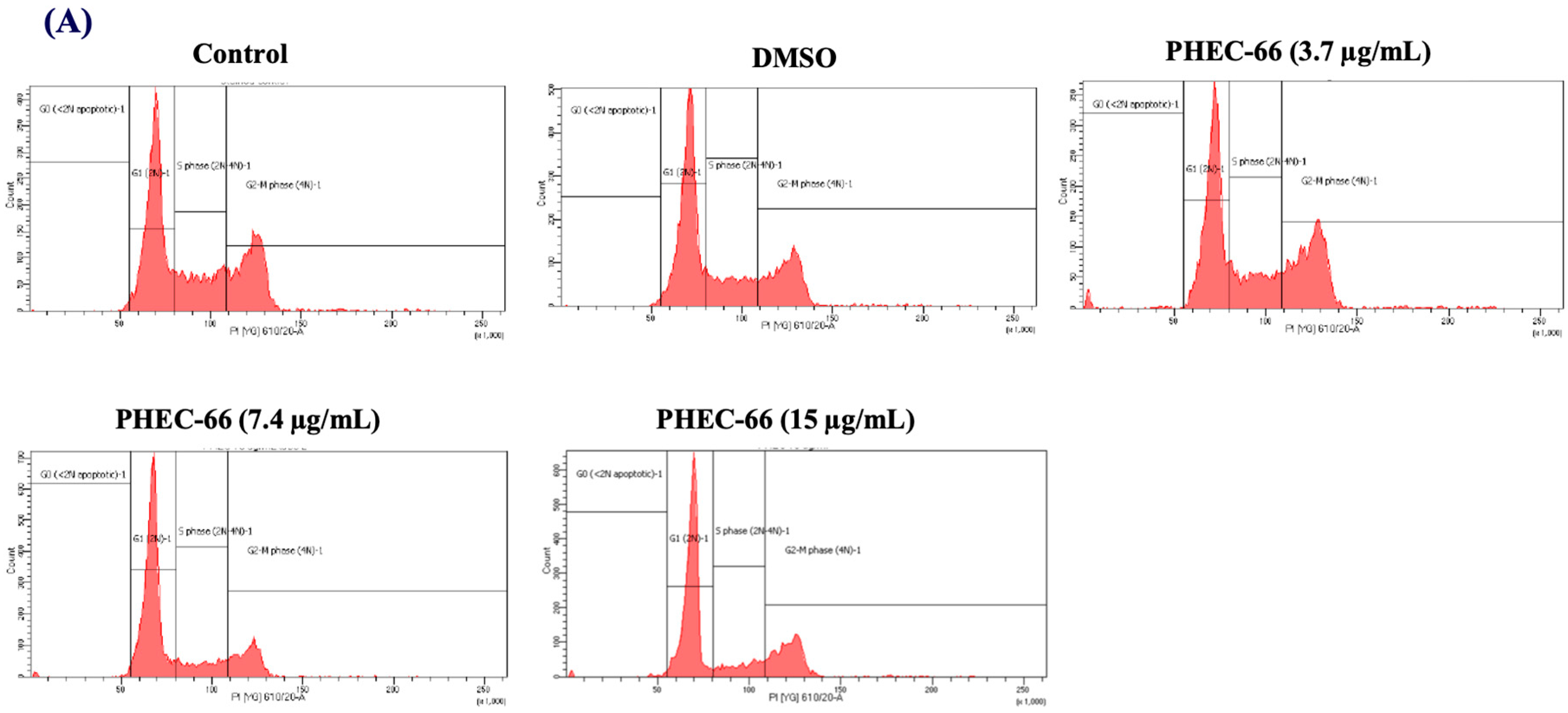
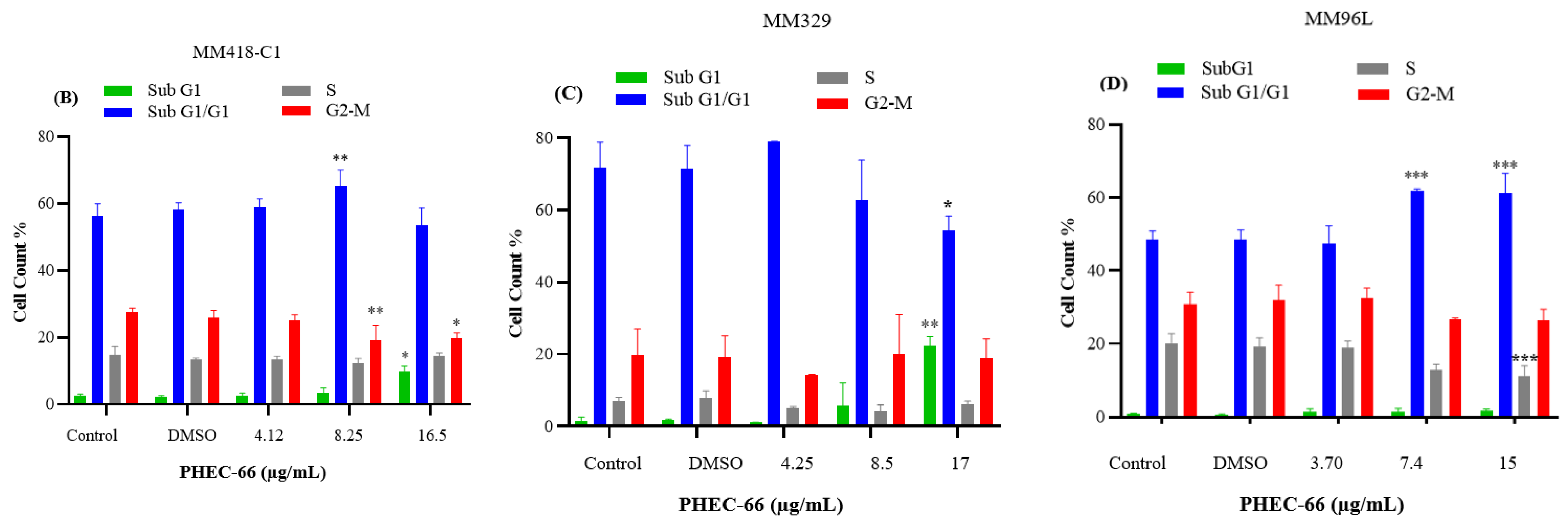
2.2.4. Cell Cycle Arrest Assay
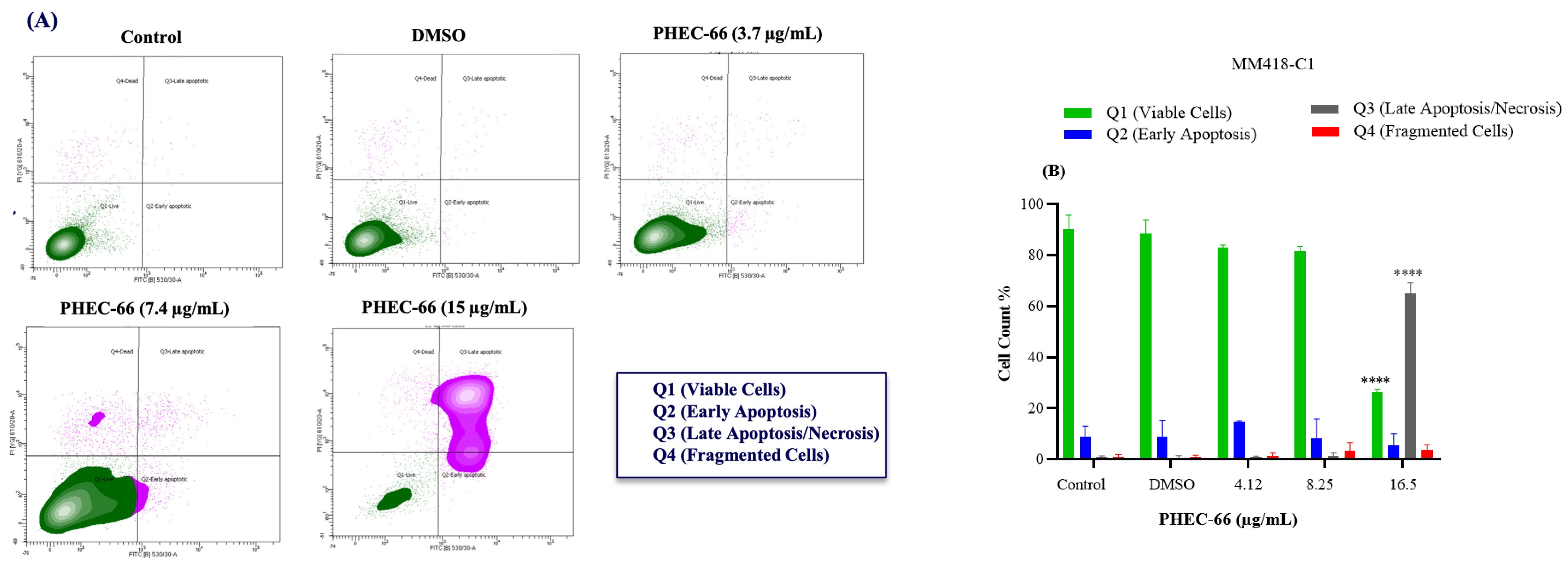
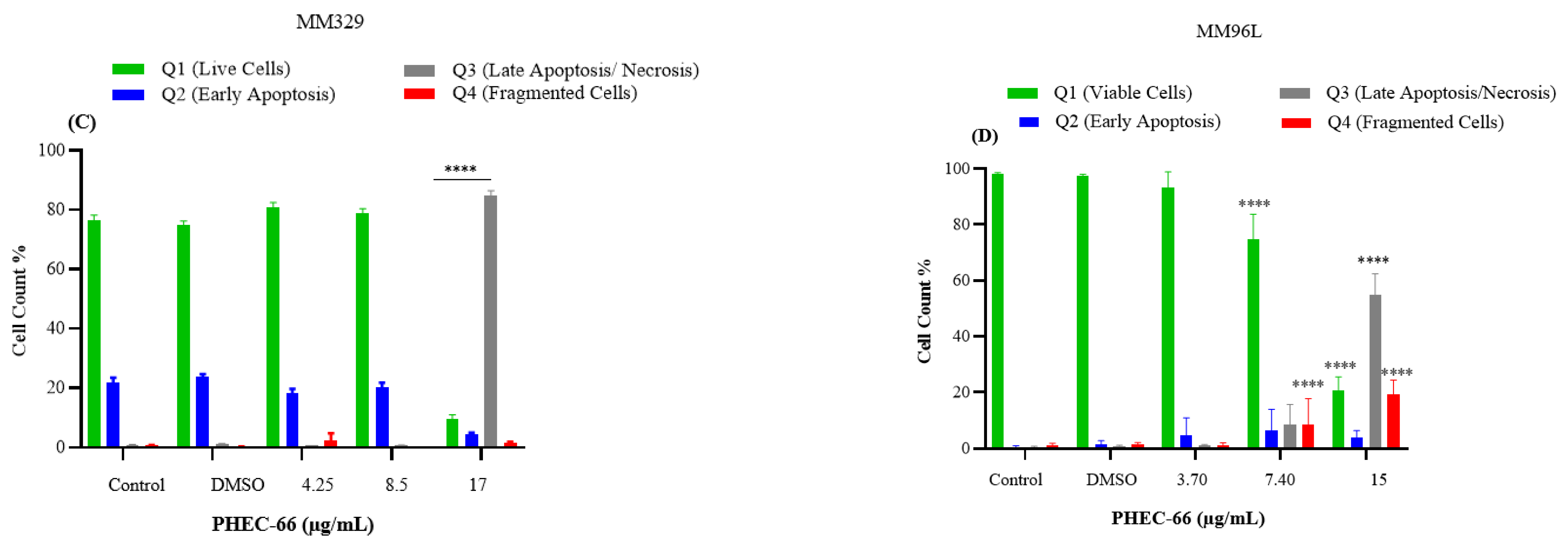
2.2.5. Annexin V-FITC/PI Staining Assay
2.3. Statistical Analysis
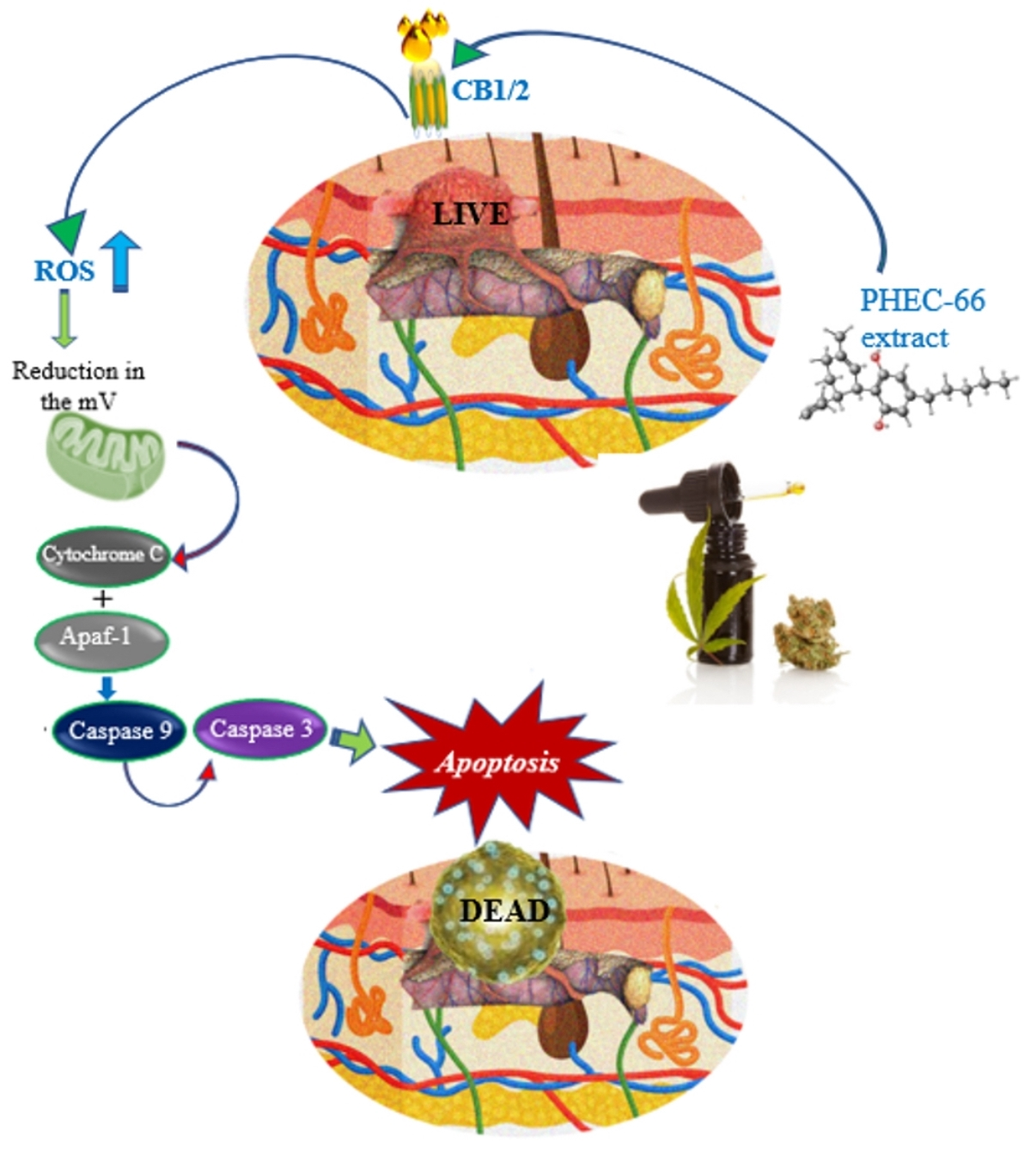
3. Results
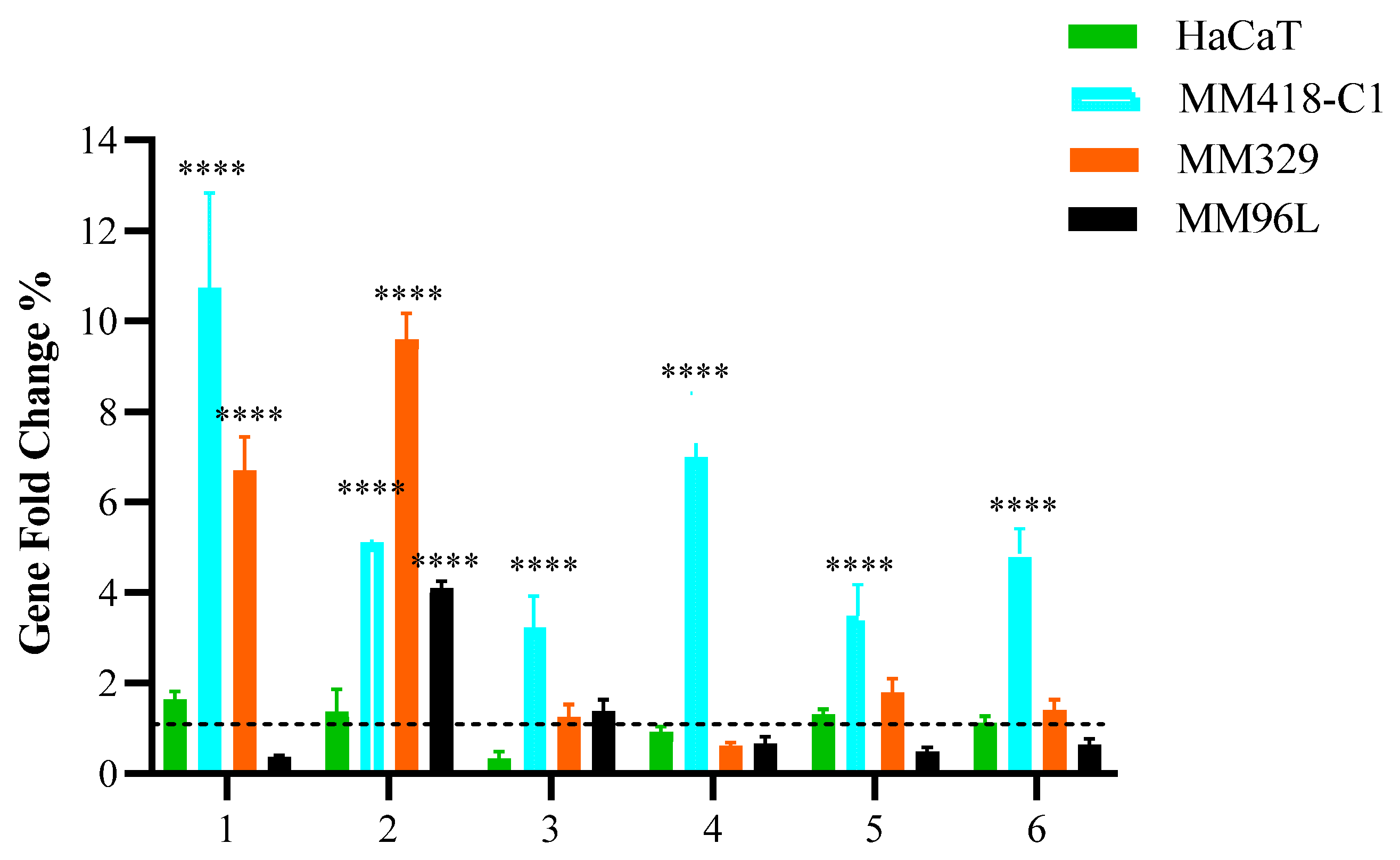
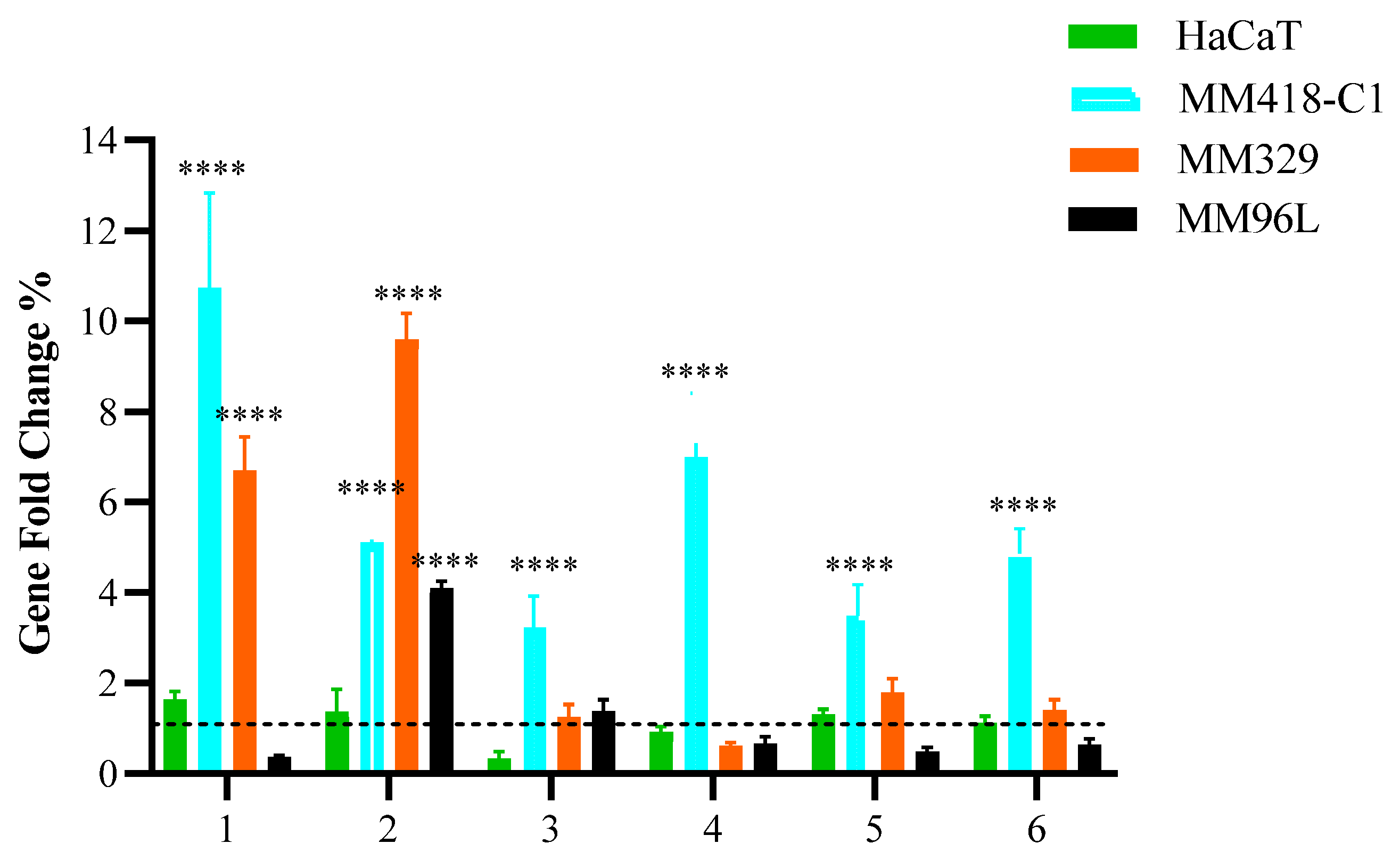
3.1. Quantitative Polymerase Chain Reaction (qPCR)
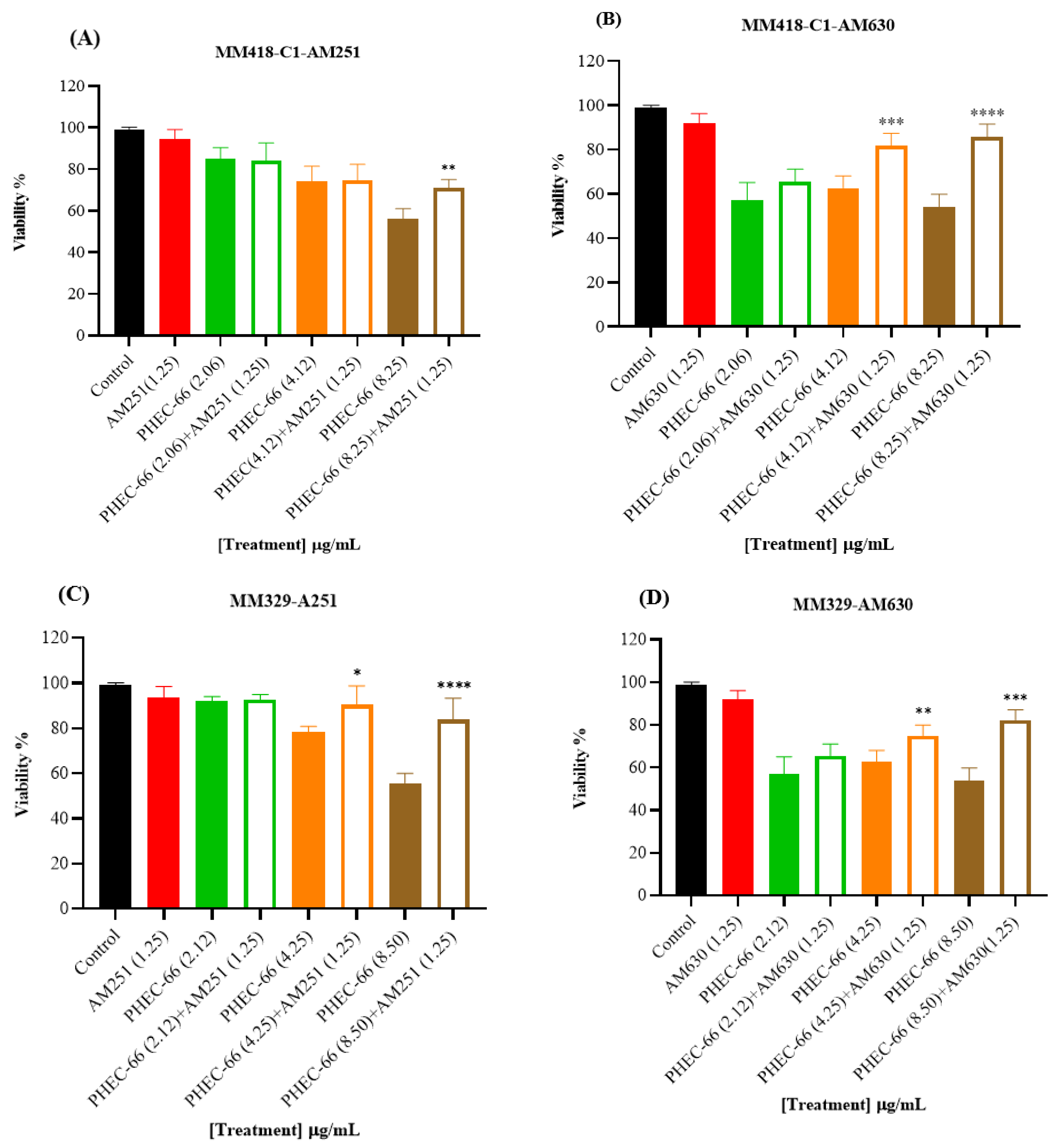
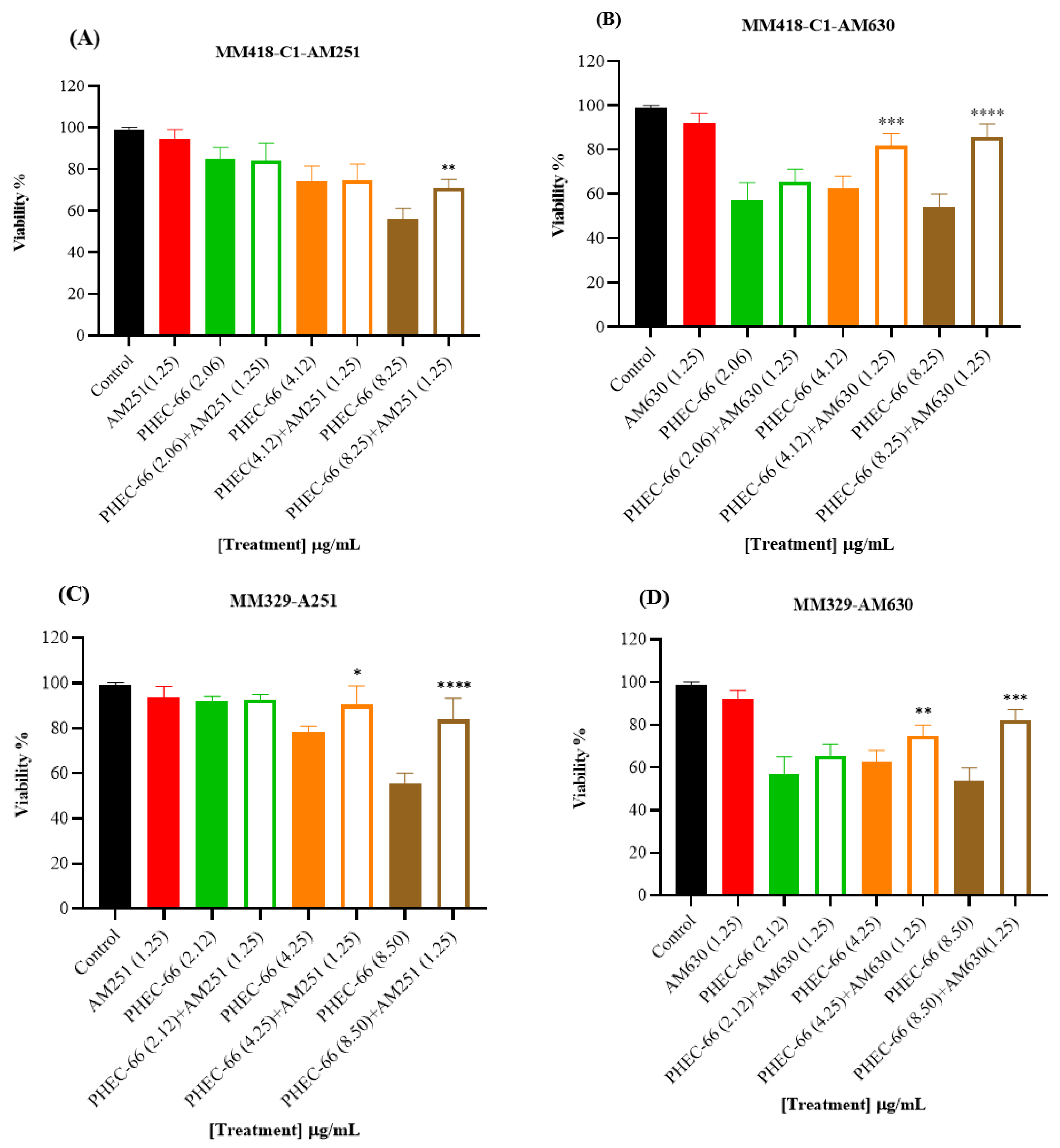
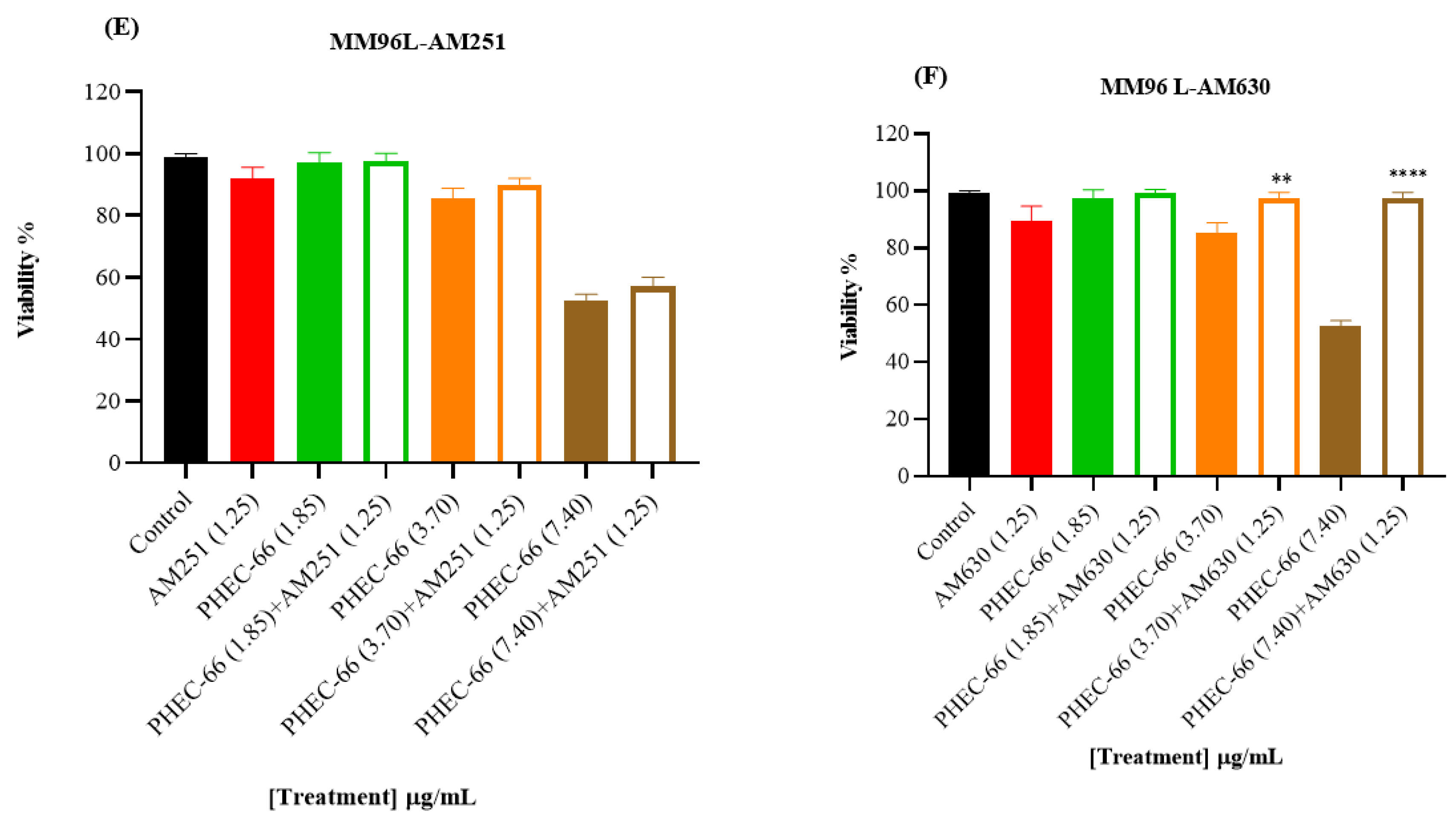
3.2. CB1 & CB2 Antagonist
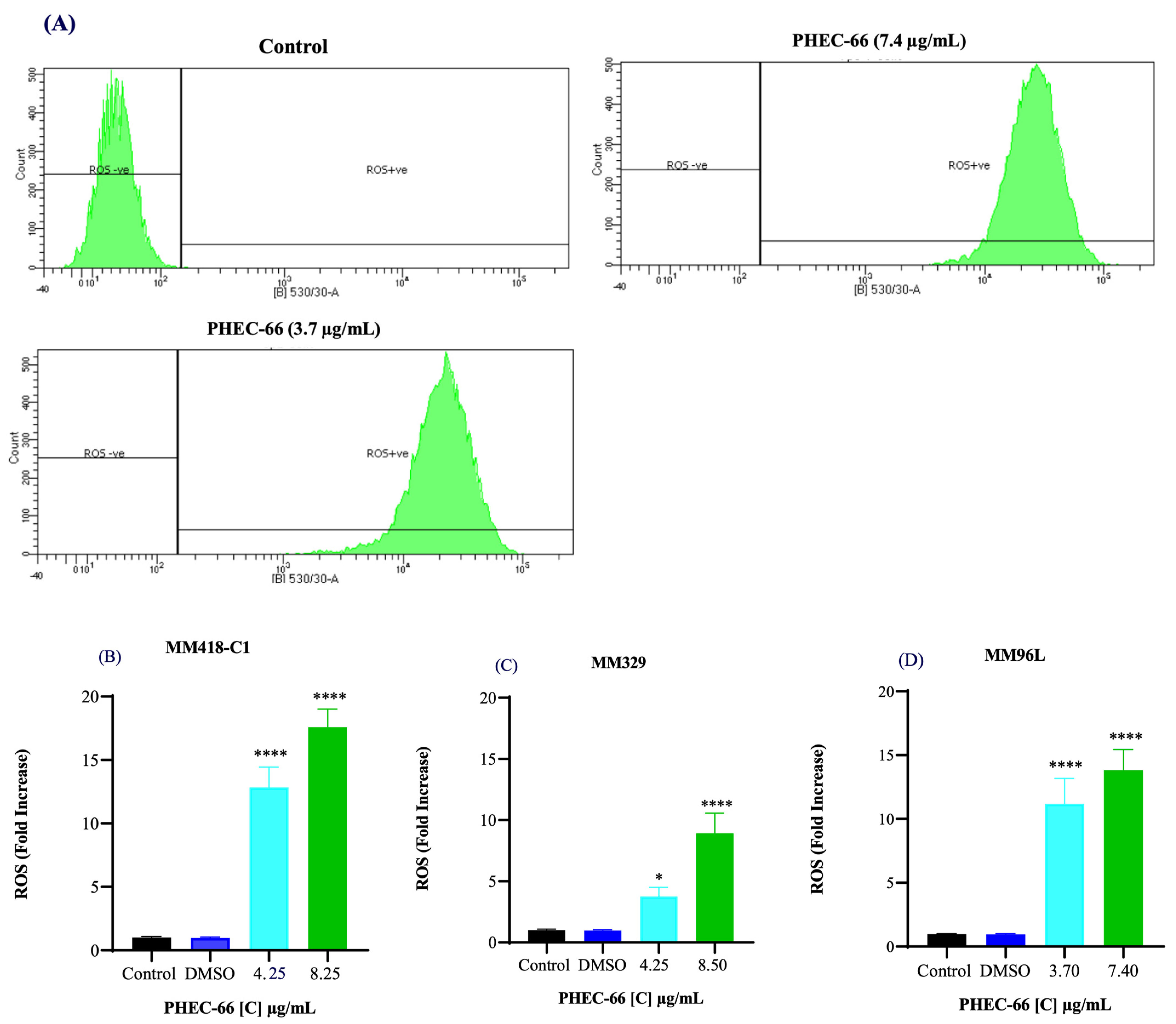
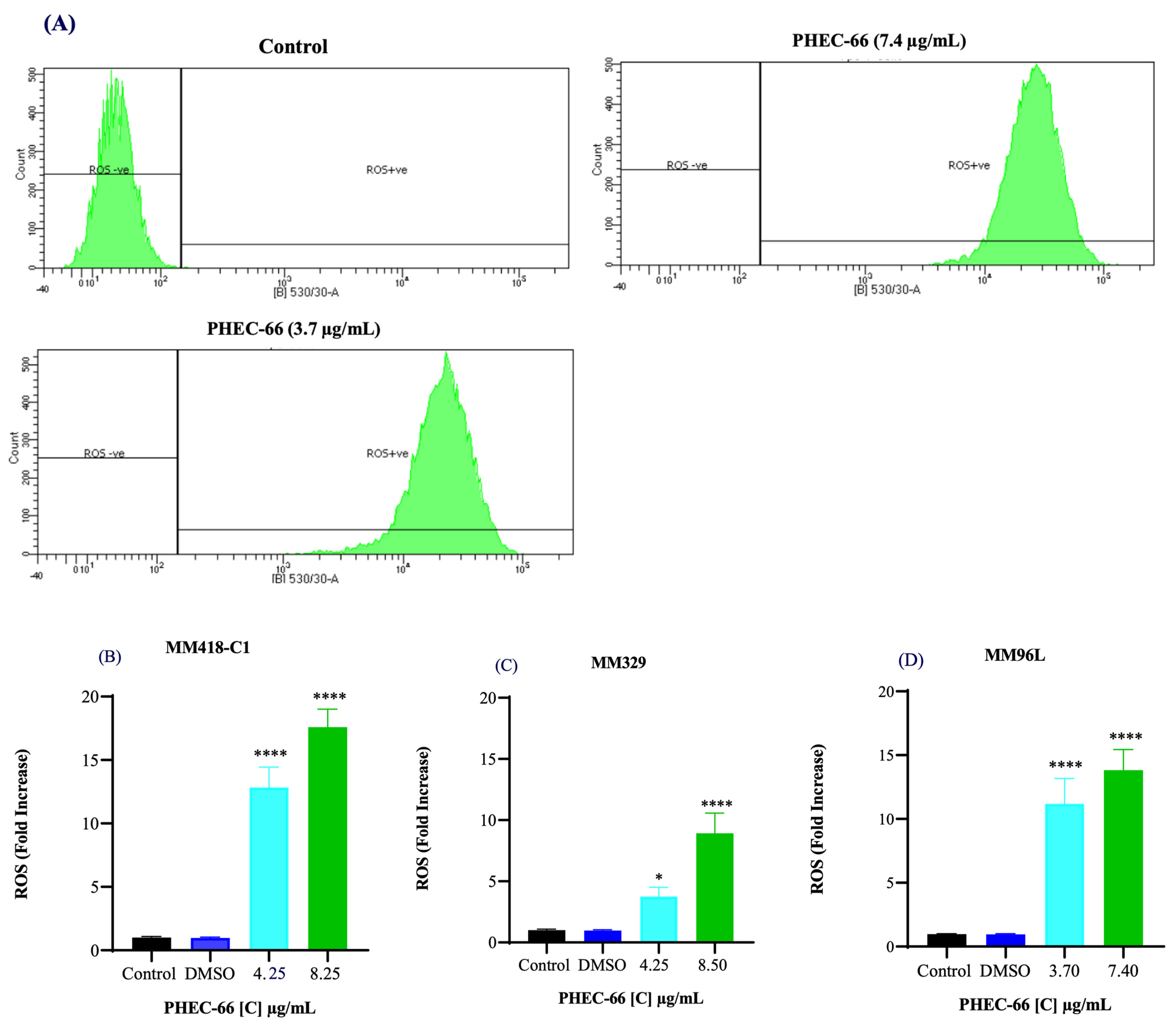
3.3. Reactive Oxygen Species (ROS)
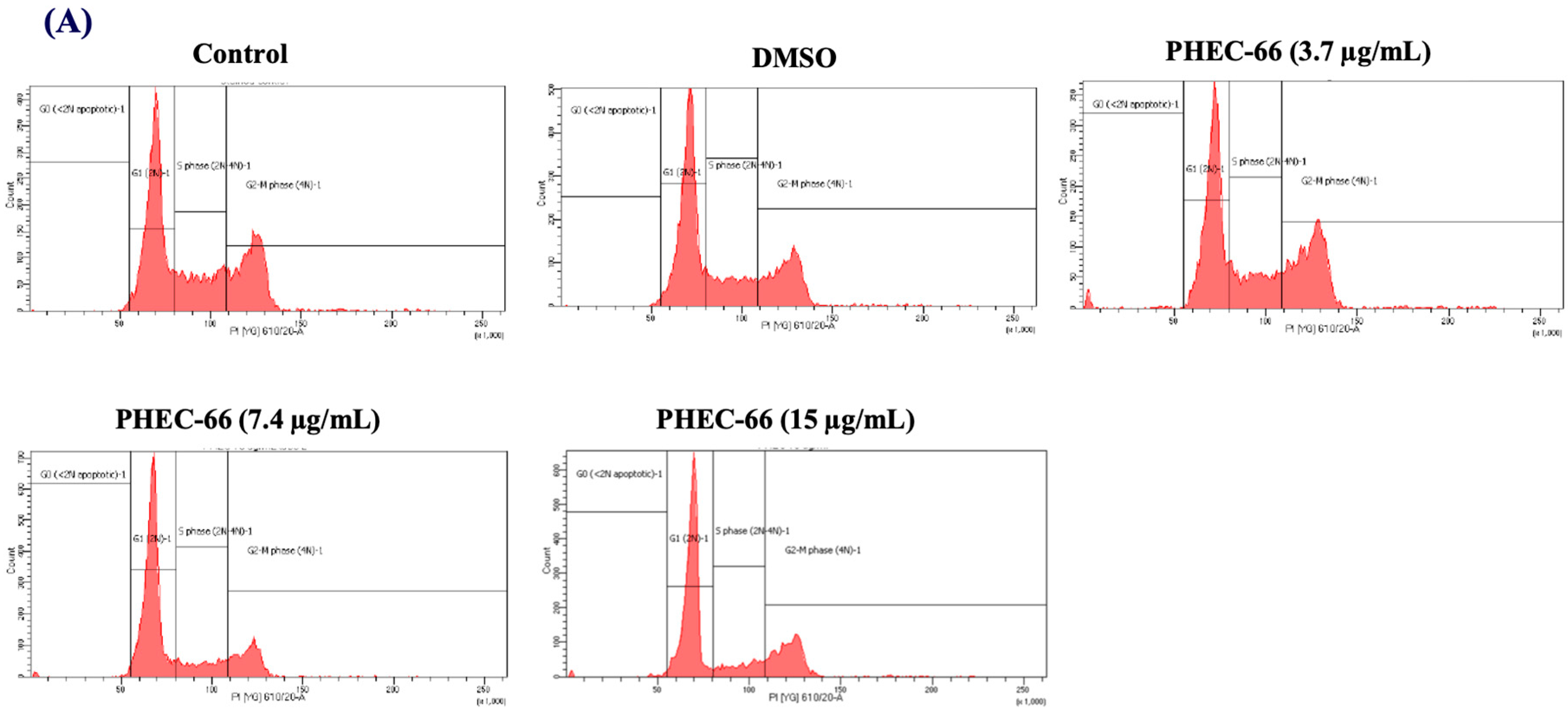
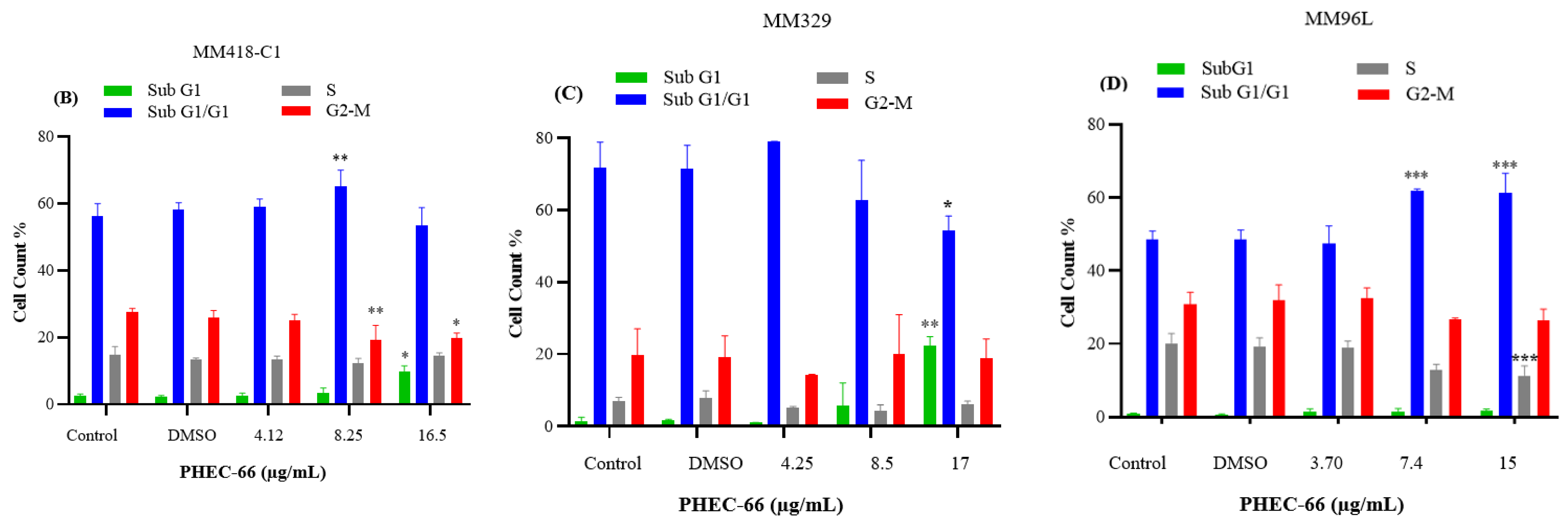
3.4. Effect of PHEC-66 on Cell Cycle Dynamics
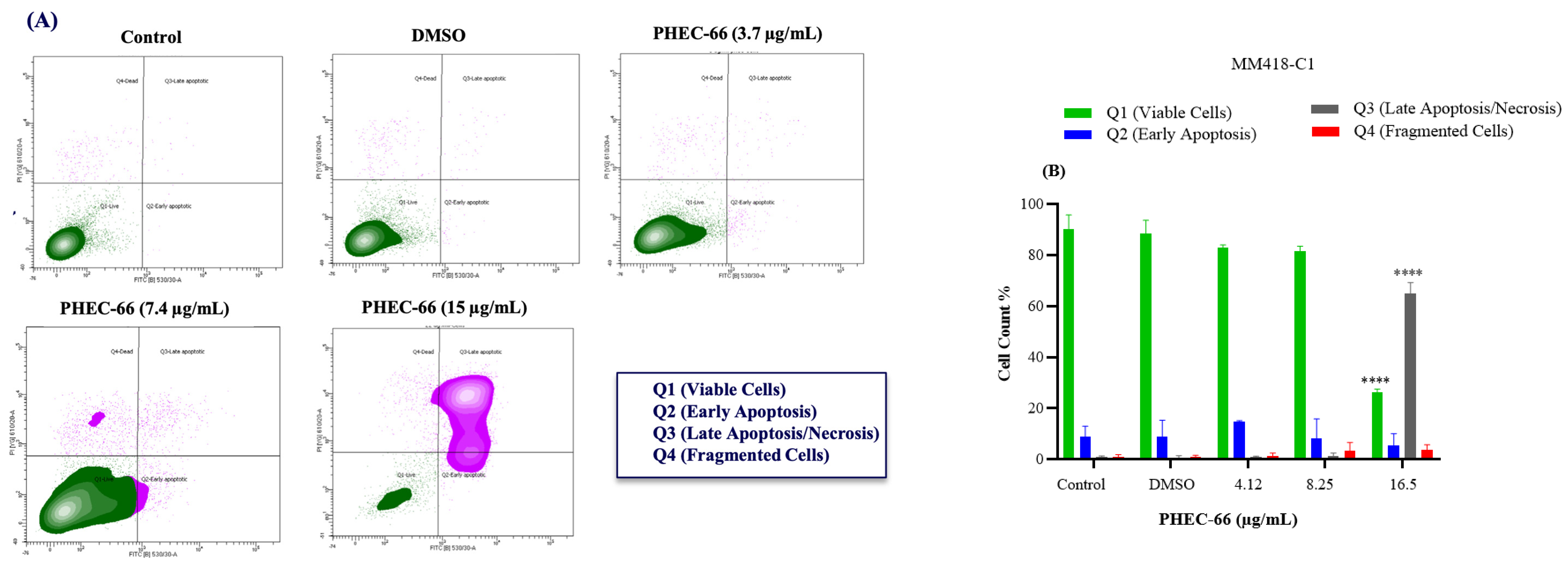
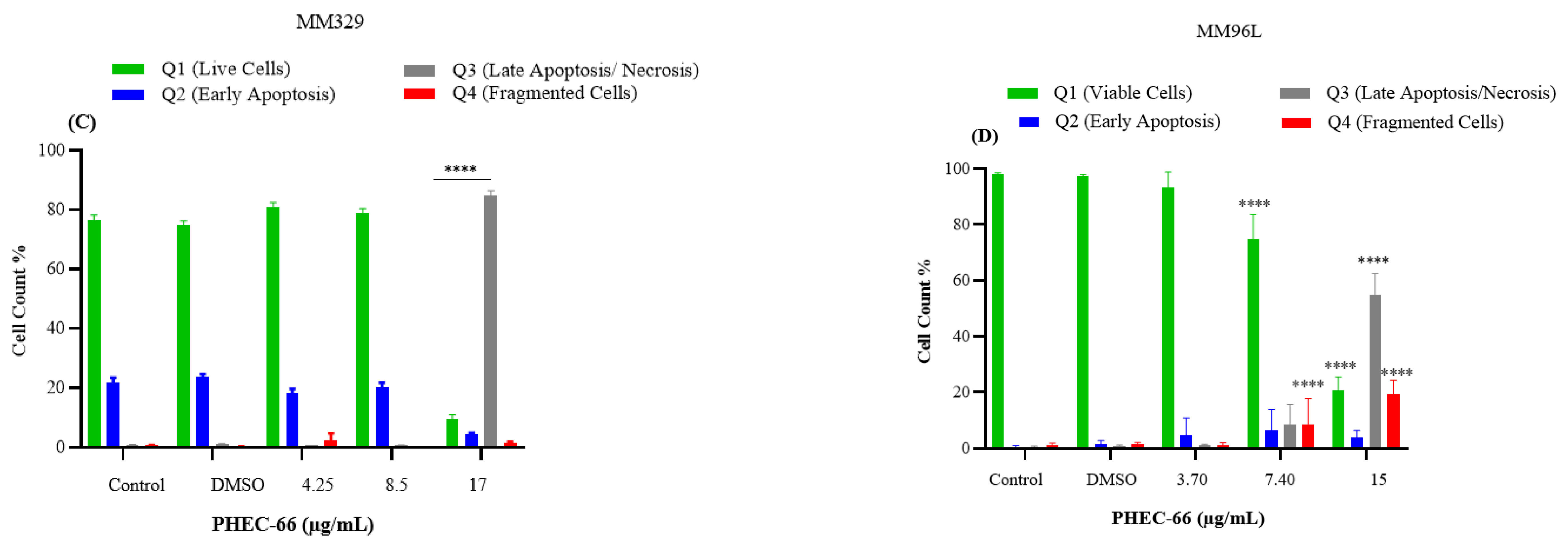
3.5. Apoptosis Assay
3.6. BAX and Bcl-2 Gene Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Fukunaga-Kalabis, M.; Li, L.; Herlyn, M. Developmental pathways activated in melanocytes and melanoma. Arch. Biochem. Biophys. 2014, 563, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Carlson, J.A.; Granese, J.; Ross, J.; Mihm, M.C., Jr.; Slominski, A. Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 2008, 3, 569–585. