
Runx3 Restoration Regresses K-Ras-Activated Mouse Lung Cancers and Inhibits Recurrence

 , , ,
, , ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Adenovirus and Tamoxifen Delivery in Mice
2.3. KPR Primary Tumor Cell Line Extract
2.4. Hematoxylin and Eosin (HE) Staining
2.5. Histology and Immunohistochemistry
2.6. DNA Exon-Seq Analysis
2.7. DNA Transfection, IP, and IB
2.8. Antibodies
2.9. RNA-Seq Analysis
2.10. Quantification and Statistical Analysis
3. Results
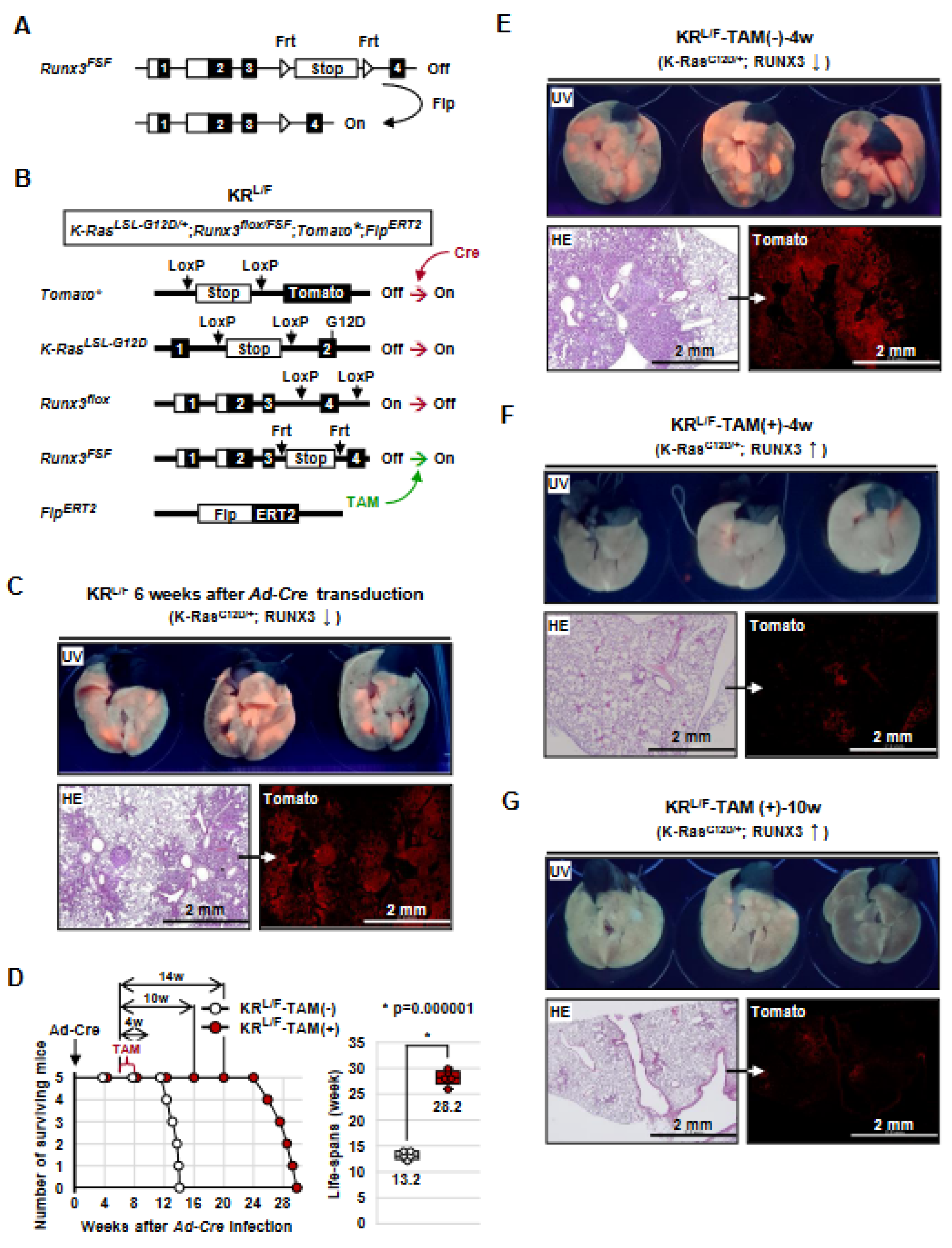
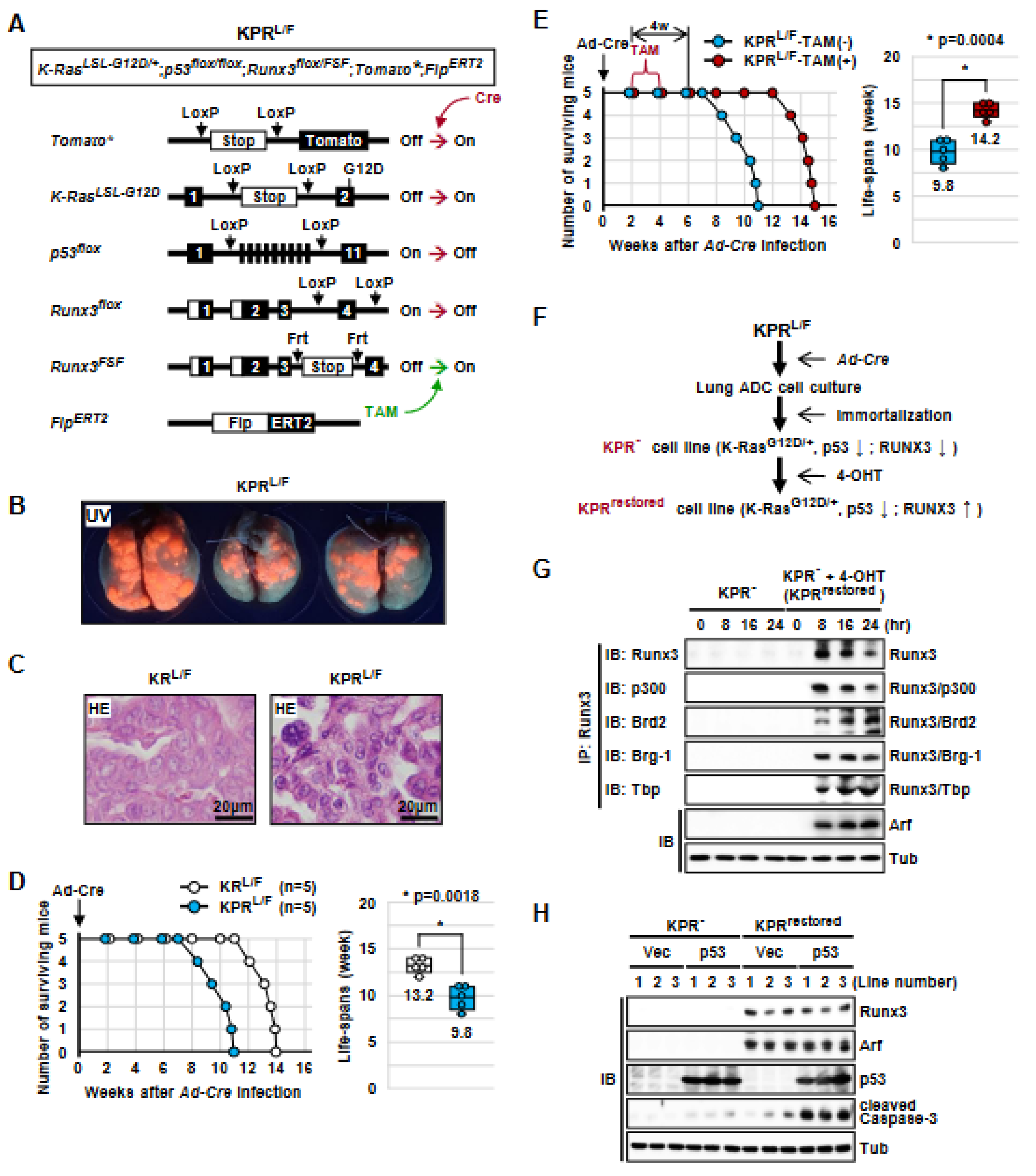
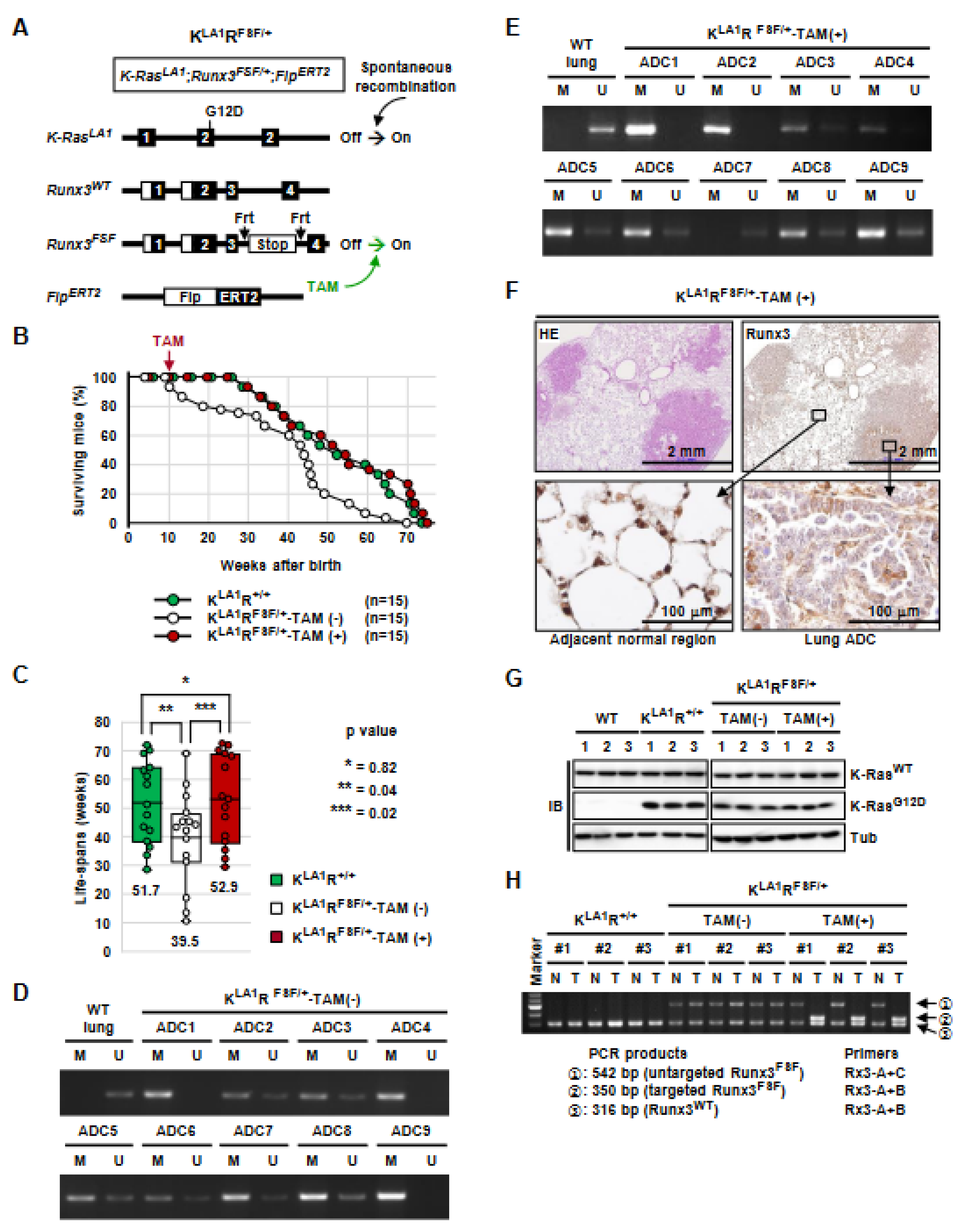
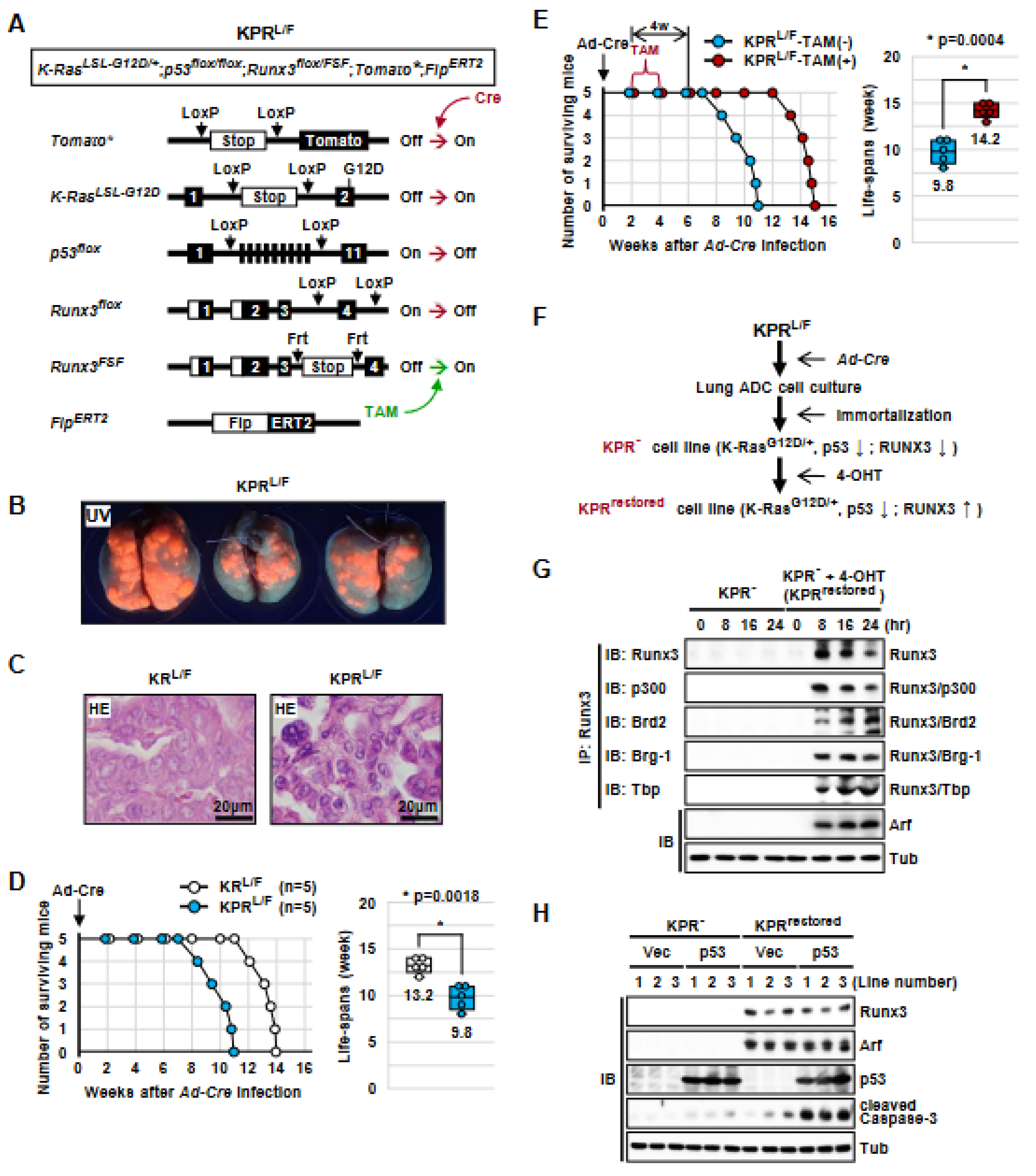
3.1. Generation of K-RasLoxP-Stop-LoxP-G12D/+, Runx3flox/FSF, Tomato*, and FlpERT2 (KRL/F) Mice
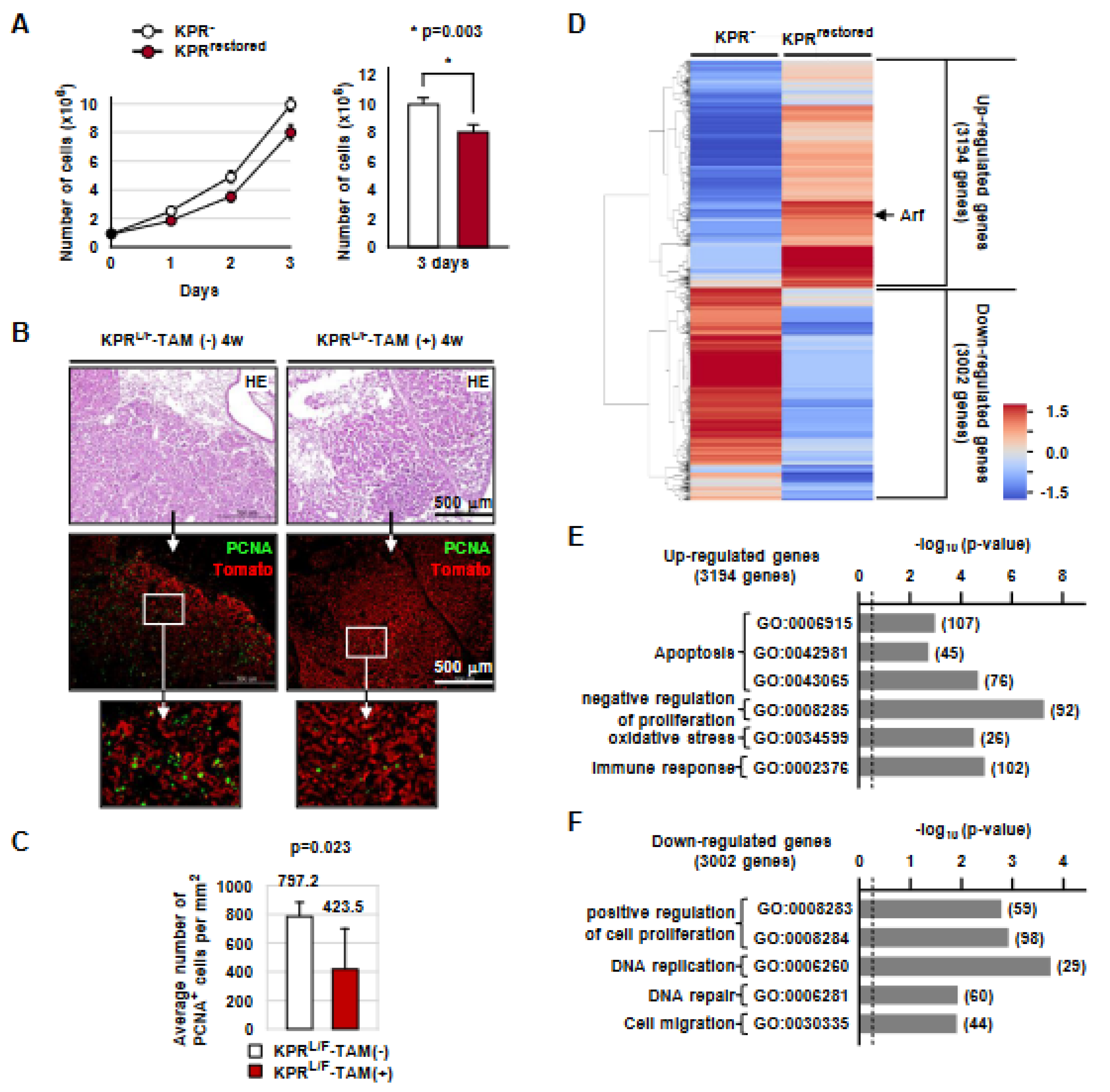
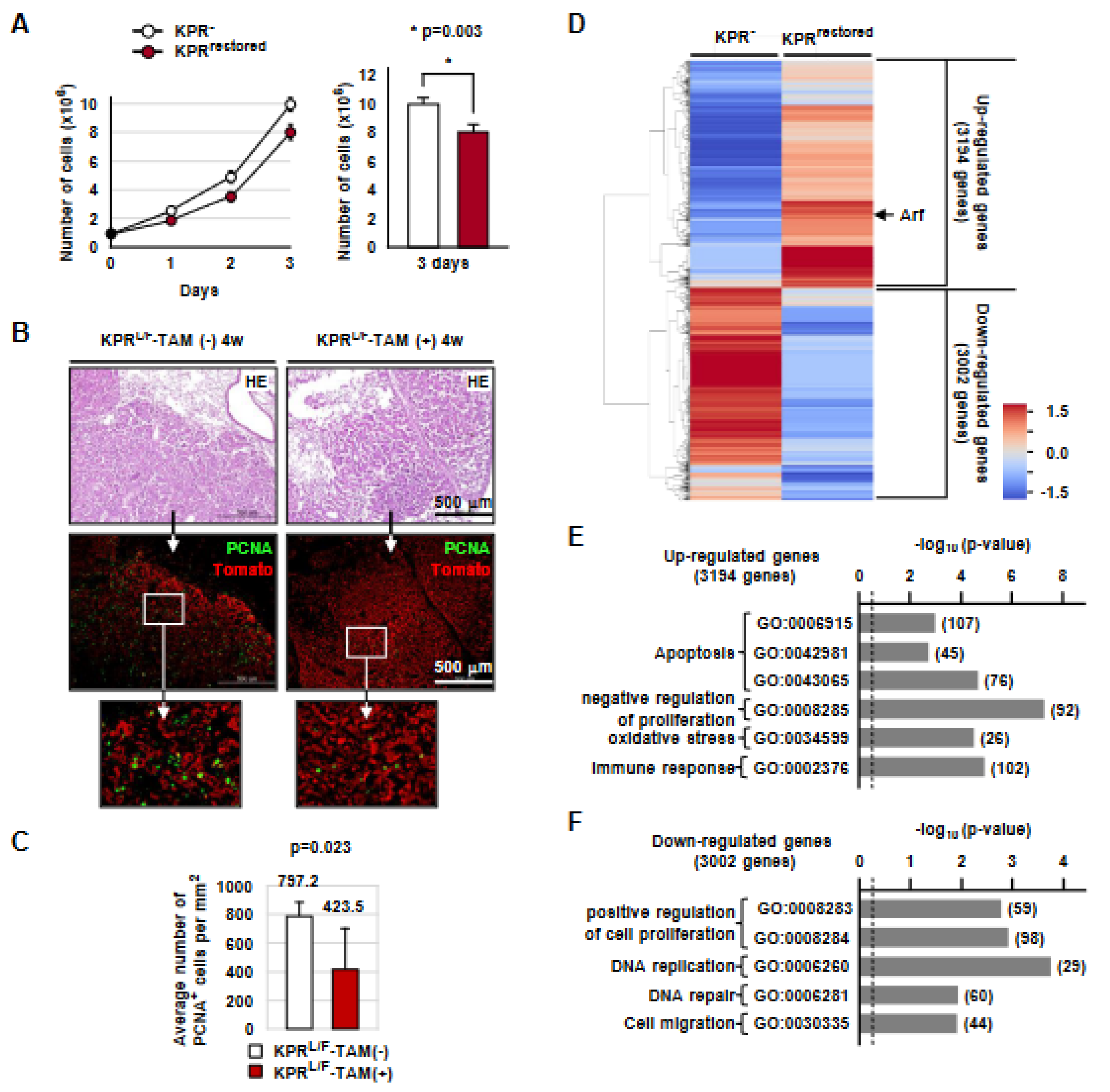
3.2. Runx3 Restoration Effectively Eliminates Established K-Ras-activated Lung Cancer Cells
