
Impact of Germline Depletion of Bonus on Chromatin State in Drosophila Ovaries

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drosophila Fly Stocks
2.2. S2 Cell Line
2.3. Protein Co-Immunoprecipitation from S2 Cells
2.4. Western Blotting
2.5. RNA Extraction and RNA-Seq Analysis
2.6. ChIP-Seq
2.7. Liquid Chromatography–Mass Spectrometry (LS–MS)
3. Results
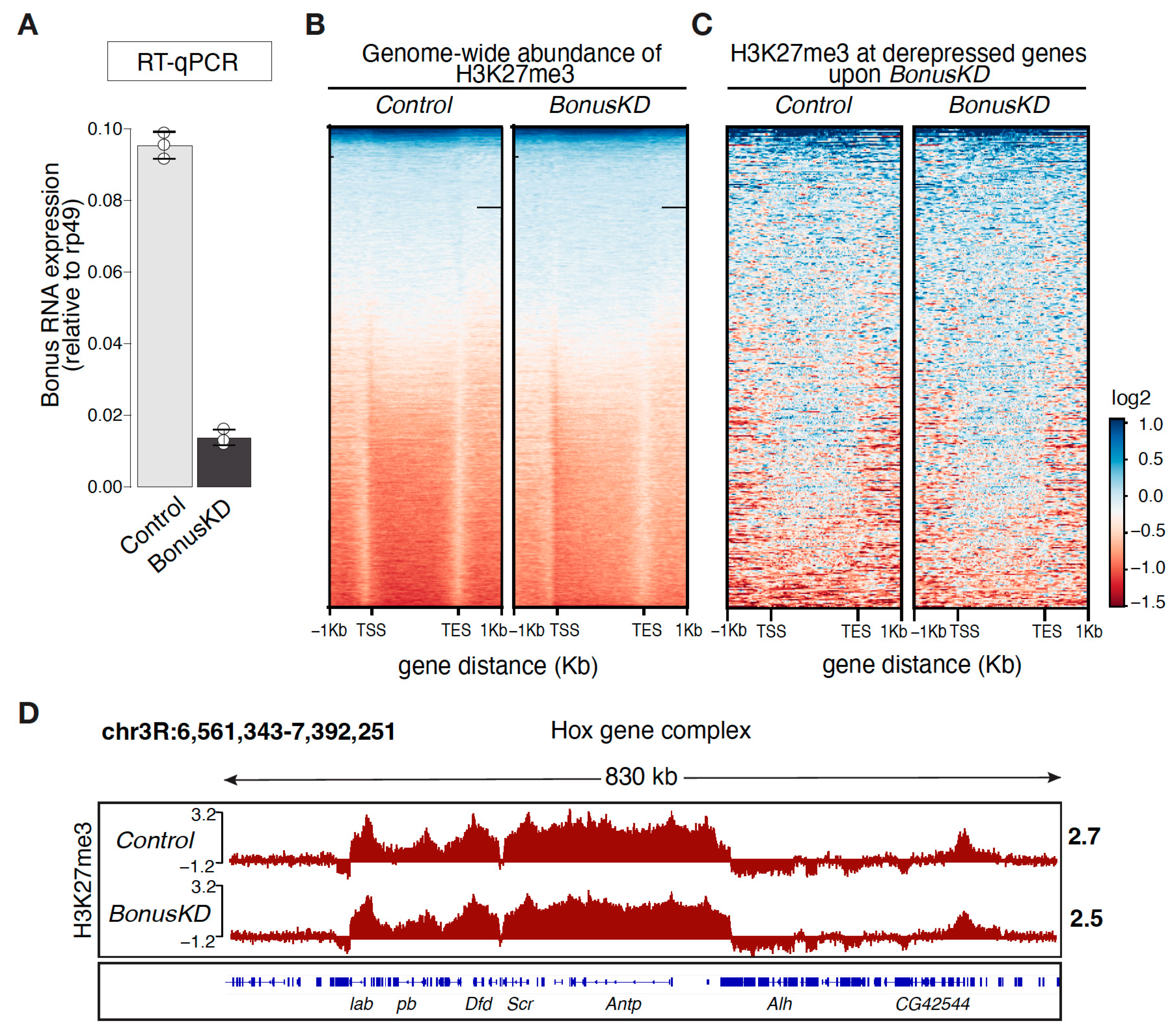
3.1. Genome-Wide Analyses of H3K27me3 Distribution in Bonus-Depleted Ovaries
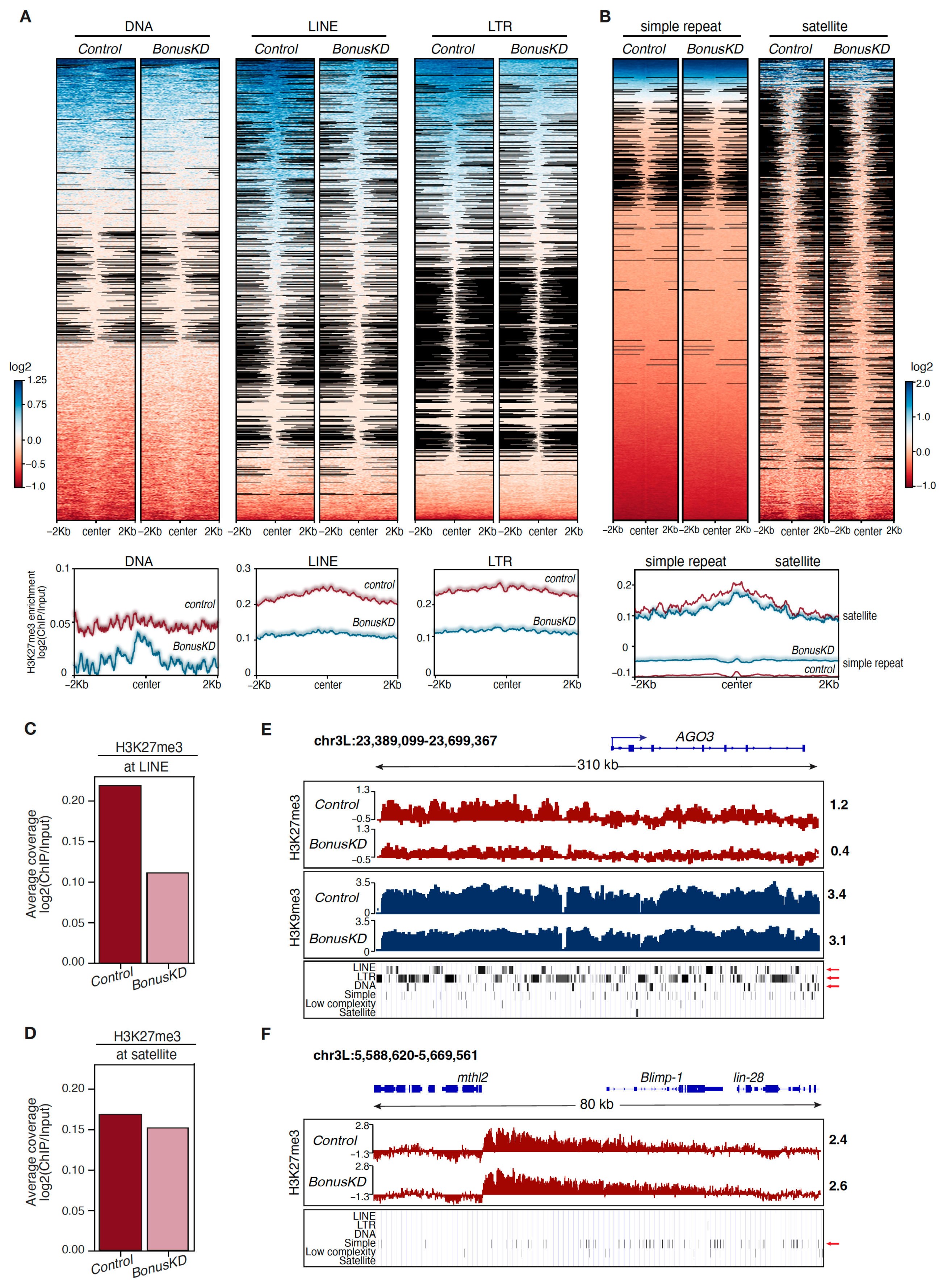
3.2. Depletion of Bonus in the Ovaries Affects the Distribution of the H3K27me3 Mark over Transposon Regions
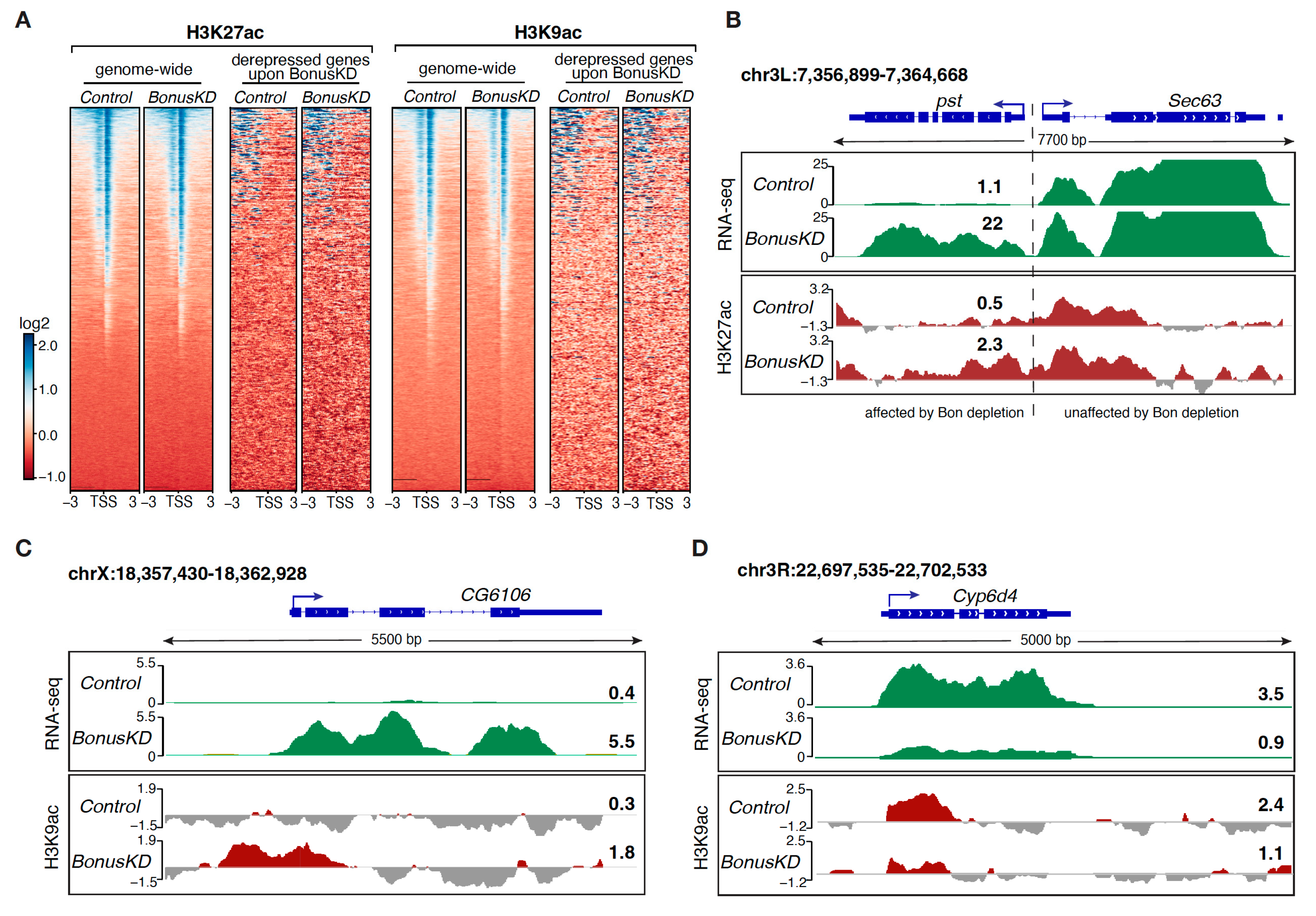
3.3. Modulation of the H3K27ac and H3K9ac Marks in Response to Bon Depletion
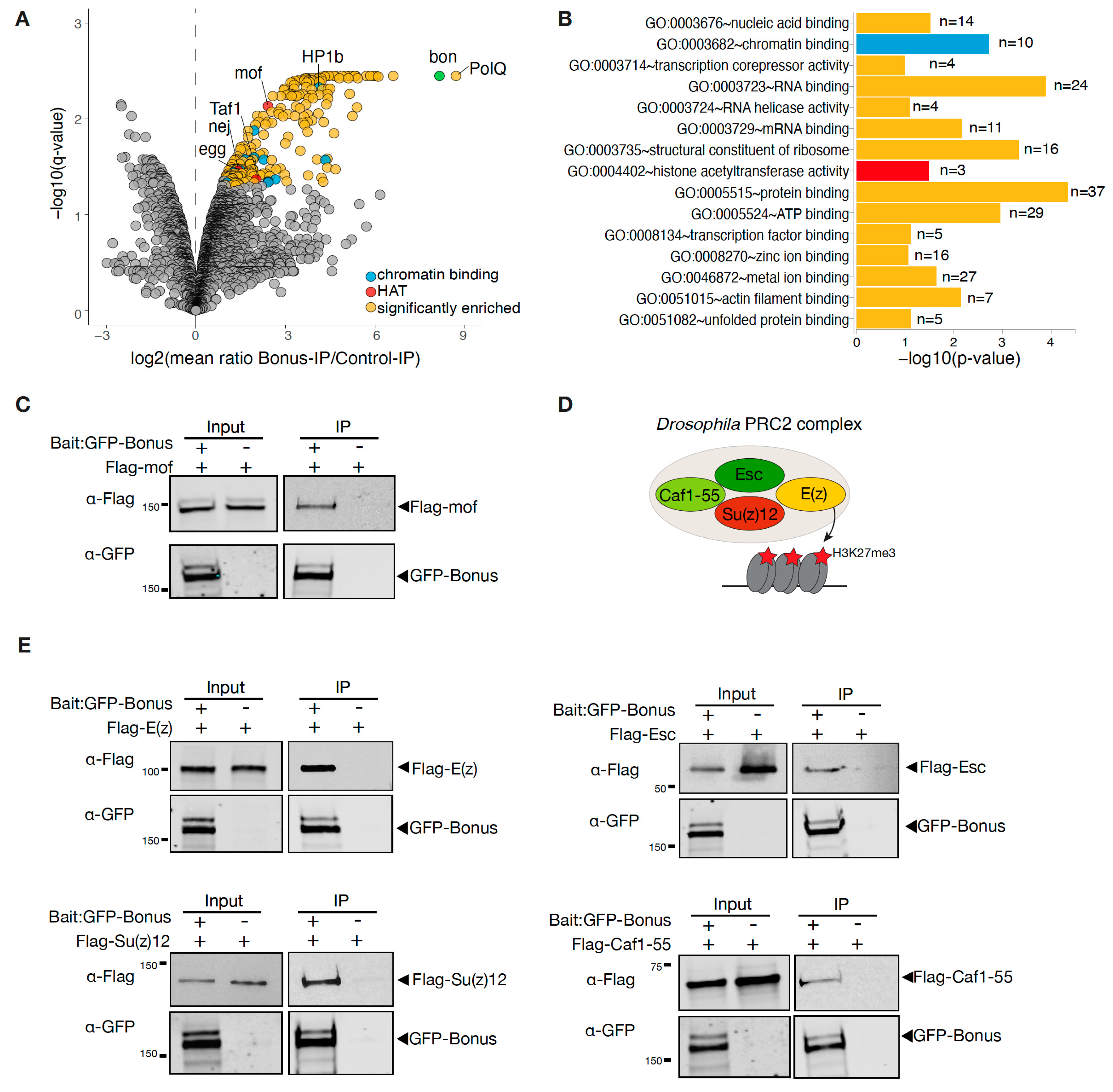
3.4. Bonus Associates with Histone Acetyltransferases and the PRC2 Complex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turner, B.M. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat. Struct. Mol. Biol. 2005, 12, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Iii, W.J.D.; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef]
- Yuan, W.; Wu, T.; Fu, H.; Dai, C.; Wu, H.; Liu, N.; Li, X.; Xu, M.; Zhang, Z.; Niu, T.; et al. Dense Chromatin Activates Polycomb Repressive Complex 2 to Regulate H3 Lysine 27 Methylation. Science 2012, 337, 971–975. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome Regulation by Polycomb and Trithorax Proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef]
