
Unravelling the Role of Cancer Cell-Derived Extracellular Vesicles in Muscle Atrophy, Lipolysis, and Cancer-Associated Cachexia


Abstract
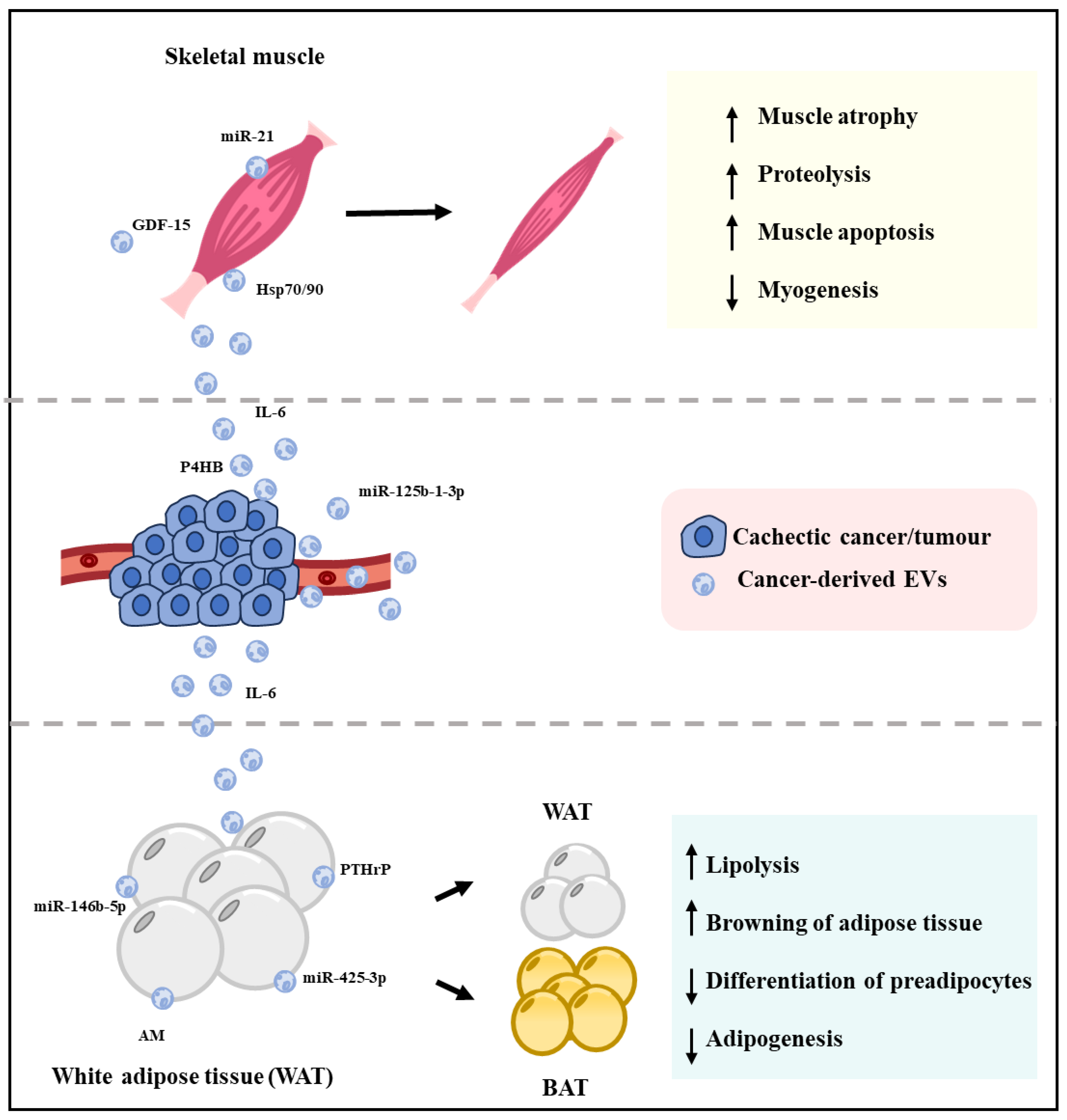
1. Cancer-Associated Cachexia
2. Skeletal Muscle Wasting during Cancer-Associated Cachexia
3. Adipose Tissue Lipolysis and Browning during Cancer-Associated Cachexia
4. Overview of Extracellular Vesicles
5. Cancer Cell-Derived EV Protein and Muscle Wasting

{kind=link}
{kind=link}
{kind=link}
| EV Content | EV Source | Cancer Type | Function | Reference |
|---|---|---|---|---|
| Proteins | ||||
| ZAG | Patient serum MAC16 | Adipose tissue browning, lipolysis | [58,59] | |
| HSP70/90 | C26 LLC | Colon cancer Lung cancer | Muscle catabolism, systemic inflammation | [65,113] |
| Adrenomedullin | Patient plasma and Panc-1 | Pancreatic cancer | Lipolysis | [66] |
| Notch-activating factor | K7M2 | Osteosarcoma cells | Impaired myogenesis | [104] |
| IL-6 | LLC | Lung cancer | Muscle catabolism, lipolysis | [105] |
| GDF15 | C26 | Colon cancer | Muscle atrophy | [114] |
| P4HB | ESCC | Oesophageal cancer | Muscle wasting | [115] |
| PTHrP | LLC | Lung cancer | Adipose tissue browning | [116] |
| ITGB1 | Panc-1 Miapaca-2 Capan-2 Patient serum | Pancreatic cancer | Lipolysis | [117] |
| ITGA6 | Panc-1 Miapaca-2 Capan-2 Patient serum | Pancreatic cancer | Lipolysis | [117] |
| miRNAs | ||||
| miR-425-3p | NSCLC | Lung cancer | Adipose tissue browning, inhibits proliferation and differentiation of preadipocytes | [118] |
| miR-146b-5p | HEK293T HCT-116 Patient tissue | Colorectal cancer | Adipose tissue browning | [119] |
| miR-410-3p | Patient plasma | Gastric cancer | Inhibits adipogenesis | [120] |
| miR-204-5p | MDMB231 SK-BR-3 4T1 E0771 | Breast cancer | Lipolysis, WAT browning | [121] |
| miR-155. | MCF-7 | Breast cancer | Lipolysis, WAT browning | [122] |
| miR-21 | PC1 Panc-2 Miapaca-2 LLC A549 | Pancreatic cancer Lung cancer | Muscle catabolism | [123] |
| miR-125b-1-3p | C26 | Colon cancer | Muscle wasting | [124] |
| miR-195a-5p | C26 | Colon cancer | Muscle wasting | [124] |
6. Cancer-Derived EV Proteins and Lipolysis
7. EV-Associated MicroRNAs and Cancer-Associated Cachexia
8. Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Carson, M.A.; Reid, J.; Hill, L.; Dixon, L.; Donnelly, P.; Slater, P.; Hill, A.; Fitzsimons, D. An exploration of the prevalence and experience of cardiac cachexia: Protocol for a mixed methods cross-sectional study. BMC Palliat. Care 2019, 18, 82. [Google Scholar] [CrossRef]
- Capozzi, L.C.; McNeely, M.L.; Lau, H.Y.; Reimer, R.A.; Giese-Davis, J.; Fung, T.S.; Culos-Reed, S.N. Patient-reported outcomes, body composition, and nutrition status in patients with head and neck cancer: Results from an exploratory randomized controlled exercise trial. Cancer 2016, 122, 1185–1200. [Google Scholar] [CrossRef]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef]
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013, 10, 90–99. [Google Scholar] [CrossRef]