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Vecchio, D.; Avci, P.; Yin, R.; Garcia-Diaz, M.; Hamblin, M.R. Melanoma resistance to photodynamic therapy: New insights. Biol. Chem. 2013, 394, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Volkovova, K.; Bilanicova, D.; Bartonova, A.; Letašiová, S.; Dusinska, M. Associations between environmental factors and incidence of cutaneous melanoma. Review. Environ. Health 2012, 11 (Suppl. 1), S12. [Google Scholar] [CrossRef]
- Nguyen, D.; Holien, J.; Dekiwadia, C.; Thrimawithana, T.; Piva, T.; Huynh, T. Momordica cochinchinensis (Gấc) Seed Extracts Induce Apoptosis and Necrosis in Melanoma Cells. Pharmaceuticals 2023, 16, 100. [Google Scholar] [CrossRef]
- Sanchez, J.N.; Wang, T.; Cohen, M.S. BRAF and MEK Inhibitors: Use and Resistance in BRAF-Mutated Cancers. Drugs 2018, 78, 549–566. [Google Scholar] [CrossRef]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role Played by Signalling Pathways in Overcoming BRAF Inhibitor Resistance in Melanoma. Int. J. Mol. Sci. 2017, 18, 1527. [Google Scholar] [CrossRef]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Raetz, J.; Karitzky, P.C.; Dinter, L.; Tietze, J.K.; Kolbe, I.; Käubler, T.; Renner, B.; Beissert, S.; Meier, F.; et al. Head-to-Head Comparison of BRAF/MEK Inhibitor Combinations Proposes Superiority of Encorafenib Plus Trametinib in Melanoma. Cancers 2022, 14, 4930. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Wayman, M. Encorafenib and Binimetinib Combination Therapy in Metastatic Melanoma. J. Adv. Pract. Oncol. 2022, 13, 450–455. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.; Elizalde, P.V.; Schillaci, R. Harnessing Tumor Necrosis Factor Alpha to Achieve Effective Cancer Immunotherapy. Cancers 2021, 13, 564. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. OncoImmunology 2016, 5, e1163462. [Google Scholar] [CrossRef]
- Faghfuri, E.; Faramarzi, M.A.; Nikfar, S.; Abdollahi, M. Nivolumab and pembrolizumab as immune-modulating monoclonal antibodies targeting the PD-1 receptor to treat melanoma. Expert Rev. Anticancer Ther. 2015, 15, 981–993. [Google Scholar] [CrossRef]
- de Azevedo, S.J.; de Melo, A.C.; Roberts, L.; Caro, I.; Xue, C.; Wainstein, A. First-line atezolizumab monotherapy in patients with advanced BRAF(V600) wild-type melanoma. Pigment Cell Melanoma Res. 2021, 34, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.P.; Hastings, K.T. Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Bachari, A.; Nassar, N.; Schanknecht, E.; Telukutla, S.; Piva, T.J.; Mantri, N. Rationalizing a prospective coupling effect of cannabinoids with the current pharmacotherapy for melanoma treatment. Wiley Interdiscip. Rev. Syst. Biol. Med. 2024, 16, e1633. [Google Scholar] [CrossRef]