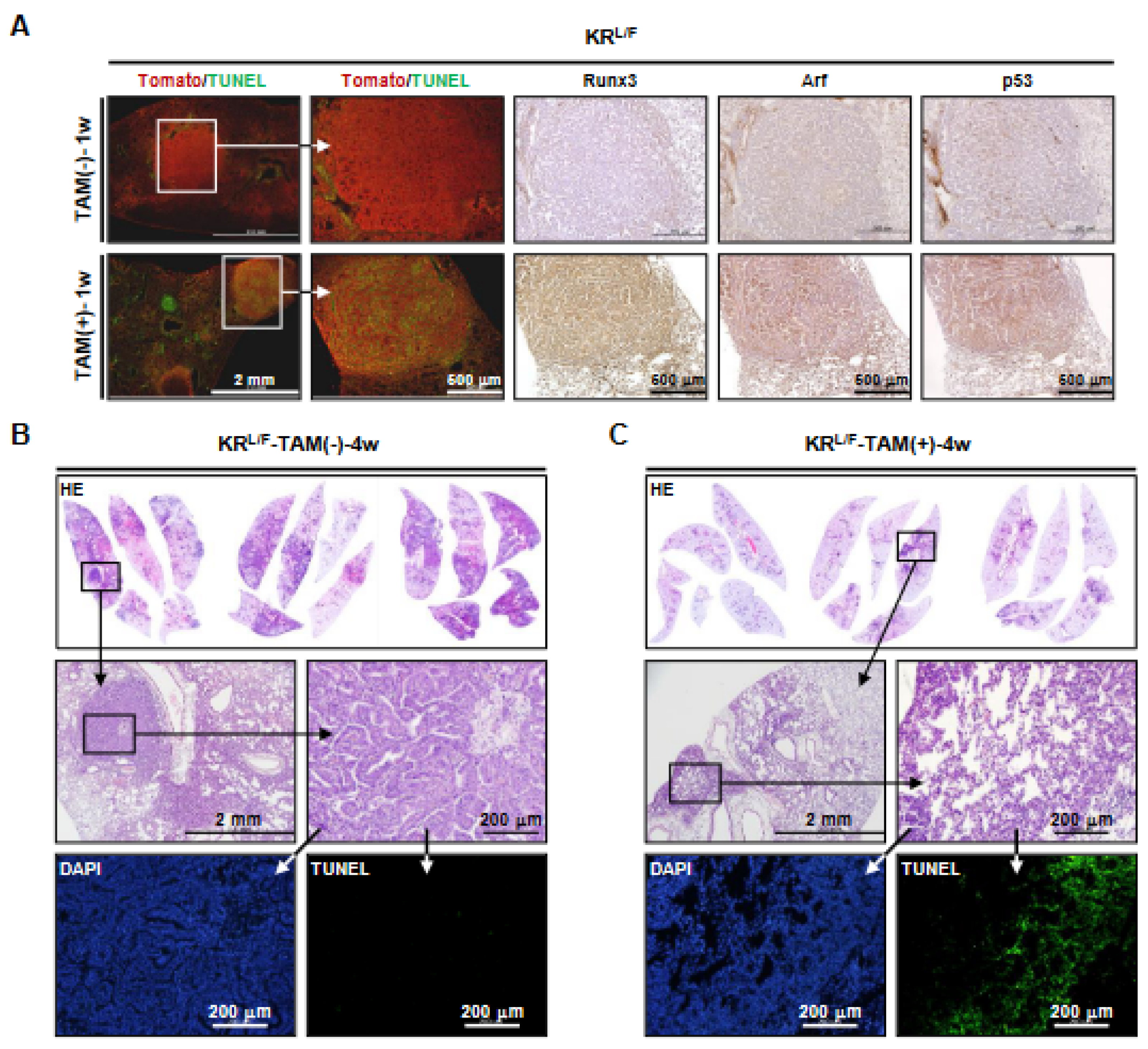
3.3. Runx3 Restoration Eliminates K-Ras-activated Lung Cancers by Inducing Apoptosis
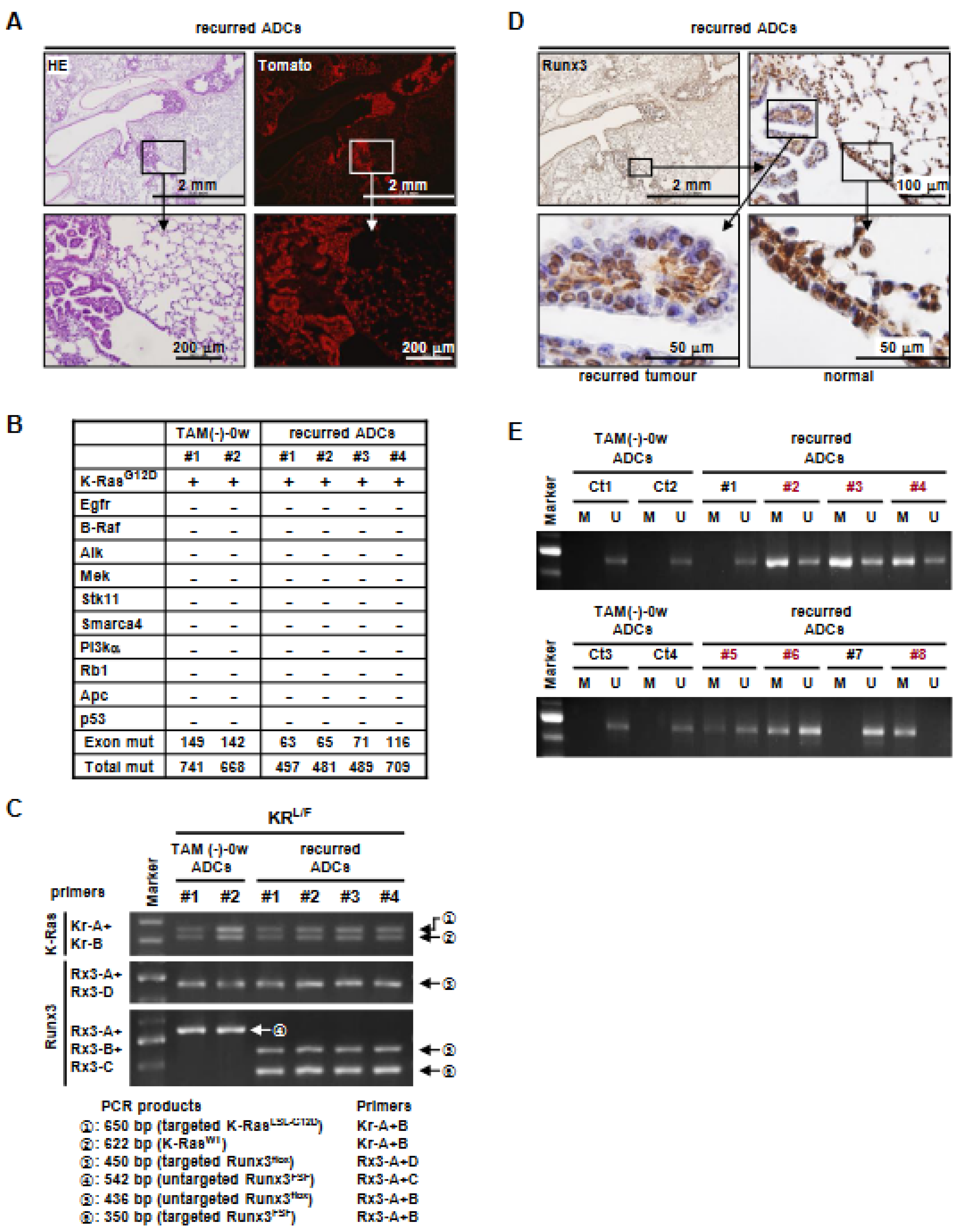
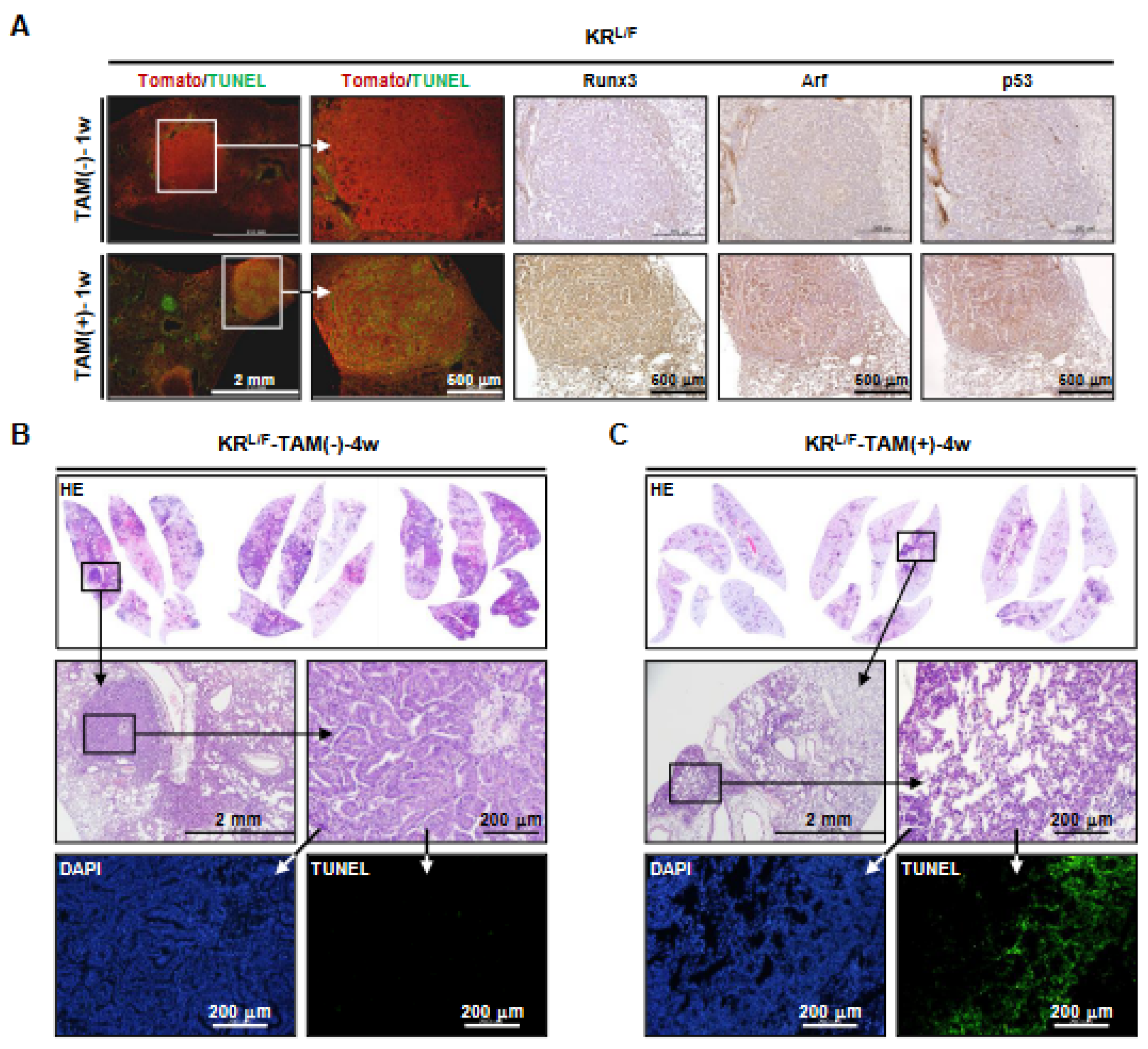
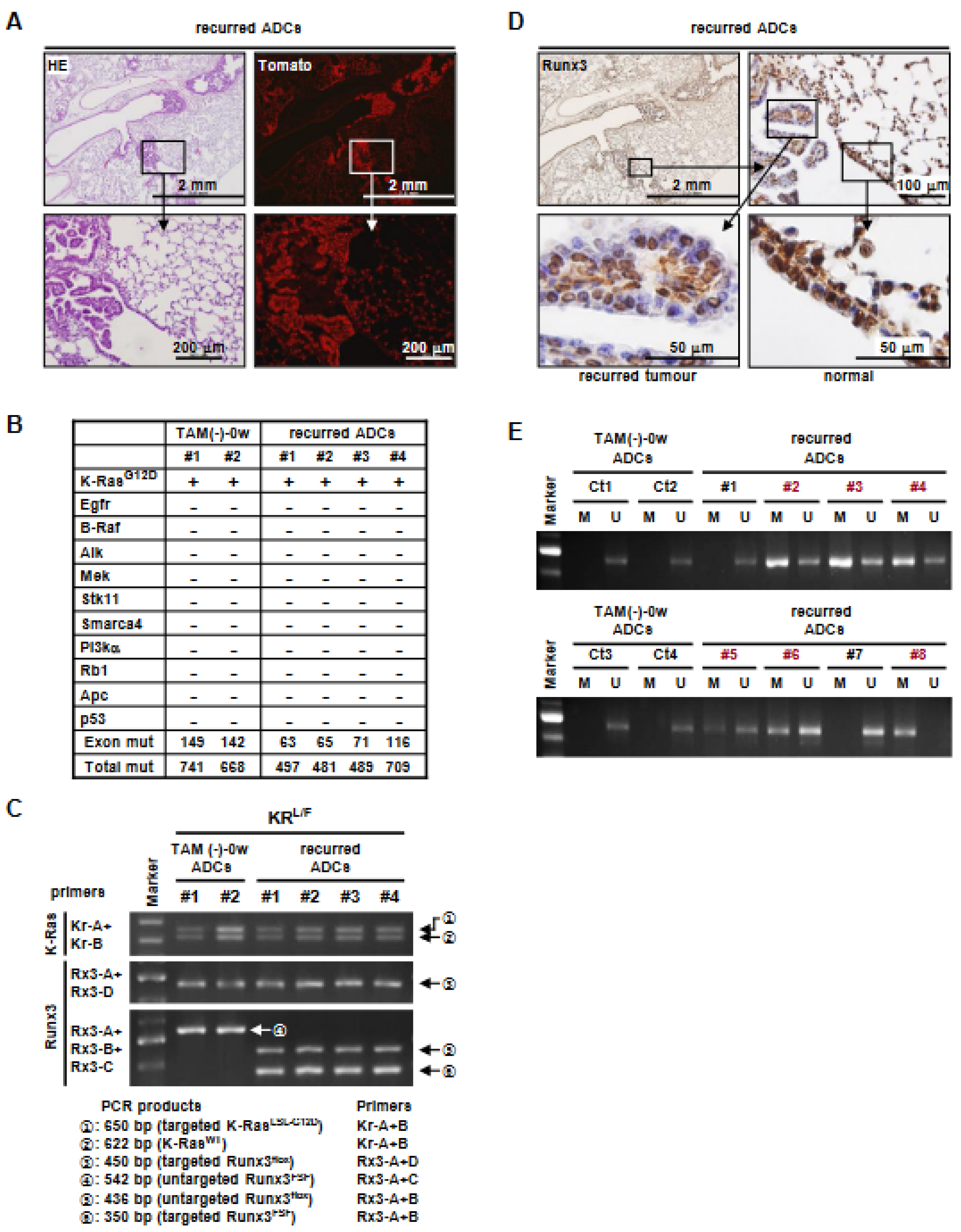
3.4. K-Ras-Activated Lung Cancer Began to Recur at 14 Weeks after Runx3 Restoration
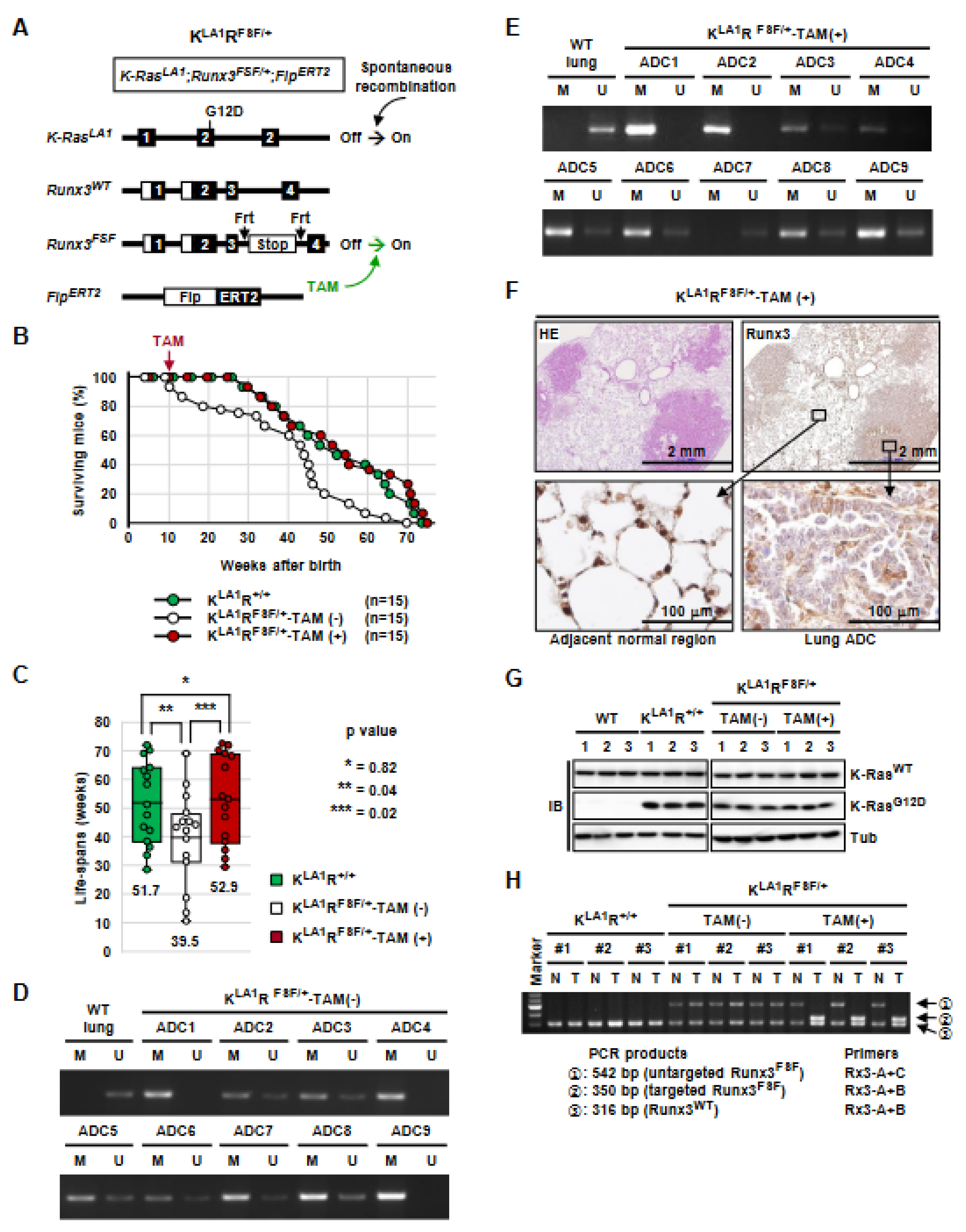
3.5. Runx3 Inactivation Is Essential for the Maintenance of K-Ras-Activated Lung Cancer
3.6. The Tumor-Suppressive Activity of Runx3 Is Largely Dependent on p53
3.7. Runx3 Restoration Recovers the R-Point and Induces Arf Expression
3.8. Runx3 Restoration Inhibits Proliferation of K-Ras-Activated Lung Tumor Cells in a p53-Independent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wistuba, I.I.; Gazdar, A.F. Lung cancer preneoplasia. Annu. Rev. Pathol. 