- Tisdale, M.J. Biology of cachexia. J. Natl. Cancer Inst. 1997, 89, 1763–1773. [Google Scholar] [CrossRef]
- Law, M.L. Cancer cachexia: Pathophysiology and association with cancer-related pain. Front. Pain Res. 2022, 3, 971295. [Google Scholar] [CrossRef]
- Ni, J.; Zhang, L. Cancer Cachexia: Definition, Staging, and Emerging Treatments. Cancer Manag. Res. 2020, 12, 5597–5605. [Google Scholar] [CrossRef]
- Dolly, A.; Dumas, J.F.; Servais, S. Cancer cachexia and skeletal muscle atrophy in clinical studies: What do we really know? J. Cachexia Sarcopenia Muscle 2020, 11, 1413–1428. [Google Scholar] [CrossRef]
- Aversa, Z.; Costelli, P.; Muscaritoli, M. Cancer-induced muscle wasting: Latest findings in prevention and treatment. Ther. Adv. Med. Oncol. 2017, 9, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Feraco, A.; Gorini, S.; Armani, A.; Camajani, E.; Rizzo, M.; Caprio, M. Exploring the Role of Skeletal Muscle in Insulin Resistance: Lessons from Cultured Cells to Animal Models. Int. J. Mol. Sci. 2021, 22, 9327. [Google Scholar] [CrossRef]
- Anthony, T.G. Mechanisms of protein balance in skeletal muscle. Domest Anim. Endocrinol. 2016, 56 (Suppl. S1), S23–S32. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Beltrá, M.; De Lucia, S.; Costelli, P. Modulating Metabolism to Improve Cancer-Induced Muscle Wasting. Oxid. Med. Cell Longev. 2018, 2018, 7153610. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Patel, H.; Patel, B. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2016, 170, 33. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef]
- Wildi, S.; Kleeff, J.; Maruyama, H.; Maurer, C.A.; Büchler, M.W.; Korc, M. Overexpression of activin A in stage IV colorectal cancer. Gut 2001, 49, 409–417. [Google Scholar] [CrossRef]
- Yang, W.; Huang, J.; Wu, H.; Wang, Y.; Du, Z.; Ling, Y.; Wang, W.; Wu, Q.; Gao, W. Molecular mechanisms of cancer cachexia-induced muscle atrophy (Review). Mol. Med. Rep. 2020, 22, 4967–4980. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Signaling pathways perturbing muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Siff, T.; Parajuli, P.; Razzaque, M.S.; Atfi, A. Cancer-Mediated Muscle Cachexia: Etiology and Clinical Management. Trends Endocrinol. Metab. 2021, 32, 382–402. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Moore-Carrasco, R.; Busquets, S.; Almendro, V.; Palanki, M.; López-Soriano, F.J.; Argilés, J.M. The AP-1/NF-kappaB double inhibitor SP100030 can revert muscle wasting during experimental cancer cachexia. Int. J. Oncol. 2007, 30, 1239–1245. [Google Scholar]
- Moylan, J.S.; Smith, J.D.; Chambers, M.A.; McLoughlin, T.J.; Reid, M.B. TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am. J. Physiol. Cell Physiol. 2008, 295, C986–C993. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef]
- Eskiler, G.G.; Bezdegumeli, E.; Ozman, Z.; Ozkan, A.D.; Bilir, C.; Kucukakca, B.N.; Ince, M.N.; Men, A.Y.; Aktas, O.; Horoz, Y.E.; et al. IL-6 mediated JAK/STAT3 signaling pathway in cancer patients with cachexia. Bratisl Lek Listy 2019, 66, 819–826. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Inaba, S.; Hinohara, A.; Tachibana, M.; Tsujikawa, K.; Fukada, S.I. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PLoS ONE 2018, 13, e0205467. [Google Scholar] [CrossRef]