- Saad, M.B.; Hong, L.; Aminu, M.; I Vokes, N.; Chen, P.; Salehjahromi, M.; Qin, K.; Sujit, S.J.; Lu, X.; Young, E.; et al. Predicting benefit from immune checkpoint inhibitors in patients with non-small-cell lung cancer by CT-based ensemble deep learning: A retrospective study. Lancet Digit. Health 2023, 5, e404–e420. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Smalley, K.S.; Eroglu, Z.; Sondak, V.K. Combination Therapies for Melanoma: A New Standard of Care? Am. J. Clin. Dermatol. 2016, 17, 99–105. [Google Scholar] [CrossRef]
- Chinembiri, T.N.; Du Plessis, L.H.; Gerber, M.; Hamman, J.H.; Du Plessis, J. Review of natural compounds for potential skin cancer treatment. Molecules 2014, 19, 11679–11721. [Google Scholar] [CrossRef]
- Bachari, A.; Piva, T.J.; Salami, S.A.; Jamshidi, N.; Mantri, N. Roles of Cannabinoids in Melanoma: Evidence from In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 6040. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.J.; Marnett, L.J. Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev. 2011, 30, 599–612. [Google Scholar] [CrossRef]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the Endocannabinoid System as a Potential Anticancer Strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2018, 11, 487. [Google Scholar] [CrossRef]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Mukhtar, H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005, 65, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Bachari, A.; Nassar, N.; Telukutla, S.; Zomer, R.; Dekiwadia, C.; Piva, T.J.; Mantri, N. In Vitro Antiproliferative Effect of Cannabis Extract PHEC-66 on Melanoma Cell Lines. Cells 2023, 12, 2450. [Google Scholar] [CrossRef]