2006, 1, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Govindan, R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Leonardi, G.C.; Metro, G.; Grignani, F.; Paglialunga, L.; Bellezza, G.; Baglivo, S.; Mencaroni, C.; Baldi, A.; Zicari, D.; et al. Targeting the KRAS variant for treatment of non-small cell lung cancer: Potential therapeutic applications. Expert Rev. Respir. Med. 2016, 10, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Nagasaka, M. Spotlight on Sotorasib (AMG 510) for KRAS (G12C) Positive Non-Small Cell Lung Cancer. Lung Cancer 2021, 12, 115–122. [Google Scholar]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Dooley, A.L.; Jacks, T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 2009, 4, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef]
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Brown Swigart, L.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571. [Google Scholar] [CrossRef]
- Berns, A. Cancer: The blind spot of p53. Nature 2010, 468, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Dorans, K.J.; Chung, K.M.; Robbins, R.; Tammela, T.; Gocheva, V.; Li, C.M.; Jacks, T. Clonal dynamics following p53 loss of heterozygosity in Kras-driven cancers. Nat. Commun. 2016, 7, 12685. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer. Chapter 8, pRb and Control of the Cell Cycle Clock; Garland Science: New York, NY, USA, 2014; pp. 275–329. [Google Scholar]
- Chi, X.Z.; Lee, J.W.; Lee, Y.S.; Park, I.Y.; Ito, Y.; Bae, S.C. Runx3 plays a critical role in restriction-point and defense against cellular transformation. Oncogene 2017, 36, 6884–6894. [Google Scholar] [CrossRef]