- Arneson, P.C.; Doles, J.D. Impaired Muscle Regeneration in Cancer-Associated Cachexia. Trends Cancer 2019, 5, 579–582. [Google Scholar] [CrossRef]
- Hogan, K.A.; Cho, D.S.; Arneson, P.C.; Samani, A.; Palines, P.; Yang, Y.; Doles, J.D. Tumor-derived cytokines impair myogenesis and alter the skeletal muscle immune microenvironment. Cytokine 2018, 107, 9–17. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Wang, G.; Biswas, A.K.; Ma, W.; Kandpal, M.; Coker, C.; Grandgenett, P.M.; Hollingsworth, M.A.; Jain, R.; Tanji, K.; Lόpez-Pintado, S.; et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 2018, 24, 770–781. [Google Scholar] [CrossRef]
- Melzer, K. Carbohydrate and fat utilization during rest and physical activity. e-SPEN Eur. e-J. Clin. Nutr. Metab. 2011, 6, e45–e52. [Google Scholar] [CrossRef]
- Church, C.; Horowitz, M.; Rodeheffer, M. WAT is a functional adipocyte? Adipocyte 2012, 1, 38–45. [Google Scholar] [CrossRef]
- Shaw, J.H.; Wolfe, R.R. Fatty acid and glycerol kinetics in septic patients and in patients with gastrointestinal cancer. The response to glucose infusion and parenteral feeding. Ann. Surg. 1987, 205, 368–376. [Google Scholar] [CrossRef]
- Drott, C.; Persson, H.; Lundholm, K. Cardiovascular and metabolic response to adrenaline infusion in weight-losing patients with and without cancer. Clin. Physiol. 1989, 9, 427–439. [Google Scholar] [CrossRef]
- Fouladiun, M.; Körner, U.; Bosaeus, I.; Daneryd, P.; Hyltander, A.G.; Lundholm, K.G. Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care—Correlations with food intake, metabolism, exercise capacity, and hormones. Cancer 2005, 103, 2189–2198. [Google Scholar] [CrossRef]
- Ebadi, M.; Mazurak, V.C. Evidence and mechanisms of fat depletion in cancer. Nutrients 2014, 6, 5280–5297. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanisms of Cancer Cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Malla, J.; Zahra, A.; Venugopal, S.; Selvamani, T.Y.; Shoukrie, S.I.; Selvaraj, R.; Dhanoa, R.K.; Hamouda, R.K.; Mostafa, J. What Role Do Inflammatory Cytokines Play in Cancer Cachexia? Cureus 2022, 14, e26798. [Google Scholar] [CrossRef]
- Okazaki, H.; Osuga, J.; Tamura, Y.; Yahagi, N.; Tomita, S.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; Kimura, S.; et al. Lipolysis in the absence of hormone-sensitive lipase: Evidence for a common mechanism regulating distinct lipases. Diabetes 2002, 51, 3368–3375. [Google Scholar] [CrossRef][Green Version]
- Dalal, S. Lipid metabolism in cancer cachexia. Ann. Palliat. Med. 2018, 8, 13–23. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid. Res. 2011, 50, 14–27. [Google Scholar] [CrossRef]
- Arora, G.K.; Gupta, A.; Narayanan, S.; Guo, T.; Iyengar, P.; Infante, R.E. Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin. JCI Insight 2018, 3, 121221. [Google Scholar] [CrossRef]