- Dariš, B.; Tancer Verboten, M.; Knez, Z.; Ferk, P. Cannabinoids in cancer treatment: Therapeutic potential and legislation. Bosn. J. Basic Med. Sci. 2019, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Heider, C.G.; Itenberg, S.A.; Rao, J.; Ma, H.; Wu, X. Mechanisms of Cannabidiol (CBD) in Cancer Treatment: A Review. Biology 2022, 11, 817. [Google Scholar] [CrossRef]
- Ramer, R.; Rohde, A.; Merkord, J.; Rohde, H.; Hinz, B. Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lung cancer cells. Pharm. Res. 2010, 27, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gibson, S.B. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy 2008, 4, 246–248. [Google Scholar] [CrossRef]
- Anis, O.; Vinayaka, A.C.; Shalev, N.; Namdar, D.; Nadarajan, S.; Anil, S.M.; Cohen, O.; Belausov, E.; Ramon, J.; Gati, E.M.; et al. Cannabis-Derived Compounds Cannabichromene and Δ9-Tetrahydrocannabinol Interact and Exhibit Cytotoxic Activity against Urothelial Cell Carcinoma Correlated with Inhibition of Cell Migration and Cytoskeleton Organization. Molecules 2021, 26, 465. [Google Scholar] [CrossRef]
- Soliman, E.; Van Dross, R. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: Receptor-independent endocannabinoid signaling. Mol. Carcinog. 2016, 55, 1807–1821. [Google Scholar] [CrossRef]