- Nielsen, A.L.; Ortiz, J.A.; You, J.; Oulad-Abdelghani, M.; Khechumian, R.; Gansmuller, A.; Chambon, P.; Losson, R. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 1999, 18, 6385–6395. [Google Scholar] [CrossRef]
- Venturini, L.; You, J.; Stadler, M.; Galien, R.; Lallemand, V.; Koken, M.H.; Mattei, M.G.; Ganser, A.; Chambon, P.; Losson, R.; et al. TIF1gamma, a novel member of the transcriptional intermediary factor 1 family. Oncogene 1999, 18, 1209–1217. [Google Scholar] [CrossRef]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Khetchoumian, K.; Teletin, M.; Mark, M.; Lerouge, T.; Cerviño, M.; Oulad-Abdelghani, M.; Chambon, P.; Losson, R. TIF1δ, a Novel HP1-interacting Member of the Transcriptional Intermediary Factor 1 (TIF1) Family Expressed by Elongating Spermatids. J. Biol. Chem. 2004, 279, 48329–48341. [Google Scholar] [CrossRef]
- Beckstead, R.; Ortiz, J.A.; Sanchez, C.; Prokopenko, S.N.; Chambon, P.; Losson, R.; Bellen, H.J. Bonus, a Drosophila homolog of TIF1 proteins, interacts with nuclear receptors and can inhibit betaFTZ-F1-dependent transcription. Mol. Cell 2001, 7, 753–765. [Google Scholar] [CrossRef]
- Le Douarin, B.; Zechel, C.; Garnier, J.; Lutz, Y.; Tora, L.; Pierrat, P.; Heery, D.; Gronemeyer, H.; Chambon, P.; Losson, R. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J. 1995, 14, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, B.; Nielsen, A.L.; Garnier, J.M.; Ichinose, H.; Jeanmougin, F.; Losson, R.; Chambon, P. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996, 15, 6701–6715. [Google Scholar] [CrossRef] [PubMed]
- Allton, K.; Jain, A.K.; Herz, H.-M.; Tsai, W.-W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef]