- Mangano, G.D.; Fouani, M.; D’Amico, D.; Di Felice, V.; Barone, R. Cancer-Related Cachexia: The Vicious Circle between Inflammatory Cytokines, Skeletal Muscle, Lipid Metabolism and the Possible Role of Physical Training. Int. J. Mol. Sci. 2022, 23, 3004. [Google Scholar] [CrossRef]
- Honors, M.A.; Kinzig, K.P. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J. Cachexia Sarcopenia Muscle 2012, 3, 5–11. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Pothuraju, R.; Jain, M.; Batra, S.K.; Nasser, M.W. Advances in cancer cachexia: Intersection between affected organs, mediators, and pharmacological interventions. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188359. [Google Scholar] [CrossRef]
- Cypess, A.M.; Kahn, C.R. The role and importance of brown adipose tissue in energy homeostasis. Curr. Opin. Pediatr. 2010, 22, 478–484. [Google Scholar] [CrossRef]
- Chan, P.C.; Hsieh, P.S. The Role and Regulatory Mechanism of Brown Adipose Tissue Activation in Diet-Induced Thermogenesis in Health and Diseases. Int. J. Mol. Sci. 2022, 23, 9448. [Google Scholar] [CrossRef]
- Beijer, E.; Schoenmakers, J.; Vijgen, G.; Kessels, F.; Dingemans, A.M.; Schrauwen, P.; Wouters, M.; van Marken Lichtenbelt, W.; Teule, J.; Brans, B. A role of active brown adipose tissue in cancer cachexia? Oncol. Rev. 2012, 6, e11. [Google Scholar] [CrossRef][Green Version]
- Tsoli, M.; Moore, M.; Burg, D.; Painter, A.; Taylor, R.; Lockie, S.H.; Turner, N.; Warren, A.; Cooney, G.; Oldfield, B.; et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. 2012, 72, 4372–4382. [Google Scholar] [CrossRef]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef]
- Wang, G.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74, 844–857.e847. [Google Scholar] [CrossRef]
- Bing, C.; Bao, Y.; Jenkins, J.; Sanders, P.; Manieri, M.; Cinti, S.; Tisdale, M.J.; Trayhurn, P. Zinc-alpha2-glycoprotein, a lipid mobilizing factor, is expressed in adipocytes and is up-regulated in mice with cancer cachexia. Proc. Natl. Acad. Sci. USA 2004, 101, 2500–2505. [Google Scholar] [CrossRef]
- Elattar, S.; Dimri, M.; Satyanarayana, A. The tumor secretory factor ZAG promotes white adipose tissue browning and energy wasting. FASEB J. 2018, 32, 4727–4743. [Google Scholar] [CrossRef]
- Mantovani, G.; Macciò, A.; Madeddu, C.; Serpe, R.; Massa, E.; Dessì, M.; Panzone, F.; Contu, P. Randomized phase III clinical trial of five different arms of treatment in 332 patients with cancer cachexia. Oncologist 2010, 15, 200–211. [Google Scholar] [CrossRef]
- Cerchietti, L.C.; Navigante, A.H.; Peluffo, G.D.; Diament, M.J.; Stillitani, I.; Klein, S.A.; Cabalar, M.E. Effects of celecoxib, medroxyprogesterone, and dietary intervention on systemic syndromes in patients with advanced lung adenocarcinoma: A pilot study. J. Pain Symptom. Manag. 2004, 27, 85–95. [Google Scholar] [CrossRef]
- Loprinzi, C.L.; Kugler, J.W.; Sloan, J.A.; Mailliard, J.A.; Krook, J.E.; Wilwerding, M.B.; Rowland, K.M., Jr.; Camoriano, J.K.; Novotny, P.J.; Christensen, B.J. Randomized comparison of megestrol acetate versus dexamethasone versus fluoxymesterone for the treatment of cancer anorexia/cachexia. J. Clin. Oncol. 1999, 17, 3299–3306. [Google Scholar] [CrossRef]