- Pagano, C.; Savarese, B.; Coppola, L.; Navarra, G.; Avilia, G.; Laezza, C.; Bifulco, M. Cannabinoids in the Modulation of Oxidative Signaling. Int. J. Mol. Sci. 2023, 24, 2513. [Google Scholar] [CrossRef]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Pozza, E.D.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharmacol. 2006, 70, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Park, S.-W.; Hah, J.H.; Oh, S.-M.; Jeong, W.-J.; Sung, M.-W. 5-lipoxygenase mediates docosahexaenoyl ethanolamide and N-arachidonoyl-L-alanine-induced reactive oxygen species production and inhibition of proliferation of head and neck squamous cell carcinoma cells. BMC Cancer 2016, 16, 458. [Google Scholar] [CrossRef]
- Vilema-Enríquez, G.; Arroyo, A.; Grijalva, M.; Amador-Zafra, R.I.; Camacho, J. Molecular and Cellular Effects of Hydrogen Peroxide on Human Lung Cancer Cells: Potential Therapeutic Implications. Oxidative Med. Cell. Longev. 2016, 2016, 1908164. [Google Scholar] [CrossRef]
- DeMorrow, S.; Francis, H.; Gaudio, E.; Ueno, Y.; Venter, J.; Onori, P.; Franchitto, A.; Vaculin, B.; Vaculin, S.; Alpini, G. Anandamide inhibits cholangiocyte hyperplastic proliferation via activation of thioredoxin 1/redox factor 1 and AP-1 activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G506–G519. [Google Scholar] [CrossRef] [PubMed]