- Beckstead, R.B.; Ner, S.S.; Hales, K.G.; Grigliatti, T.A.; Baker, B.S.; Bellen, H.J. Bonus, a Drosophila TIF1 homolog, is a chromatin-associated protein that acts as a modifier of position-effect variegation. Genetics 2005, 169, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Sato, K.; Koganezawa, M.; Ote, M.; Matsumoto, K.; Hama, C.; Yamamoto, D. Fruitless Recruits Two Antagonistic Chromatin Factors to Establish Single-Neuron Sexual Dimorphism. Cell 2012, 149, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Moberg, K.H.; Veraksa, A. Hippo pathway and Bonus control developmental cell fate decisions in the Drosophila eye. Dev. Cell 2023, 58, 416–434.e12. [Google Scholar] [CrossRef]
- Godneeva, M.B.; Ninova, M.; Tóth, K.F.; Aravin, A.A. SUMOylation of Bonus, the Drosophila homolog of Transcription Intermediary Factor 1, safeguards germline identity by recruiting repressive chromatin complexes to silence tissue-specific genes. eLife 2023, 12, RP89493. [Google Scholar] [CrossRef]
- Ninova, M.; Chen, Y.-C.A.; Godneeva, B.; Rogers, A.K.; Luo, Y.; Tóth, K.F.; Aravin, A.A. Su(var)2-10 and the SUMO Pathway Link piRNA-Guided Target Recognition to Chromatin Silencing. Mol. Cell 2020, 77, 556–570.e6. [Google Scholar] [CrossRef]
- Le Thomas, A.; Stuwe, E.; Li, S.; Du, J.; Marinov, G.; Rozhkov, N.; Chen, Y.-C.A.; Luo, Y.; Sachidanandam, R.; Toth, K.F.; et al. Transgenerationally inherited piRNAs trigger piRNA biogenesis by changing the chromatin of piRNA clusters and inducing precursor processing. Genes Dev. 2014, 28, 1667–1680. [Google Scholar] [CrossRef]