- Anderson, L.J.; Albrecht, E.D.; Garcia, J.M. Update on Management of Cancer-Related Cachexia. Curr. Oncol. Rep. 2017, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Tazi, E.; Errihani, H. Treatment of cachexia in oncology. Indian J. Palliat Care 2010, 16, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef]
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T.; et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 2016, 65, 1165–1174. [Google Scholar] [CrossRef]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016, 126, 1139–1143. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Kalra, H.; Drummen, G.P.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, P.; Marzan, A.L.; Mathivanan, S. Introduction to the Community of Extracellular Vesicles. In New Frontiers: Extracellular Vesicles; Mathivanan, S., Fonseka, P., Nedeva, C., Atukorala, I., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 3–18. [Google Scholar] [CrossRef]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Peng, X.; Yang, L.; Ma, Y.; Li, Y.; Li, H. Focus on the morphogenesis, fate and the role in tumor progression of multivesicular bodies. Cell Commun. Signal 2020, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30, sup pp. 11–13. [Google Scholar] [CrossRef]
- Stein, J.M.; Luzio, J.P. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. The sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem. J. 1991, 274 Pt 2, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Fordjour, F.K.; Guo, C.; Ai, Y.; Daaboul, G.G.; Gould, S.J. A shared, stochastic pathway mediates exosome protein budding along plasma and endosome membranes. J. Biol. Chem. 2022, 298, 102394. [Google Scholar] [CrossRef]
- Mathieu, M.; Névo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021, 12, 4389. [Google Scholar] [CrossRef]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef]
- Liang, H.; Ma, X.; Zhang, Y.; Liu, Y.; Liu, N.; Zhang, W.; Chen, J.; Liu, B.; Du, W.; Liu, X.; et al. The formation of migrasomes is initiated by the assembly of sphingomyelin synthase 2 foci at the leading edge of migrating cells. Nat. Cell Biol. 2023, 25, 1173–1184. [Google Scholar] [CrossRef]
- Banasiak, K.; Turek, M.; Pokrzywa, W. Preparation of Caenorhabditis elegans for Scoring of Muscle-derived Exophers. Bio Protoc. 2023, 13, e4586. [Google Scholar] [CrossRef] [PubMed]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef]
- Cram, E.J. Exophers to feed them all. EMBO Rep. 2021, 22, e53265. [Google Scholar] [CrossRef]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.J.; Cavallini, L.; Ciardiello, C.; Reis Sobreiro, M.; Morello, M.; et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 2015, 6, 11327–11341. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140203. [Google Scholar] [CrossRef]