- Murad, H.; Hawat, M.; Ekhtiar, A.; AlJapawe, A.; Abbas, A.; Darwish, H.; Sbenati, O.; Ghannam, A. Induction of G1-phase cell cycle arrest and apoptosis pathway in MDA-MB-231 human breast cancer cells by sulfated polysaccharide extracted from Laurencia papillosa. Cancer Cell Int. 2016, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Lu, B.; Gan, J.; Wang, S.; Jiang, X.; Li, H. Apoptosis detection: A purpose-dependent approach selection. Cell Cycle 2021, 20, 1033–1040. [Google Scholar] [CrossRef]
- Limas, J.C.; Cook, J.G. Preparation for DNA replication: The key to a successful S phase. FEBS Lett. 2019, 593, 2853–2867. [Google Scholar] [CrossRef]
- McKenna, S.; García-Gutiérrez, L.; Matallanas, D.; Fey, D. BAX and SMAC regulate bistable properties of the apoptotic caspase system. Sci. Rep. 2021, 11, 3272. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Broman, K.; Jonas, E.A. Oxidative stress battles neuronal Bcl-xL in a fight to the death. Neural Regen. Res. 2021, 16, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | IC50 (μg/mL) |
|---|---|
| HaCaT | 13.37 ± 1.90 |
| MM418-C1 | 8.21 ± 0.75 |
| MM329 | 8.47 ± 0.14 |
| MM96L | 7.41 ± 0.94 |
| Cell Lines | BAX | Bcl-2 | Ratio of BAX/Bcl-2 |
|---|---|---|---|
| HaCaT | 0.62 | 2.27 | 0.27 |
| MM418-C1 | 5.25 | 0.84 | 6.25 |
| MM329 | 1.90 | 0.79 | 2.40 |
| MM96L | 0.90 | 0.61 | 1.47 |
| Effect | MM418-C1 | MM329 | MM96L |