- Schwartz, Y.B.; Kahn, T.G.; Nix, D.A.; Li, X.-Y.; Bourgon, R.; Biggin, M.; Pirrotta, V. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet. 2006, 38, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, D.; Leleu, M.; Splinter, E.; Rougemont, J.; De Laat, W.; Duboule, D. The Dynamic Architecture of Hox Gene Clusters. Science 2011, 334, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Frapporti, A.; Pina, C.M.; Arnaiz, O.; Holoch, D.; Kawaguchi, T.; Humbert, A.; Eleftheriou, E.; Lombard, B.; Loew, D.; Sperling, L.; et al. The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat. Commun. 2019, 10, 2710. [Google Scholar] [CrossRef] [PubMed]
- Miró-Pina, C.; Charmant, O.; Kawaguchi, T.; Holoch, D.; Michaud, A.; Cohen, I.; Humbert, A.; Jaszczyszyn, Y.; Chevreux, G.; Del Maestro, L.; et al. Paramecium Polycomb repressive complex 2 physically interacts with the small RNA-binding PIWI protein to repress transposable elements. Dev. Cell 2022, 57, 1037–1052.e8. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.A.; Tanizawa, Y.; Galik, B.; Wang, N.; Ito, T.; Mochizuki, T.; Akimcheva, S.; Bowman, J.L.; Cognat, V.; Maréchal-Drouard, L.; et al. Chromatin Organization in Early Land Plants Reveals an Ancestral Association between H3K27me3, Transposons, and Constitutive Heterochromatin. Curr. Biol. 2020, 30, 573–588.e7. [Google Scholar] [CrossRef]
- Zhao, X.; Xiong, J.; Mao, F.; Sheng, Y.; Chen, X.; Feng, L.; Dui, W.; Yang, W.; Kapusta, A.; Feschotte, C.; et al. RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena. Genes Dev. 2019, 33, 348–364. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Lavarone, E.; Barbieri, C.M.; Pasini, D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Commun. 2019, 10, 1679. [Google Scholar] [CrossRef]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef]