- Li, S.; Lin, Z.; Jiang, X.; Yu, X. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Tian, T.; Zhu, Y.-L.; Hu, F.-H.; Wang, Y.-Y.; Huang, N.-P.; Xiao, Z.-D. Dynamics of exosome internalization and trafficking. J. Cell. Physiol. 2013, 228, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Karampoga, A.; Tzaferi, K.; Koutsakis, C.; Kyriakopoulou, K.; Karamanos, N.K. Exosomes and the extracellular matrix: A dynamic interplay in cancer progression. Int. J. Dev. Biol. 2022, 66, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.P.; Janssen, E.; Pegtel, D.M.; Crudden, C. The forces driving cancer extracellular vesicle secretion. Neoplasia 2021, 23, 149–157. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef]
- Chang, W.H.; Cerione, R.A.; Antonyak, M.A. Extracellular Vesicles and Their Roles in Cancer Progression. Methods Mol. Biol. 2021, 2174, 143–170. [Google Scholar] [CrossRef]
- Liu, Y.; Mao, D.; Wang, H.; Che, X.; Chen, Y. Formation of pre-metastatic niches induced by tumor extracellular vesicles in lung metastasis. Pharmacol. Res. 2023, 188, 106669. [Google Scholar] [CrossRef]
- Kong, J.; Tian, H.; Zhang, F.; Zhang, Z.; Li, J.; Liu, X.; Li, X.; Liu, J.; Li, X.; Jin, D.; et al. Extracellular vesicles of carcinoma-associated fibroblasts creates a pre-metastatic niche in the lung through activating fibroblasts. Mol. Cancer 2019, 18, 175. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, H.; Yin, X.; Yang, M.; Wei, H.; Chen, Q.; Feng, F.; Liu, Y.; Xu, W.; Li, Y. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J. Exp. Clin. Cancer Res. 2019, 38, 81. [Google Scholar] [CrossRef]
- Chen, W.T.; Yang, Y.J.; Zhang, Z.D.; An, Q.; Li, N.; Liu, W.; Yang, B. MiR-1307 promotes ovarian cancer cell chemoresistance by targeting the ING5 expression. J. Ovarian Res. 2017, 10, 1. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Guigni, B.A.; van der Velden, J.; Kinsey, C.M.; Carson, J.A.; Toth, M.J. Effects of conditioned media from murine lung cancer cells and human tumor cells on cultured myotubes. Am. J. Physiol. Endocrinol. Metab. 2020, 318, e22–e32. [Google Scholar] [CrossRef] [PubMed]
- Chitti, S.V.; Kang, T.; Fonseka, P.; Marzan, A.L.; Stewart, S.; Shahi, S.; Bramich, K.; Ang, C.S.; Pathan, M.; Gummadi, S.; et al. Proteomic analysis of the small extracellular vesicles and soluble secretory proteins from cachexia inducing and non-inducing cancer cells. Proteomics 2023, 23, e2100314. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Agarwal, R.; March, D.; Rothenberg, A.; Voigt, C.; Tebbets, J.; Huard, J.; Weiss, K. Notch Signaling Mediates Skeletal Muscle Atrophy in Cancer Cachexia Caused by Osteosarcoma. Sarcoma 2016, 2016, 3758162. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ru, Z.; Zhou, Y.; Xiao, W.; Sun, R.; Zhang, S.; Gao, Y.; Li, X.; Zhang, X.; Yang, H. Lung cancer-derived extracellular vesicles induced myotube atrophy and adipocyte lipolysis via the extracellular IL-6-mediated STAT3 pathway. Biochim Biophys Acta Mol. Cell Biol. Lipids 2019, 1864, 1091–1102. [Google Scholar] [CrossRef]
- Chitti, S.V.; Fonseka, P.; Mathivanan, S. Emerging role of extracellular vesicles in mediating cancer cachexia. Biochem. Soc. Trans. 2018, 46, 1129–1136. [Google Scholar] [CrossRef]