|---|---|---|---|
| CB1 Expression CB2 Expression | ↑ ↑ | ↑ ↑ | ↓ ↑ |
| CB1 Antagonist CB2 Antagonist | + + | + + | − + |
| Cell Cycle Arrest | Sub G1 Damage and fragmented DNA | Sub G1 Damage and fragmented DNA | Sub G1-G1 |
| ROS Level | ↑ | ↑ | ↑ |
| Apoptosis Effect | Apoptosis | Apoptosis | Apoptosis/Necrosis |
| BAX Expression Bcl-2 Expression | ↑ ↓ | ↑ ↓ | = = |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachari, A.; Nassar, N.; Telukutla, S.; Zomer, R.; Piva, T.J.; Mantri, N. Evaluating the Mechanism of Cell Death in Melanoma Induced by the Cannabis Extract PHEC-66. Cells 2024, 13, 268. https://doi.org/10.3390/cells13030268
Bachari A, Nassar N, Telukutla S, Zomer R, Piva TJ, Mantri N. Evaluating the Mechanism of Cell Death in Melanoma Induced by the Cannabis Extract PHEC-66. Cells. 2024; 13(3):268. https://doi.org/10.3390/cells13030268
Chicago/Turabian StyleBachari, Ava, Nazim Nassar, Srinivasareddy Telukutla, Roby Zomer, Terrence J. Piva, and Nitin Mantri. 2024. "Evaluating the Mechanism of Cell Death in Melanoma Induced by the Cannabis Extract PHEC-66" Cells 13, no. 3: 268. https://doi.org/10.3390/cells13030268
APA StyleBachari, A., Nassar, N., Telukutla, S., Zomer, R., Piva, T. J., & Mantri, N. (2024). Evaluating the Mechanism of Cell Death in Melanoma Induced by the Cannabis Extract PHEC-66. Cells, 13(3), 268. https://doi.org/10.3390/cells13030268