- Boyd, J.B.; Sakaguchi, K.; Harris, P.V. mus308 mutants of Drosophila exhibit hypersensitivity to DNA cross-linking agents and are defective in a deoxyribonuclease. Genetics 1990, 125, 813–819. [Google Scholar] [CrossRef]
- Leonhardt, E.A.; Henderson, D.S.; Rinehart, J.E.; Boyd, J.B. Characterization of the mus308 gene in Drosophila melanogaster. Genetics 1993, 133, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Marini, F.; Wood, R.D. POLQ (Pol), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003, 31, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Parisky, K.; Celotto, A.M.; Reenan, R.A.; Graveley, B.R. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc. Natl. Acad. Sci. USA 2004, 101, 15974–15979. [Google Scholar] [CrossRef]
- Herold, N.; Will, C.L.; Wolf, E.; Kastner, B.; Urlaub, H.; Lührmann, R. Conservation of the Protein Composition and Electron Microscopy Structure of Drosophila melanogaster and Human Spliceosomal Complexes. Mol. Cell. Biol. 2009, 29, 281–301. [Google Scholar] [CrossRef]
- Akhtar, A.; Becker, P.B. Activation of Transcription through Histone H4 Acetylation by MOF, an Acetyltransferase Essential for Dosage Compensation in Drosophila. Mol. Cell 2000, 5, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef]
- Tie, F.; Banerjee, R.; Conrad, P.A.; Scacheri, P.C.; Harte, P.J. Histone Demethylase UTX and Chromatin Remodeler BRM Bind Directly to CBP and Modulate Acetylation of Histone H3 Lysine 27. Mol. Cell. Biol. 2012, 32, 2323–2334. [Google Scholar] [CrossRef]