- Marzan, A.L.; Nedeva, C.; Mathivanan, S. Extracellular Vesicles in Metabolism and Metabolic Diseases. In New Frontiers: Extracellular Vesicles; Mathivanan, S., Fonseka, P., Nedeva, C., Atukorala, I., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 393–410. [Google Scholar] [CrossRef]
- Busatto, S.; Morad, G.; Guo, P.; Moses, M.A. The role of extracellular vesicles in the physiological and pathological regulation of the blood-brain barrier. FASEB Bioadv. 2021, 3, 665–675. [Google Scholar] [CrossRef]
- Marzan, A.L.; Stewart, S.E. Elucidating the Role of Extracellular Vesicles in Pancreatic Cancer. Cancers 2021, 13, 5669. [Google Scholar] [CrossRef]
- Pin, F.; Beltrà, M.; Garcia-Castillo, L.; Pardini, B.; Birolo, G.; Matullo, G.; Penna, F.; Guttridge, D.; Costelli, P. Extracellular vesicles derived from tumour cells as a trigger of energy crisis in the skeletal muscle. J. Cachexia Sarcopenia Muscle 2022, 13, 481–494. [Google Scholar] [CrossRef]
- Fan, M.; Sun, W.; Gu, X.; Lu, S.; Shen, Q.; Liu, X.; Zhang, X. The critical role of STAT3 in biogenesis of tumor-derived exosomes with potency of inducing cancer cachexia in vitro and in vivo. Oncogene 2022, 41, 1050–1062. [Google Scholar] [CrossRef]
- AlSudais, H.; Rajgara, R.; Saleh, A.; Wiper-Bergeron, N. C/EBPβ promotes the expression of atrophy-inducing factors by tumours and is a central regulator of cancer cachexia. J. Cachexia Sarcopenia Muscle 2022, 13, 743–757. [Google Scholar]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734.e726. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, W.; Gu, X.; Miao, C.; Feng, L.; Shen, Q.; Liu, X.; Zhang, X. GDF-15 in tumor-derived exosomes promotes muscle atrophy via Bcl-2/caspase-3 pathway. Cell Death Discov. 2022, 8, 162. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Y.; Lu, F.; Chen, X.; Yang, D.; Cao, Y.; Zhang, W.; Chen, J.; Zheng, L.; Wang, G.; et al. Extracellular vesicles derived from oesophageal cancer containing P4HB promote muscle wasting via regulating PHGDH/Bcl-2/caspase-3 pathway. J. Extracell Vesicles 2021, 10, e12060. [Google Scholar] [CrossRef]
- Hu, W.; Xiong, H.; Ru, Z.; Zhao, Y.; Zhou, Y.; Xie, K.; Xiao, W.; Xiong, Z.; Wang, C.; Yuan, C.; et al. Extracellular vesicles-released parathyroid hormone-related protein from Lewis lung carcinoma induces lipolysis and adipose tissue browning in cancer cachexia. Cell Death Dis. 2021, 12, 134. [Google Scholar] [CrossRef]
- Shibata, C.; Otsuka, M.; Seimiya, T.; Kishikawa, T.; Ishigaki, K.; Fujishiro, M. Lipolysis by pancreatic cancer-derived extracellular vesicles in cancer-associated cachexia via specific integrins. Clin. Transl. Med. 2022, 12, e1089. [Google Scholar] [CrossRef]
- Liu, A.; Pan, W.; Zhuang, S.; Tang, Y.; Zhang, H. Cancer cell-derived exosomal miR-425-3p induces white adipocyte atrophy. Adipocyte 2022, 11, 487–500. [Google Scholar] [CrossRef]
- Di, W.; Zhang, W.; Zhu, B.; Li, X.; Tang, Q.; Zhou, Y. Colorectal cancer prompted adipose tissue browning and cancer cachexia through transferring exosomal miR-146b-5p. J. Cell. Physiol. 2021, 236, 5399–5410. [Google Scholar] [CrossRef]