- Lee, J.-W.; Kim, D.-M.; Jang, J.-W.; Park, T.-G.; Song, S.-H.; Lee, Y.-S.; Chi, X.-Z.; Park, I.Y.; Hyun, J.-W.; Ito, Y.; et al. RUNX3 regulates cell cycle-dependent chromatin dynamics by functioning as a pioneer factor of the restriction-point. Nat. Commun. 2019, 10, 1897. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, T.G.; Bae, S.C. Involvement of RUNX and BRD Family Members in Restriction Point. Mol. Cells 2019, 42, 836–839. [Google Scholar]
- Lee, J.W.; Bae, S.C. Role of RUNX Family Members in G1 Restriction Point Regulation. Mol. Cells 2020, 43, 182. [Google Scholar] [PubMed]
- Lee, J.W.; Lee, Y.S.; Kim, M.K.; Chi, X.Z.; Kim, D.; Bae, S.C. Role of RUNX3 in Restriction Point Regulation. Cells 2023, 12, 708. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lee, Y.S.; Lee, J.M.; Ito, K.; Cinghu, S.; Kim, J.H.; Jang, J.W.; Li, Y.H.; Goh, Y.M.; Chi, X.Z.; et al. Runx3 is required for the differentiation of lung epithelial cells and suppression of lung cancer. Oncogene 2010, 29, 3349–3361. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, J.W.; Jang, J.W.; Chi, X.Z.; Kim, J.H.; Li, Y.H.; Kim, M.K.; Kim, D.M.; Choi, B.S.; Kim, E.G.; et al. Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell 2013, 24, 603–616. [Google Scholar] [CrossRef]
- Simon, A. FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 June 2021).
- Hannon-Lab. FASTX Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 30 June 2021).
- Bushnell, B. BBMap. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 30 June 2021).
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- R-Development-Core-Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, J.Y.; Song, S.H.; Kim, D.M.; Lee, J.W.; Chi, X.Z.; Ito, Y.; Bae, S.C. K-Ras-Activated Cells Can Develop into Lung Tumors When Runx3-Mediated Tumor Suppressor Pathways Are Abrogated. Mol. Cells 2020, 43, 889–897. [Google Scholar] [PubMed]
- Efeyan, A.; Serrano, M. p53, guardian of the genome and policeman of the oncogenes. Cell Cycle 2007, 6, 1006–1010. [Google Scholar] [CrossRef]
- Podsypanina, K.; Politi, K.; Beverly, L.J.; Varmus, H.E. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc. Natl. Acad. Sci. USA 2008, 105, 5242–5247. [Google Scholar] [CrossRef]
- Janne, P.A.; Gray, N.; Settleman, J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat. Rev. Drug Discov. 2009, 8, 709–723. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-Y.; Lee, J.-W.; Park, T.-G.; Han, S.-H.; Yoo, S.-Y.; Jung, K.-M.; Kim, D.-M.; Lee, O.-J.; Kim, D.; Chi, X.-Z.; et al. Runx3 Restoration Regresses K-Ras-Activated Mouse Lung Cancers and Inhibits Recurrence. Cells 2023, 12, 2438. https://doi.org/10.3390/cells12202438