- Birve, A.; Sengupta, A.K.; Beuchle, D.; Larsson, J.; Kennison, J.A.; Rasmuson-Lestander, A.; Müller, J. Su(z)12, a novel Drosophila Polycomb group gene that is conserved in vertebrates and plants. Development 2001, 128, 3371–3379. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- McAvera, R.M.; Crawford, L.J. TIF1 Proteins in Genome Stability and Cancer. Cancers 2020, 12, 2094. [Google Scholar] [CrossRef]
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative analysis of metazoan chromatin organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, S.; Kagey, M.H.; Frampton, G.M.; Rahl, P.B.; Young, R.A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009, 23, 2484–2489. [Google Scholar] [CrossRef]
- Voigt, P.; LeRoy, G.; Drury, W.J.; Zee, B.M.; Son, J.; Beck, D.B.; Young, N.L.; Garcia, B.A.; Reinberg, D. Asymmetrically Modified Nucleosomes. Cell 2012, 151, 181–193. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xi, Y.; Li, W.; McCarthy, R.L.; Stratton, S.A.; Zou, W.; Dent, S.Y.; Jain, A.K.; Barton, M.C. TRIM28 interacts with EZH2 and SWI/SNF to activate genes that promote mammosphere formation. Oncogene 2017, 36, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Ren, X.; Kerppola, T.K. KAP1 Represses Differentiation-Inducible Genes in Embryonic Stem Cells through Cooperative Binding with PRC1 and Derepresses Pluripotency-Associated Genes. Mol. Cell. Biol. 2014, 34, 2075–2091. [Google Scholar] [CrossRef]