- Sun, D.; Ding, Z.; Shen, L.; Yang, F.; Han, J.; Wu, G. miR-410-3P inhibits adipocyte differentiation by targeting IRS-1 in cancer-associated cachexia patients. Lipids Health Dis. 2021, 20, 115. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Chen, Y.; Zhang, X.; Zhou, H.; Hu, S.; Li, X.; Li, M.; Li, J.; Cheng, S.; et al. Cancer-cell-secreted miR-204-5p induces leptin signalling pathway in white adipose tissue to promote cancer-associated cachexia. Nat. Commun. 2023, 14, 5179. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Z.; Yao, F.; Sun, K.; Li, Z.; Sun, S.; Li, C. Breast cancer cell-derived exosome-delivered microRNA-155 targets UBQLN1 in adipocytes and facilitates cancer cachexia-related fat loss. Hum. Mol. Genet. 2023, 32, 2219–2228. [Google Scholar] [CrossRef]
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529. [Google Scholar] [CrossRef]
- Miao, C.; Zhang, W.; Feng, L.; Gu, X.; Shen, Q.; Lu, S.; Fan, M.; Li, Y.; Guo, X.; Ma, Y.; et al. Cancer-derived exosome miRNAs induce skeletal muscle wasting by Bcl-2-mediated apoptosis in colon cancer cachexia. Mol. Ther. Nucleic Acids 2021, 24, 923–938. [Google Scholar] [CrossRef]
- Xiong, H.; Ye, J.; Xie, K.; Hu, W.; Xu, N.; Yang, H. Exosomal IL-8 derived from Lung Cancer and Colon Cancer cells induced adipocyte atrophy via NF-κB signaling pathway. Lipids Health Dis. 2022, 21, 147. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.G.; Finn, S.P.; Cuffe, S.; O’Byrne, K.J.; Barr, M.P. The emerging role of microRNAs in resistance to lung cancer treatments. Cancer Treat. Rev. 2015, 41, 160–169. [Google Scholar] [CrossRef]
- Zhou, X.; Hu, S.; Zhang, Y.; Du, G.; Li, Y. The mechanism by which noncoding RNAs regulate muscle wasting in cancer cachexia. Precis Clin. Med. 2021, 4, 136–147. [Google Scholar] [CrossRef]
- Liu, S.L.; Sun, P.; Li, Y.; Liu, S.S.; Lu, Y. Exosomes as critical mediators of cell-to-cell communication in cancer pathogenesis and their potential clinical application. Transl. Cancer Res. 2019, 8, 298–311. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzan, A.L.; Chitti, S.V. Unravelling the Role of Cancer Cell-Derived Extracellular Vesicles in Muscle Atrophy, Lipolysis, and Cancer-Associated Cachexia. Cells 2023, 12, 2598. https://doi.org/10.3390/cells12222598
Marzan AL, Chitti SV. Unravelling the Role of Cancer Cell-Derived Extracellular Vesicles in Muscle Atrophy, Lipolysis, and Cancer-Associated Cachexia. Cells. 2023; 12(22):2598. https://doi.org/10.3390/cells12222598
Chicago/Turabian StyleMarzan, Akbar L., and Sai V. Chitti. 2023. "Unravelling the Role of Cancer Cell-Derived Extracellular Vesicles in Muscle Atrophy, Lipolysis, and Cancer-Associated Cachexia" Cells 12, no. 22: 2598. https://doi.org/10.3390/cells12222598
APA StyleMarzan, A. L., & Chitti, S. V. (2023). Unravelling the Role of Cancer Cell-Derived Extracellular Vesicles in Muscle Atrophy, Lipolysis, and Cancer-Associated Cachexia. Cells, 12(22), 2598. https://doi.org/10.3390/cells12222598