Lee J-Y, Lee J-W, Park T-G, Han S-H, Yoo S-Y, Jung K-M, Kim D-M, Lee O-J, Kim D, Chi X-Z, et al. Runx3 Restoration Regresses K-Ras-Activated Mouse Lung Cancers and Inhibits Recurrence. Cells. 2023; 12(20):2438. https://doi.org/10.3390/cells12202438
Chicago/Turabian StyleLee, Ja-Yeol, Jung-Won Lee, Tae-Geun Park, Sang-Hyun Han, Seo-Yeong Yoo, Kyoung-Mi Jung, Da-Mi Kim, Ok-Jun Lee, Dohun Kim, Xin-Zi Chi, and et al. 2023. "Runx3 Restoration Regresses K-Ras-Activated Mouse Lung Cancers and Inhibits Recurrence" Cells 12, no. 20: 2438. https://doi.org/10.3390/cells12202438
APA StyleLee, J.-Y., Lee, J.-W., Park, T.-G., Han, S.-H., Yoo, S.-Y., Jung, K.-M., Kim, D.-M., Lee, O.-J., Kim, D., Chi, X.-Z., Kim, E.-G., Lee, Y.-S., & Bae, S.-C. (2023). Runx3 Restoration Regresses K-Ras-Activated Mouse Lung Cancers and Inhibits Recurrence. Cells, 12(20), 2438. https://doi.org/10.3390/cells12202438