- Carré, C.; Szymczak, D.; Pidoux, J.; Antoniewski, C. The Histone H3 Acetylase dGcn5 Is a Key Player in Drosophila melanogaster Metamorphosis. Mol. Cell. Biol. 2005, 25, 8228–8238. [Google Scholar] [CrossRef]
- Agricola, E.; Randall, R.A.; Gaarenstroom, T.; Dupont, S.; Hill, C.S. Recruitment of TIF1γ to Chromatin via Its PHD Finger-Bromodomain Activates Its Ubiquitin Ligase and Transcriptional Repressor Activities. Mol. Cell 2011, 43, 85–96. [Google Scholar] [CrossRef]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- White, D.E.; Negorev, D.; Peng, H.; Ivanov, A.V.; Maul, G.G.; Rauscher, F.J. KAP1, a Novel Substrate for PIKK Family Members, Colocalizes with Numerous Damage Response Factors at DNA Lesions. Cancer Res. 2006, 66, 11594–11599. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godneeva, B.; Fejes Tóth, K.; Quan, B.; Chou, T.-F.; Aravin, A.A. Impact of Germline Depletion of Bonus on Chromatin State in Drosophila Ovaries. Cells 2023, 12, 2629. https://doi.org/10.3390/cells12222629
Godneeva B, Fejes Tóth K, Quan B, Chou T-F, Aravin AA. Impact of Germline Depletion of Bonus on Chromatin State in Drosophila Ovaries. Cells. 2023; 12(22):2629. https://doi.org/10.3390/cells12222629
Chicago/Turabian StyleGodneeva, Baira, Katalin Fejes Tóth, Baiyi Quan, Tsui-Fen Chou, and Alexei A. Aravin. 2023. "Impact of Germline Depletion of Bonus on Chromatin State in Drosophila Ovaries" Cells 12, no. 22: 2629. https://doi.org/10.3390/cells12222629
APA StyleGodneeva, B., Fejes Tóth, K., Quan, B., Chou, T.-F., & Aravin, A. A. (2023). Impact of Germline Depletion of Bonus on Chromatin State in Drosophila Ovaries. Cells, 12(22), 2629. https://doi.org/10.3390/cells12222629